

Abstract
Well-characterized promoters are essential tools for metabolic engineering and synthetic biology. In Streptomyces coelicolor, the native kasOp is a temporally expressed promoter strictly controlled by two regulators, ScbR and ScbR2. In this work, first, kasOp was engineered to remove a common binding site of ScbR and ScbR2 upstream of its core region, thus generating a stronger promoter, kasOp3. Second, another ScbR binding site internal to the kasOp3 core promoter region was abolished by random mutation and screening of the mutant library to obtain the strongest promoter, kasOp* (where the asterisk is used to distinguish the engineered promoter from the native promoter). The activities of kasOp* were compared with those of two known strong promoters, ermEp* and SF14p, in three Streptomyces species. kasOp* showed the highest activity at the transcription and protein levels in all three hosts. Furthermore, relative to ermEp* and SF14p, kasOp* was shown to confer the highest actinorhodin production level when used to drive the expression of actII-ORF4 in S. coelicolor. Therefore, kasOp* is a simple and well-defined strong promoter useful for gene overexpression in streptomycetes.
INTRODUCTION
The genus Streptomyces is known for its ability to produce antibiotics and for its complex morphology (1, 2). It is not only an ideal model to study bacterial differentiation but is also considered a good host for antibiotic production. In the past decades, many genetic tools have been developed for streptomycetes (3, 4). They have greatly facilitated genetic manipulations of these organisms. However, for gene expression, only a limited number of promoters, such as the constitutive promoters ermEp* (where the asterisk signifies the presence of a one-base-pair mutation) and SF14p (5, 6) and the inducible promoters tipAp and nitAp (7, 8), are available. Among these promoters, only ermEp* is widely used for the overexpression of target genes. But even ermEp* has not been completely characterized and sometimes gives undesirable results in some Streptomyces species (9).
In modern metabolic engineering and synthetic biology practices, the fine-tuning of gene expression by well-characterized promoters is necessary (10, 11). In recent years, great efforts have been made to develop useful promoters or promoter libraries in several model organisms (12–14). However, for streptomycetes, such efforts and advances are lagging. This may be due to the intricate regulatory networks of streptomycetes, which contain hundreds of regulatory proteins and dozens of sigma factors (15, 16). This regulatory complexity is reflected by the degeneracy of reported promoter sequences (17). Among the 139 Streptomyces promoters previously compiled, only about 20% showed conserved core promoter sequences similar to those recognized by Escherichia coli σ70 (18). Due to the lack of understanding of these diverse promoters in streptomycetes, few native promoters could be easily applied for controlled gene expression. The widely used ermEp*, which is a heterogenous promoter from Saccharopolyspora erythraea, has multiple −10 and −35 sites and bidirectional promoter activities (5). Such structural complexity hinders its characterization and evaluation (9, 19). Therefore, developing simple and well-characterized promoters is necessary for metabolic engineering in streptomycetes. Streptomyces coelicolor is the best genetically characterized species of streptomycetes. Among the 65 sigma factors in its genome, several have been characterized (20); for example, BldN is involved in the regulation of aerial growth (21) and WhiG is involved in spore formation and maturation (22). Other sigma factors are specifically used in response to various stresses, such as SigR, which is involved in the regulation of the oxidative stress response (23). Among them, HrdB was identified as the housekeeping sigma factor responsible for the transcription of essential genes (24). It recognizes promoters with the consensus sequence TTGACN (−35)—17 nucleotides (nt)—TAGAPuT (−10) (18). The strength of bacterial promoters is determined not only by the core −35 and −10 region but also by two flanking regions: 5′ of −35 and 3′ of the transcription start site (TSS) (25). However, the intrinsic strength of a promoter (transcription activity conferred by the core RNA polymerase in a manner independent of other transcriptional factors) is strongly correlated with the core promoter sequences which are recognized by the housekeeping or an alternative sigma factor in bacteria (26, 27). Hence, in S. coelicolor, promoters recognized by housekeeping sigma factor HrdB, which is normally highly expressed during growth, are the preferred candidates for promoter development.
According to previous research, kasO (also known as cpkO and SCO6280), encodes a SARP family regulator and is an activator of a cryptic type I polyketide synthase gene cluster responsible for coelimycin P1 production in S. coelicolor A3 (2, 28, 29). The upstream region of the kasO promoter (kasOp) spans nearly 400 bp of DNA, and its core promoter sequence is similar to the HrdB-recognized consensus sequence (30) (Fig. 1A). Our previous work indicated that kasOp is rigorously regulated by ScbR and ScbR2; the former is the γ-butyrolactone (GBL, SCB) receptor in S. coelicolor, whereas ScbR2 shows great similarity to ScbR but could not bind GBL and thus was called a “pseudo”-GBL receptor (31). kasO transcripts are undetectable during rapid growth, but the gene is sharply turned on and off in a small time window at the transition phase (2, 30). The regulatory mechanism is most likely determined by the interplay between ScbR and ScbR2 (32). The onset of kasO transcription is probably initiated by SCBs by derepression of the GBL receptor protein ScbR (30). The pseudo-γ-butyrolactone receptor ScbR2 binds the upstream region of kasOp, at a time after ScbR derepression, to shut down kasO expression, thus giving rise to the pulse-expression pattern of kasO (32). The endogenous antibiotics actinorhodin (Act) and undecylprodigiosin could also play a role in the control of kasOp expression by interaction with ScbR2 (31). These known interactions between kasOp and its regulators make it an ideal candidate for promoter engineering.
Fig 1.

Evaluation of kasOp activity in E. coli. (A) Nucleotide sequences of kasOp. The sequence is numbered on the left. The TSS is indicated by a bent arrow and bigger letters. The sequences of site OA and site OB are underlined. The putative −10 and −35 sites of kasOp are marked by dashed frames. The translation start codon of kasO is marked in gray letters. Translated amino acids are given below the nucleotide sequence. The truncated sites of kasOp1, kasOp2, kasOp3, and kasOp4 are indicated by thick arrows. (B) Growth (OD600) and bioluminescence of E. coli DH5α containing pK-Lux. RLU, relative light units. (C) Increase of kasOp activity in response to induced expression of HrdB in E. coli BL21(DE3) containing pK-Lux and pHrdB. Data are expressed as average values obtained from three independent experiments. Error bars indicate means ± standard deviations (SDs).
Strong promoters could be obtained by many approaches. A simple method is to use a shotgun clone method with native promoters from genomic DNA. The promoter SF14p was discovered this way from a fragment of Streptomyces ghanaensis phage I19 (6). Recently, synthetic promoter libraries have been adopted as a means to obtain useful promoters and many synthetic promoters have been generated this way (12, 33, 34). Seghezzi et al. constructed a promoter library mimicking Streptomyces vegetative promoters with a relatively fixed −35 region (TTGACN) and a more variable −10 box(es) (TASVDT) and obtained a collection of promoters with various strengths (12). However, none of these promoters showed higher activity than ermEp*. The reasons for this are unknown (12). Promoter engineering is another strategy to develop and obtain desired promoters (35). In this work, we chose kasOp for rational engineering. After two engineering steps, a strong promoter, kasOp*, was obtained. The performance of this promoter was evaluated in E. coli and several Streptomyces strains by different methods. Our results indicate that it is the strongest simple promoter currently reported for streptomycetes.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
The bacterial strains used in this study are listed in Table 1. E. coli strains were grown aerobically at 37°C in Luria-Bertani medium (36). For the spore suspension preparation and kanamycin resistance assay, Streptomyces coelicolor M145 and Streptomyces avermitilis NRRL8165 and their derivatives were cultivated on mannitol soy flour medium (MS) agar plates, whereas Streptomyces venezuelae strains were cultivated on a malt extract-yeast extract-maltose medium (MYM) agar plate (3). To isolate RNA and to assay XylE activity, S. coelicolor strains were cultivated in liquid minimal medium supplemented with 0.2% Casamino Acids (SMM), while S. avermitilis and S. venezuelae were cultivated in yeast extract-malt extract (YEME) medium at 28°C and 250 rpm (3, 32). For Act production in S. coelicolor, both SMM and R2YE were used (3). Antibiotics were added appropriately as follows: kanamycin at 50 μg/ml, hygromycin at 50 μg/ml, ampicillin at 50 μg/ml, and chloramphenicol at 25 μg/ml.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotypea | Reference or source |
|---|---|---|
| Strains | ||
| S. coelicolor M145 | Prototrophic derivative of S. coelicolor A3(2) | 3 |
| S. venezuelae ISP5230 | Wild type | 28 |
| S. avermitilis NRRL 8165 | Wild type | ATCC |
| S. coelicolor actII-K*OE | pK*-actII-integrated S. coelicolor M145 | This study |
| S. coelicolor actII-SOE | pS-actII-integrated S. coelicolor M145 | This study |
| S. coelicolor actII-EOE | pE-actII-integrated S. coelicolor M145 | This study |
| S. coelicolor control | pDR4-integrated S. coelicolor M145 | This study |
| E. coli JM109 | General cloning host for plasmid manipulation | Novagen |
| E. coli ET12567(pUZ8002) | Donor strain for conjugation between E. coli and Streptomycetes | 3 |
| E. coli DH5α | Host for reporter system | Novagen |
| E. coli BL21(DE3) | Host for expression plasmids with T7-derived promoter | Novagen |
| Plasmids | ||
| pCS26-Pac | Kanr, promoterless luxCDABE reporter | 33 |
| pCDFDuet-1 | Smr, containing two multiple cloning sites preceded by a T7lac promoter and ribosome binding site (rbs), CloDF13-derived CDF replicon | Novagen |
| pHrdB | Smr; insertion of hrdB gene into pCDFDuet-1 | This study |
| pK-Lux | Kanr, pCS26-Pac containing the kasOp | This study |
| pK1-Lux | Kanr, pCS26-Pac containing the kasOp1 | This study |
| pK2-Lux | Kanr, pCS26-Pac containing the kasOp2 | This study |
| pK3-Lux | Kanr, pCS26-Pac containing the kasOp3 | This study |
| pK4-Lux | Kanr, pCS26-Pac containing the kasOp4 | This study |
| pACYC184 | Cmr, Tcr, repp15A | NEB |
| pScbR2 | Cmr; for ScbR2 expression in the reporter system | 27 |
| pET23b::scbR | For recombinant expression of ScbR protein | 27 |
| pET23b::scbR2 | For recombinant expression of ScbR2 protein | 27 |
| pScbR | Cmr; for ScbR expression in the reporter system | This study |
| pDR2 | Double-reporter vector containing a xylE-neo cassette | 34 |
| pDR3 | Multiple-cloning site inserted in pDR2 | This study |
| pIJ963 | Contains the hygromycin resistance gene | Presented by K. F. Chater |
| pDR4 | Apramycin resistance gene replaced by hygromycin resistance gene in pDR3 | This study |
| pDR4-K | pDR4 inserting kasOp | This study |
| pDR4-K1 | pDR4 inserting kasOp314 | This study |
| pDR4-K* | pDR4 inserting kasOp361 (kasOp*) | This study |
| pDR4-K3 | pDR4 inserting kasOp382 | This study |
| pDR4-K4 | pDR4 inserting kasOp3154 | This study |
| pDR4-E | pDR4 inserting ermEp* | This study |
| pDR4-S | pDR4 inserting SF14p | This study |
| pK*-actII | actII-orf4 replacing xylE-neo in pDR4-K* | This study |
| pS-actII | actII-orf4 replacing xylE-neo in pDR4-KS | This study |
| pE-actII | actII-orf4 replacing xylE-neo in pDR4-KE | This study |
Cm, chloramphenicol; Kan, kanamycin; Sm, spectinomycin; Tc, tetracycline.
Evaluation of kasOp activity in E. coli.
All primers and synthetic oligonucleotides used in this work are listed in Table S1 in the supplemental material. Standard techniques for nucleic acid manipulation were used as described by Sambrook and Russell (36). The kasOp-driving Lux reporter plasmid was constructed as follows. Promoter kasOp was amplified from genomic DNA of S. coelicolor M145 by the use of primers kasOpF and kasOpR (Table S1). The PCR product was digested by XhoI and BamHI and then inserted into pCS26-Pac (37) digested with the same enzymes to get pK-Lux. The growth and bioluminescence of an E. coli DH5α transformant with pK-Lux were measured simultaneously. Growth was measured as optical density at 600 nm (OD600), and the bioluminescence was measured using the same culture and a 20/20-n single tube luminometer (Turner Biosystems). For the construction of pHrdB, hrdB was amplified with primers HrdB-F and HrdB-R (Table S1) from genomic DNA of S. coelicolor M145. The PCR product and pCDFDuet-1 were both digested by NdeI and BglII and then ligated to create pHrdB. It was used to express HrdB upon isopropyl-β-d-thiogalactopyranoside (IPTG) induction. For evaluating the effect of HrdB on kasOp activity, pHrdB and pK-Lux were transformed into E. coli BL21(DE3). Bioluminescence was measured as described above.
Serial truncation of kasOp and evaluation of kasOp variants in E. coli.
Promoters kasOp1, kasOp2, kasOp3, and kasOp4 were amplified with the corresponding forward primers kasOp1F, kasOp2F, kasOp3F, and kasOp4F and the same reverse primer, kasOpR (see Table S1 in the supplemental material), from genomic DNA of S. coelicolor M145. Similar to kasOp, they were introduced to XhoI-BamHI-cut pCS26-Pac to yield plasmids pK1-Lux, pK2-Lux, pK3-Lux, and pK4-Lux, respectively. For the construction of pScbR, an scbR gene with an introduced Shine-Dalgarno sequence was amplified from S. coelicolor genomic DNA by using a pair of primers, scbRF and scbRR (Table S1). The BamHI-digested scbR was then inserted into pACYC184 digested with EcoRV and BamHI to obtain pScbR. To analyze the repression effect of ScbR and ScbR2 on engineered kasOp3 in vivo, pScbR and the previously constructed pScbR2 (31) were transformed into DH5α bearing pK3-Lux, respectively. After 12 h of incubation, the bioluminescence of E. coli cultures was measured.
Constructing and screening of a random site OA library of kasOp3.
To construct a site OA mutant library of kasOp3, overlap extension PCR was used with the degenerated primers kasOp3nF and kasOp3nR (see Table S1 in the supplemental material). The PCR fragments were digested by XhoI and BamHI and inserted into the promoterless Lux reporter plasmid pCS26-Pac to obtain the site OA mutant library. Afterwards, the site OA mutant library was transformed into DH5α bearing pScbR. Clones that bioluminesce similarly to DH5α harboring only pK3-Lux were selected and further sequenced.
Electrophoretic mobility shift assay of engineered kasOp promoters.
His6-tagged ScbR2 and ScbR were purified from E. coli BL21(DE3) harboring pET23b::scbR2 and pET23b::ScbR, as described by Xu et al. (31). Promoters kasOp3, kasOp314, kasOp361, kasOp382, and kasOp3154 were amplified with the same pair of primers (KF and kasOpR) (see Table S1 in the supplemental material) using their corresponding templates, respectively. The subsequent binding experiments were performed using a modified gel mobility shift assay described previously (32). The DNA probe (5 ng) was incubated with various concentrations of purified ScbR2 or ScbR at 25°C for 30 min in 20 μl of buffer containing 20 mM Tris base (pH 7.5), 2 mM dithiothreitol, 5 mM MgCl2, 0.5 mg/ml calf bovine serum albumin (BSA), and 5% (vol/vol) glycerol. After incubation and electrophoresis, the nondenaturing 4% (wt/vol) polyacrylamide gels were stained with SYBR Gold nucleic acid gel stain (Invitrogen) for 30 min in TBE (89 mM Tris base, 89 mM boric acid, 1 mM EDTA, pH 8.0) buffer and photographed under a UV transilluminator using a Bio-Rad Gel Doc XR system.
Evaluation of engineered kasOp promoters in streptomycetes by a double-reporter method.
The xylE-neo double-reporter cassette was cloned into pDR2 previously in our laboratory (38). Plasmid pDR3 was derived from pDR2 by inserting the synthetic multiple cloning site MCS1 (Table S1) into the NotI-digested and blunt-ended site. Following that, plasmid pDR4 was derived from pDR3 by replacing the original apramycin resistance gene with a hygromycin resistance gene. The 1,883-bp hygromycin resistance gene was amplified from pIJ963 with a pair of primers, hygF and hygR (see Table S1 in the supplemental material). The amplified PCR products were then digested by appropriate enzymes and inserted between ApaLI and NheI sites of pDR3 to generate pDR4.
To evaluate the activities of different promoters in streptomycetes, promoters kasOp314, kasOp361, kasOp382, and kasOp3154 were all amplified with the primer pair KF and KR (see Table S1 in the supplemental material), whereas the native kasOp was amplified with the primer pair KOF and KR (Table S1). The PCR products were digested with BamHI and SpeI and then inserted into the corresponding site of pDR4 to create pDR4-K1, pDR4-K*, pDR4-K3, pDR4-K4, and pDR4-K, respectively. The five resulting plasmids were transformed into E. coli ET12567 (pUZ8002) and stably integrated into the chromosome of S. coelicolor M145 by site-specific recombination at the phage C31 attachment site (attB) to obtain the promoter-reporter strains via conjugation.
To compare the activities of promoters kasOp*, SF14p, and ermEp* in streptomycetes, SF14p and ermEp* were amplified with two pairs of primers, primer pair SF and SR and primer pair EF and ER (see Table S1 in the supplemental material). The PCR products were digested by the corresponding enzymes listed in Table S1 and inserted into pDR4 digested with the same enzymes to construct pDR4-S and pDR4-E, respectively. The two plasmids along with pDR4-K* were then introduced into S. coelicolor M145, S. venezuelae ISP5230, and S. avermitilis NRRL 8165 by E. coli/Streptomyces conjugation to obtain the respective promoter-reporter strains.
RNA isolation and real-time PCR.
The promoter-reporter strains were cultivated as described above and sampled at different time points for the S. coelicolor strains (24, 36, 48, and 60 h), S. venezuelae strains (12, 24, and 36 h), and S. avermitilis strains (48, 72, 96, and 120 h). Total RNA was isolated per a standard procedure (3). The RNA samples were then treated with RNase-free DNase (Progema) and checked by PCR to eliminate the possibility of chromosomal DNA contamination.
First-strand cDNA synthesis was carried out using 2 μg of each RNA sample and a SuperScript III cDNA synthesis kit (Invitrogen), following the manufacturer's instructions. Real-time PCR was performed using selected genes and an LC-480 II real-time PCR detection system (Roche) and an Ultra SYBR mixture (with Rox). Ten percent of the cDNA synthesis reaction mixture was used as a template for each subsequent PCR using primers KMF and KMR (see Table S1 in the supplemental material) for the detection of the kanamycin restriction gene (neo), primers hrdBFv and hrdBRv (Table S1) for the detection of the hrdB gene in S. venezuelae, and primers hrdBFc and hrdBRc (Table S1) for the detection of the hrdB gene in S. coelicolor and in S. avermitilis. Real-time quantitative PCR (qPCR) parameters were set as follows: 95°C for 10 min followed by 40 two-step amplification cycles consisting of 15 s of denaturation at 95°C and 60 s of annealing and extension at 60°C. The results were analyzed by the use of LC-480 II software v2.0.1, and the relative expression levels of target genes were normalized internally to the hrdB level. Relative transcript levels were quantified by the 2−ΔΔCT method (39) and are shown as relative fold changes. All sample assays were conducted in triplicate.
Evaluation of kanamycin resistance level and XylE activity.
About 105 spores of promoter-reporter strains were spread on plates containing 50 μg/ml, 100 μg/ml, 200 μg/ml, 400 μg/ml, 600 μg/ml, and 800 μg/ml kanamycin and 50 μg/ml hygromycin and then incubated at 28°C for 4 days. The kanamycin resistance levels were chosen in accordance with the kanamycin concentration that resulted in a plate with just no perceptible growth where the plate corresponding to the next-lowest kanamycin concentration showed poor growth.
Quantitative measurement of total catechol-2,3-dioxygenase activity (XylE) in cell extracts was performed by a method described previously (40). The XylE activity was tested at the stationary phase: 48 h for S. coelicolor, 24 h for S. venezuelae, and 96 h for S. avermitilis. The XylE activity was calculated as the rate of change in optical density at 375 nm per minute per milligram of protein.
Measuring the Act production level.
The 768-bp actII-ORF4 fragment was amplified with primers ActF and ActR (see Table S1 in the supplemental material) and digested by SpeI and KpnI. The reporter plasmids pDR4-K*, pDR4-S, and pDR4-E were cut by SpeI and KpnI to remove the xylE-neo reporter cassette and used as a backbone. The digested actII-ORF4 fragment was then joined with the three plasmid backbones mentioned above to create actII-ORF4 overexpression plasmids pK*-actII, pS-actII, and pE-actII, respectively. The three plasmids along with pDR4 were conjugated into S. coelicolor M145 as mentioned above to generate strains S. coelicolor actII-K*OE, S. coelicolor actII-SOE, S. coelicolor actII-EOE, and an S. coelicolor control, respectively.
About 4 × 108 spores of the actII-ORF4 overexpression strains and the control strain which contained pDR4 were incubated in 250-ml flasks containing 100 ml SMM at 28°C and 250 rpm for 60 h. In parallel, about 2 × 108 spores were cultured in 250-ml flasks containing 100 ml R2YE liquid medium at 28°C and 250 rpm for 48 h when Act but not Red was produced. Act production levels under both sets of growth conditions were assayed as described by Hopwood et al. (3).
RESULTS
kasOp is recognized by HrdB in E. coli.
The putative TSS of kasOp, which is 35 nucleotides (nt) upstream of the kasO translation start site, was determined previously by high-resolution S1 nuclease protection analysis (30) (Fig. 1A). Upstream of the kasO TSS, two binding sites for GBL receptor ScbR, designated sites OA and OB, were identified. Site OA lies at −15 to −33 nt overlapping the core promoter region, whereas site OB lies at −222 nt to −242 nt (30) (Fig. 1A). The pseudo-GBL receptor ScbR2 binds only the OB site (32). To evaluate kasOp's activity without influence from ScbR and ScbR2, it was inserted into the Lux reporter plasmid and transformed into E. coli DH5α. The bioluminescence curve indicated that, in contrast to the expression pattern in S. coelicolor (see Fig. S1 in the supplemental material), kasOp exhibited continuous expression in heterologous host E. coli (Fig. 1B). To verify that kasOp was recognized by HrdB, a hrdB expression plasmid was constructed and transformed into E. coli BL21(DE3) harboring the kasOp reporter plasmid pK-Lux. As shown in Fig. 1C, the bioluminescence of the resulting transformants was significantly boosted after IPTG induction of HrdB expression. The results indicate that kasOp is indeed recognized by HrdB.
Identification of engineered promoters not repressed by ScbR2.
To abolish the binding site of ScbR2 and determine the optimal length of kasOp, four 5′-truncated promoters of kasOp, designated kasOp1, kasOp2, kasOp3, and kasOp4, were amplified (Fig. 1A). The four truncated promoters were inserted into the Lux reporter plasmid and transformed into DH5α. The bioluminescence of those transformants was measured. As shown in Fig. 2A, four truncated promoters (without site OB) all displayed higher activities than the full-length promoter kasOp, especially the 97-bp kasOp3, which showed activity that was nearly 40 times higher. To confirm that the binding of ScbR2 had been abolished in vivo, plasmid pScbR2 (31), which could constitutively express ScbR2, was transformed into the DH5α bearing the kasOp3 reporter plasmid. The bioluminescence of the resulting strain was unaffected by the expression of ScbR2, thus verifying that kasOp3 was no longer controlled by ScbR2 (Fig. 2B). In contrast, in DH5α harboring both the kasOp3 reporter plasmid and ScbR expression plasmid pScbR, bioluminescence was obviously repressed (Fig. 2B), indicating that the kasOp3 was still repressed by ScbR. In contrast to the kasOp3 results, the bioluminescence of pK-Lux transformants was unaffected in the presence of the pACYC184 plasmid but was severely repressed by plasmid expressing ScbR or ScbR2 (Fig. 2B). The conclusions mentioned above were further supported by the results of an in vitro electrophoretic mobility shift assay (EMSA), which showed that ScbR2 could not bind kasOp3 whereas ScbR could still bind kasOp3 (see Fig. S2 in the supplemental material).
Fig 2.

Truncation of kasOp to remove the binding site OB and obtain the optimal upstream length. Bioluminescence levels were detected in E. coli DH5α containing various plasmid combinations. All values are in relative light units (RLU) and represent the averages of the results of at least three independent readings. Error bars indicate means ± SDs.
Identification of kasOp3 mutant promoters not repressed by ScbR.
The ScbR binding OA site overlaps the −10 and −35 regions of kasOp. Besides the core promoter region, the sequence between the −10 and −35 regions is also important for promoter activity. To abolish the binding of ScbR while preserving the strength of the promoter, a random-site OA mutant library of kasOp3 was constructed. The mutants were created by using a pair of degenerate primers and then inserted into the Lux pCS26-Pac reporter plasmid to form a promoter library. The promoter library was transformed into DH5α bearing pScbR (Fig. 3A). Facilitated by the two-plasmid reporter system, the promoter library was conveniently screened. Four kasOp3 mutants (designated kasOp314, kasOp361, kasOp382, and kasOp3154) showed strong luminescence unaffected by the presence of ScbR (Fig. 3B). The four mutants were subsequently sequenced, confirming that the OA sites were indeed altered (Fig. 3C). Furthermore, in vitro, EMSA was performed to verify that these mutants had lost the ability to bind ScbR or ScbR2 (see Fig. S3 in the supplemental material for the kasOp361 EMSA result).
Fig 3.

Reporter system for the screening of randomized kasOp3 mutants not repressed by ScbR. (A) Schematic representation of the reporter system, which bears two plasmids: a ScbR expression plasmid and a reporter plasmid. The promoter library was constructed by inserting the mutant promoters into the reporter plasmid. The bioluminescence of the reporter indicates whether the mutant promoters were regulated by ScbR. (B) Bioluminescence levels of the strains in which ScbR repression was abolished screened by the two-plasmid reporter system at the time of the stationary phase. (C) Sequences of the site OA region of native kasOp and four mutants not repressed by ScbR. Promoter kasOp361 is renamed kasOp*. RBR is the abbreviation of Random mutational Binding Region. The gray letters represent the sequence of the partial −35 and −10 region. The dashed arrows represent the palindromic sequence of the OA site.
Evaluation of the strength of different engineered promoters in Streptomycetes.
The engineering and evaluation of kasOp-based promoters mentioned above were carried out in E. coli. To evaluate their performance in streptomycetes, kasOp314, kasOp361, kasOp382, and kasOp3154 as well as the native kasOp were cloned in front of the xylE-neo double-reporter cassette and integrated into S. coelicolor M145 by conjugation. XylE activities of these transformants were monitored in SMM after 36 h of growth. The results revealed that the four engineered promoters show much higher activities than the native one (see Fig. S4 in the supplemental material). Similar to the results obtained in E. coli, kasOp361 exhibited the highest activity in S. coelicolor M145 (Fig. S4). So we chose kasOp361 for further evaluation and designated it kasOp*.
To systematically compare the strength of kasOp* with that of ermEp* and SF14p in streptomycetes, their activities were evaluated in three Streptomyces strains. These promoters were inserted into a xylE-neo reporter plasmid (pDR4) and integrated into the genomes of S. coelicolor, S. venezuelae, and S. avermitilis. First, the kanamycin resistance levels of these strains were evaluated. As shown in Table 2, kasOp* conferred much higher kanamycin resistance than ermEp* and SF14p, especially in S. venezuelae. Also, XylE (the second reporter) activity assay showed a similar trend of higher expression levels under the control of kasOp* (see Fig. S5 in the supplemental material). Furthermore, at the transcription level, real-time qPCR was performed and showed that the mRNA levels of neo were markedly higher under the control of kasOp* in all three different Streptomyces hosts during both the exponential (Fig. 4A) and stationary growth phases (Fig. 4B).
Table 2.
Kanamycin resistance levels conferred by different promoters in different Streptomyces strains
| Strain | Promoter | Growth result at indicated kanamycin concn (μg/ml)a |
|||||
|---|---|---|---|---|---|---|---|
| 50 | 100 | 200 | 400 | 600 | 800 | ||
| S. coelicolor | kasOp* | + | + | + | + | + | + |
| SF14p | + | + | + | + | − | − | |
| ermEp* | + | + | + | + | − | − | |
| S. venezuelae | kasOp* | + | + | + | + | + | + |
| SF14p | + | + | − | − | − | − | |
| ermEp* | + | + | − | − | − | − | |
| S. avermitilis | kasOp* | + | + | + | + | + | + |
| SF14p | + | + | + | + | + | − | |
| ermEp* | + | + | − | − | − | − | |
The symbols + and − indicate growth and no growth, respectively.
Fig 4.
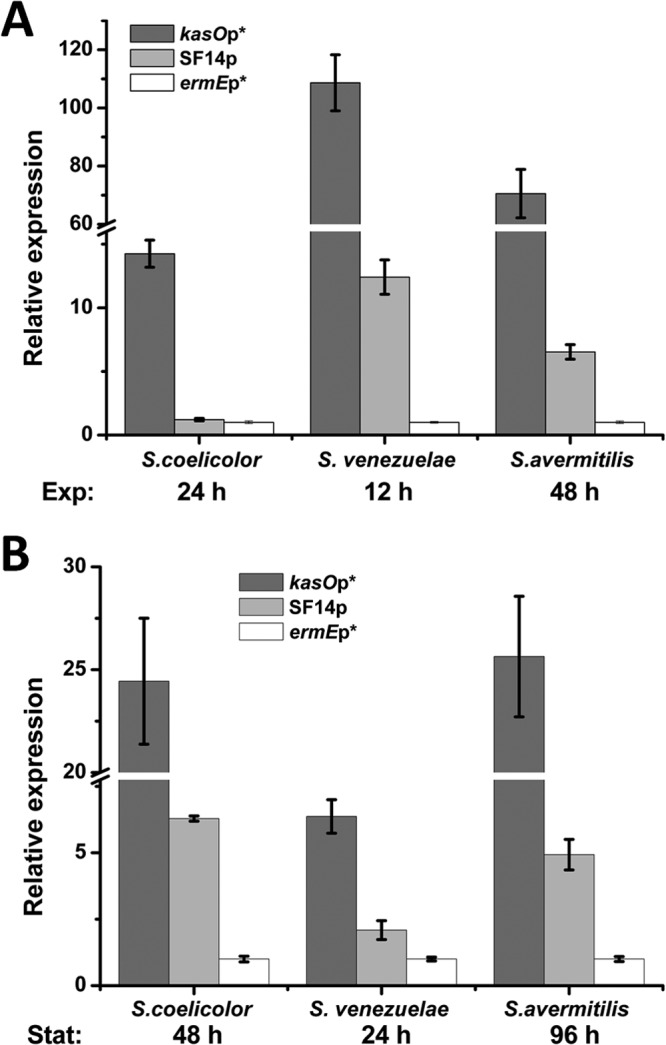
Quantitative analysis of the transcriptional profiles of xylE-neo double reporter genes in S. coelicolor M145, S. venezuelae ISP5230, and S. avermitilis NRRL8165 by real-time qPCR. Panels A and B show the relative expression levels of the neo gene in the exponential phase and stationary phase, respectively. Relative values were obtained using hrdB as the internal reference. The relative values of ermEp*-directed expression were arbitrarily assigned as 1 in the respective three strains. Error bars indicate means ± SDs.
To compare the expression dynamics of kasOp* with those of the known strong promoter SF14p and ermEp*, the relative expression levels of reporter genes under the control of kasOp*, SF14p, ermEp*, and native kasOp were profiled by qPCR during growth of S. coelicolor M145 containing the corresponding plasmids in liquid medium (see Fig. S1 in the supplemental material). As expected, the activity of native kasOp rapidly declined below the expression level of ermEp* after 24 h, whereas kasOp* constantly exhibited a higher level of expression than ermEp* and SF14p at all time points. The increases of kasOp* activity relative to kasOp activity were significant at all time points: a 1.8-fold increase at the peak level (24 h), an 85-fold increase at 12 h, a 69-fold increase at 36 h, a 112-fold increase at 48 h, and an 82-fold increase at 60 h. In addition, it is worth noting that the reporter expression level under the control of all promoters did not remain constant.
Evaluation of kasOp* performance in overexpressing Act.
Act is an aromatic polyketide produced by S. coelicolor during the stationary phase (41). The pathway-specific activator of the Act biosynthesis gene cluster, actII-ORF4, controls the onset of Act biosynthesis (42). Overexpression of actII-ORF4 on a multicopy plasmid resulted in increased Act production (42). Manipulation of regulatory genes that govern secondary metabolite production is an effective strategy to improve antibiotic production levels (43). Here, the performance of kasOp* in increasing Act production by overexpressing actII-ORF4 was tested and compared with that of SF14p and ermEp*. As shown in Fig. 5, in two different fermentation media, the highest Act production levels was detected in strain actII-K*OE, which contains the kasOp*-based overexpression plasmid. The Act production in R2YE with strain actII-K*OE was significantly higher than the levels seen with the ermEp* and SF14p overexpression strains in SMM.
Fig 5.
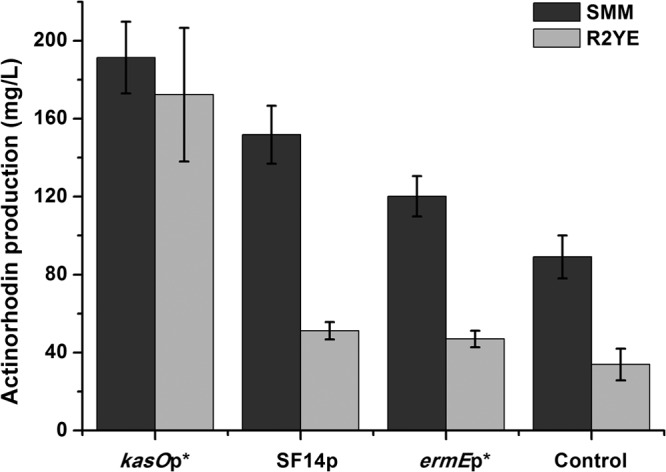
Act production levels in the S. coelicolor strains overexpressing actII-ORF4 under the control of different promoters. The dark gray bars display the production of Act in SMM at 60 h, and the light gray bars show the production of Act in R2YE medium at 48 h. Data are expressed as average values and standard deviations (SD) of the results of three parallel studies. The results are all statistically significant (P < 0.05).
DISCUSSION
In S. coelicolor, kasOp is a relatively well-characterized promoter with a core promoter region highly similar to the consensus sequence recognized by the housekeeping sigma factor HrdB (30). Previously, in vitro reconstitution of E. coli RNA polymerase core enzyme activity with Streptomyces sigma factors (including a HrdB homologue) was reported (44, 45), indicating that E. coli RNA polymerase could recognize Streptomyces sigma factors. Here, the increased bioluminescence of E. coli containing pK-Lux in response to HrdB expression also suggests that the E. coli RNA polymerase core enzyme can recognize HrdB. Two rational steps were taken to remove or abolish ScbR and ScbR2 binding sites in kasOp. The first step was removing the ScbR/ScbR2 binding site at −222 nt to −242 nt by truncating kasOp. As shown in Fig. 2A, the activity of the shortened promoters exhibited an continuous increase until the optimal length was reached. The second step was to abolish the ScbR binding site overlapping the −35-to-−10 region while still preserving promoter strength. This could not be easily accomplished because the promoter strength was also dependent on this region. Therefore, a kasOp3 mutant library with random sequences in the ScbR binding site was constructed and screened to obtain the best promoter. Several ScbR-deregulated promoters were identified; among them, kasOp* (kasOp361) showed the highest activity (Fig. 3B).
A good reporter system is extremely useful for promoter evaluation, particularly for screening a promoter library. As shown in Fig. 3A, our reporter system contains a Lux reporter plasmid and a protein expression plasmid, which has greatly facilitated the screening and identification of mutant promoters no longer repressed by ScbR. Moreover, it is applicable in the verification of other promoter-regulator interactions as long as the promoter is recognized by the E. coli RNA polymerase holoenzyme. E. coli worked well as a heterologous host for the evaluation of Streptomyces promoters recognized by the essential sigma factor HrdB in this instance: it provided a relatively clean background without the noise presented by Streptomyces. The promoter activity detected in E. coli showed good agreement with the results obtained in Streptomyces (see Fig. S4 in the supplemental material), which is probably due to the fact that the core promoter recognized by the essential sigma factor HrdB of S. coelicolor is very similar to those recognized by the housekeeping sigma factor (σ70) of E. coli. However, promoters recognized by other Streptomyces sigma factors may not be recognized in E. coli.
Characterization of the strength and host range of promoters in vivo is a critical step before application in metabolic engineering and synthetic biology (10, 11). To our knowledge, although many promoters have been well defined by S1 nuclease mapping over a time course, none has been dynamically evaluated by real-time PCR in a whole growth phase in different Streptomyces. In this work, the temporal expression profiles of three different promoters were compared in three streptomycetes strains: S. coelicolor, S. venezuelae, and S. avermitilis (Fig. 4; see also Fig. S5 in the supplemental material). The evaluations at both the transcription and protein levels demonstrated that kasOp* is the strongest promoter followed by SF14p and ermEp*. This is most likely due to the differences in promoter sequences and structures. As mentioned earlier, both ermEp* and SF14p are heterogenous promoters; thus, their core promoter sequences may not be recognized by HrdB efficiently, or they are recognized by totally different sigma factors. Additionally, both ermEp* and SF14p contain two overlapping core promoter regions (5, 6), which could result in sterical hindrance for the binding of RNA polymerase holoenzymes and hence affect transcription efficiency depending on the spacing between the two regions (46). In contrast, kasOp is an innate promoter of Streptomyces, whose core promoter with upstream and downstream regions is relatively optimized to allow efficient interaction with HrdB and RNA polymerase. It had been shown that these regions have a significant influence on transcription efficiency (47, 48). Preservation of the regions upstream of −35 and downstream of −10 in kasOp may help kasOp* perform consistently in various genetic contexts of streptomycetes. Although the optimal upstream sequence was selected in E. coli, the resulting 97-bp kasOp3 also displayed the strongest activity in S. coelicolor (see Fig. S4 in the supplemental material). The fact that kasOp3 showed higher activity than the 77-bp kasOp4 in E. coli clearly demonstrates that the 20-bp region 5′ of the −35 site is important for transcription efficiency. This region is reported to be involved in the interaction with the carboxy-terminal domain of the RNA polymerase holoenzyme α subunit (49). Another critical factor determining promoter strength is the spacer length between the −35 and −10 sites. For ermEp*, the lengths of the two spacers between two putative −35 and −10 sites are 14 and 17 bp, whereas for SF14p, the lengths are 17 and 19 bp, respectively (5, 6). They are different from the 18-bp spacer of kasOp*. So 18 bp may be the optimal spacer length for HrdB-recognized promoters, but this needs further investigation.
Moreover, the transcription of xylE-neo reporter genes driven by the three promoters kasOp*, SF14p, and ermEp* was dynamically evaluated in S. coelicolor (see Fig. S1 in the supplemental material). To our surprise, the generally accepted constitutive promoter, ermEp*, was not constantly expressed during growth according to the temporal profile of reporter gene neo. Similarly, kasOp* and SF14p also peaked at the exponential phase and declined thereafter, showing obvious growth-dependent activity (see Fig. S1 in the supplemental material). If we take an open view of the concept of constitutive expression, these results are not surprising at all. Recent work revealed that the expression of essential sigma factor HrdB in Streptomyces is also growth phase dependent (44, 50); thus, the activities of promoters recognized by HrdB should follow a similar pattern. To our knowledge, a real constitutive promoter should drive gene expression constantly throughout a growth phase; this may demand a multiplexer promoter that could be continuously transcribed by different sigma factors. The rRNA promoter(s) in S. coelicolor may be such a promoter, having four transcriptional start sites probably initiated by different sigma factor and core RNA polymerase combinations (51). Similarly, in Bacillus subtilis, the widely used constitutive promoter P43 contains two tandem core promoters recognized by σ55 during growth and by σ37 during the stationary phase, respectively (52).
Act production was greatly increased in S. coelicolor when actII-ORF4 was overexpressed by kasOp* compared to that seen with overexpression by SF14p and ermEp*, again indicating that kasOp* was a stronger promoter. However, the increase in Act production did not correlate with the increase in expression levels reflected by qPCR results, where kasOp* shows remarkable superiority relative to the other two promoters (Fig. 4 and 5). This could be due to an imbalance between metabolic flux and gene expression levels. This explanation is supported by the Act fermentation results in different media, which may supply different levels of biosynthetic precursors for Act.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Ministry of Science and Technology of China, grants 2009CB118905 and 2013CB734001.
Footnotes
Published ahead of print 17 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00985-13.
REFERENCES
- 1. Hopwood DA, Chater KF, Bibb MJ. 1995. Genetics of antibiotic production in Streptomyces coelicolor A3(2), a model streptomycete. Biotechnology 28:65–102 [DOI] [PubMed] [Google Scholar]
- 2. Nieselt K, Battke F, Herbig A, Bruheim P, Wentzel A, Jakobsen OM, Sletta H, Alam MT, Merlo ME, Moore J, Omara WA, Morrissey ER, Juarez-Hermosillo MA, Rodriguez-Garcia A, Nentwich M, Thomas L, Iqbal M, Legaie R, Gaze WH, Challis GL, Jansen RC, Dijkhuizen L, Rand DA, Wild DL, Bonin M, Reuther J, Wohlleben W, Smith MC, Burroughs NJ, Martin JF, Hodgson DA, Takano E, Breitling R, Ellingsen TE, Wellington EM. 2010. The dynamic architecture of the metabolic switch in Streptomyces coelicolor. BMC Genomics 11:10. 10.1186/1471-2164-11-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. The John Innes Foundation, Norwich, United Kingdom [Google Scholar]
- 4. Medema MH, Breitling R, Takano E. 2011. Synthetic biology in Streptomyces bacteria. Methods Enzymol. 497:485–502 [DOI] [PubMed] [Google Scholar]
- 5. Bibb MJ, Janssen GR, Ward JM. 1985. Cloning and analysis of the promoter region of the erythromycin resistance gene (ermE) of Streptomyces erythraeus. Gene 38:215–226 [DOI] [PubMed] [Google Scholar]
- 6. Labes G, Bibb M, Wohlleben W. 1997. Isolation and characterization of a strong promoter element from the Streptomyces ghanaensis phage I19 using the gentamicin resistance gene (aacC1) of Tn1696 as reporter. Microbiology 143(Pt 5):1503–1512 [DOI] [PubMed] [Google Scholar]
- 7. Takano E, White J, Thompson CJ, Bibb MJ. 1995. Construction of thiostrepton-inducible, high-copy-number expression vectors for use in Streptomyces spp. Gene 166:133–137 [DOI] [PubMed] [Google Scholar]
- 8. Herai S, Hashimoto Y, Higashibata H, Maseda H, Ikeda H, Omura S, Kobayashi M. 2004. Hyper-inducible expression system for streptomycetes. Proc. Natl. Acad. Sci. U. S. A. 101:14031–14035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou X, Wu H, Li Z, Bai L, Deng Z. 2011. Over-expression of UDP-glucose pyrophosphorylase increases validamycin A but decreases validoxylamine A production in Streptomyces hygroscopicus var. jinggangensis 5008. Metab. Eng. 13:768–776 [DOI] [PubMed] [Google Scholar]
- 10. Keasling JD. 2012. Synthetic biology and the development of tools for metabolic engineering. Metab. Eng. 14:189–195 [DOI] [PubMed] [Google Scholar]
- 11. Dehli T, Solem C, Jensen PR. 2012. Tunable promoters in synthetic and systems biology. Subcell. Biochem. 64:181–201 [DOI] [PubMed] [Google Scholar]
- 12. Seghezzi N, Amar P, Koebmann B, Jensen PR, Virolle MJ. 2011. The construction of a library of synthetic promoters revealed some specific features of strong Streptomyces promoters. Appl. Microbiol. Biotechnol. 90:615–623 [DOI] [PubMed] [Google Scholar]
- 13. Blount BA, Weenink T, Vasylechko S, Ellis T. 2012. Rational diversification of a promoter providing fine-tuned expression and orthogonal regulation for synthetic biology. PLoS One 7:e33279. 10.1371/journal.pone.0033279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hartner FS, Ruth C, Langenegger D, Johnson SN, Hyka P, Lin-Cereghino GP, Lin-Cereghino J, Kovar K, Cregg JM, Glieder A. 2008. Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res. 36:e76. 10.1093/nar/gkn369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147 [DOI] [PubMed] [Google Scholar]
- 16. Buttner MJ. 1989. RNA polymerase heterogeneity in Streptomyces coelicolor A3(2). Mol. Microbiol. 3:1653–1659 [DOI] [PubMed] [Google Scholar]
- 17. Kang JG, Hahn MY, Ishihama A, Roe JH. 1997. Identification of sigma factors for growth phase-related promoter selectivity of RNA polymerases from Streptomyces coelicolor A3(2). Nucleic Acids Res. 25:2566–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Strohl WR. 1992. Compilation and analysis of DNA sequences associated with apparent streptomycete promoters. Nucleic Acids Res. 20:961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wagner N, Osswald C, Biener R, Schwartz D. 2009. Comparative analysis of transcriptional activities of heterologous promoters in the rare actinomycete Actinoplanes friuliensis. J. Biotechnol. 142:200–204 [DOI] [PubMed] [Google Scholar]
- 20. Mazurakova V, Sevcikova B, Rezuchova B, Kormanec J. 2006. Cascade of sigma factors in streptomycetes: identification of a new extracytoplasmic function sigma factor σJ that is under the control of the stress-response sigma factor σH in Streptomyces coelicolor A3(2). Arch. Microbiol. 186:435–446 [DOI] [PubMed] [Google Scholar]
- 21. Bibb MJ, Buttner MJ. 2003. The Streptomyces coelicolor developmental transcription factor σBldN is synthesized as a proprotein. J. Bacteriol. 185:2338–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chater KF. 2001. Regulation of sporulation in Streptomyces coelicolor A3(2): a checkpoint multiplex? Curr. Opin. Microbiol. 4:667–673 [DOI] [PubMed] [Google Scholar]
- 23. Paget MS, Molle V, Cohen G, Aharonowitz Y, Buttner MJ. 2001. Defining the disulphide stress response in Streptomyces coelicolor A3(2): identification of the σR regulon. Mol. Microbiol. 42:1007–1020 [DOI] [PubMed] [Google Scholar]
- 24. Shiina T, Tanaka K, Takahashi H. 1991. Sequence of hrdB, an essential gene encoding sigma-like transcription factor of Streptomyces coelicolor A3(2): homology to principal sigma factors. Gene 107:145–148 [DOI] [PubMed] [Google Scholar]
- 25. Lanzer M, Bujard H. 1988. Promoters largely determine the efficiency of repressor action. Proc. Natl. Acad. Sci. U. S. A. 85:8973–8977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Straney R, Krah R, Menzel R. 1994. Mutations in the −10 TATAAT sequence of the gyrA promoter affect both promoter strength and sensitivity to DNA supercoiling. J. Bacteriol. 176:5999–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar A, Malloch RA, Fujita N, Smillie DA, Ishihama A, Hayward RS. 1993. The minus 35-recognition region of Escherichia coli sigma 70 is inessential for initiation of transcription at an “extended minus 10” promoter. J. Mol. Biol. 232:406–418 [DOI] [PubMed] [Google Scholar]
- 28. Pawlik K, Kotowska M, Chater KF, Kuczek K, Takano E. 2007. A cryptic type I polyketide synthase (cpk) gene cluster in Streptomyces coelicolor A3(2). Arch. Microbiol. 187:87–99 [DOI] [PubMed] [Google Scholar]
- 29. Gomez-Escribano JP, Song L, Fox DJ, Yeo V, Bibb MJ, Challis GL. 2012. Structure and biosynthesis of the unusual polyketide alkaloid coelimycin P1, a metabolic product of the cpk gene cluster of Streptomyces coelicolor M145. Chem. Sci. 3:2716–2720 [Google Scholar]
- 30. Takano E, Kinoshita H, Mersinias V, Bucca G, Hotchkiss G, Nihira T, Smith CP, Bibb M, Wohlleben W, Chater K. 2005. A bacterial hormone (the SCB1) directly controls the expression of a pathway-specific regulatory gene in the cryptic type I polyketide biosynthetic gene cluster of Streptomyces coelicolor. Mol. Microbiol. 56:465–479 [DOI] [PubMed] [Google Scholar]
- 31. Xu G, Wang J, Wang L, Tian X, Yang H, Fan K, Yang K, Tan H. 2010. “Pseudo” gamma-butyrolactone receptors respond to antibiotic signals to coordinate antibiotic biosynthesis. J. Biol. Chem. 285:27440–27448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Wang W, Wang L, Zhang G, Fan K, Tan H, Yang K. 2011. A novel role of ‘pseudo' gamma-butyrolactone receptors in controlling gamma-butyrolactone biosynthesis in Streptomyces. Mol. Microbiol. 82:236–250 [DOI] [PubMed] [Google Scholar]
- 33. Hammer K, Mijakovic I, Jensen PR. 2006. Synthetic promoter libraries—tuning of gene expression. Trends Biotechnol. 24:53–55 [DOI] [PubMed] [Google Scholar]
- 34. Rud I, Jensen PR, Naterstad K, Axelsson L. 2006. A synthetic promoter library for constitutive gene expression in Lactobacillus plantarum. Microbiology 152:1011–1019 [DOI] [PubMed] [Google Scholar]
- 35. Alper H, Fischer C, Nevoigt E, Stephanopoulos G. 2005. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. U. S. A. 102:12678–12683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sambrook J, Russell DW. 2006. The condensed protocols from molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 37. Tahlan K, Ahn SK, Sing A, Bodnaruk TD, Willems AR, Davidson AR, Nodwell JR. 2007. Initiation of actinorhodin export in Streptomyces coelicolor. Mol. Microbiol. 63:951–961 [DOI] [PubMed] [Google Scholar]
- 38. Xiang S, Li J, Yin H, Zheng J, Yang X, Wang H, Luo J, Bai H, Yang K. 2009. Application of a double-reporter-guided mutant selection method to improve clavulanic acid production in Streptomyces clavuligerus. Metab. Eng. 11:310–318 [DOI] [PubMed] [Google Scholar]
- 39. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 40. Ingram C, Brawner M, Youngman P, Westpheling J. 1989. xylE functions as an efficient reporter gene in Streptomyces spp.: use for the study of galP1, a catabolite-controlled promoter. J. Bacteriol. 171:6617–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rudd BA, Hopwood DA. 1979. Genetics of actinorhodin biosynthesis by Streptomyces coelicolor A3(2). J. Gen. Microbiol. 114:35–43 [DOI] [PubMed] [Google Scholar]
- 42. Gramajo HC, Takano E, Bibb MJ. 1993. Stationary-phase production of the antibiotic actinorhodin in Streptomyces coelicolor A3(2) is transcriptionally regulated. Mol. Microbiol. 7:837–845 [DOI] [PubMed] [Google Scholar]
- 43. Chen Y, Smanski MJ, Shen B. 2010. Improvement of secondary metabolite production in Streptomyces by manipulating pathway regulation. Appl. Microbiol. Biotechnol. 86:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Otani H, Higo A, Nanamiya H, Horinouchi S, Ohnishi Y. 2013. An alternative sigma factor governs the principal sigma factor in Streptomyces griseus. Mol. Microbiol. 87:1223–1236 [DOI] [PubMed] [Google Scholar]
- 45. Takano H, Obitsu S, Beppu T, Ueda K. 2005. Light-induced carotenogenesis in Streptomyces coelicolor A3(2): identification of an extracytoplasmic function sigma factor that directs photodependent transcription of the carotenoid biosynthesis gene cluster. J. Bacteriol. 187:1825–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shearwin KE, Callen BP, Egan JB. 2005. Transcriptional interference—a crash course. Trends Genet. 21:339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chan CL, Gross CA. 2001. The anti-initial transcribed sequence, a portable sequence that impedes promoter escape, requires σ70 for function. J. Biol. Chem. 276:38201–38209 [DOI] [PubMed] [Google Scholar]
- 48. Davis JH, Rubin AJ, Sauer RT. 2011. Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res. 39:1131–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Estrem ST, Ross W, Gaal T, Chen ZW, Niu W, Ebright RH, Gourse RL. 1999. Bacterial promoter architecture: subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase alpha subunit. Genes Dev. 13:2134–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Craney A, Hohenauer T, Xu Y, Navani NK, Li Y, Nodwell J. 2007. A synthetic luxCDABE gene cluster optimized for expression in high-GC bacteria. Nucleic Acids Res. 35:e46. 10.1093/nar/gkm086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Wezel GP, Vijgenboom E, Bosch L. 1991. A comparative study of the ribosomal RNA operons of Streptomyces coelicolor A3(2) and sequence analysis of rrnA. Nucleic Acids Res. 19:4399–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang P-Z, Doi RH. 1984. Overlapping promoters transcribed by Bacillus subtilis σ55 and σ37 RNA polymerase holoenzymes during growth and stationary phases. J. Biol. Chem. 259:8619–8625 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.