

Abstract
Ralstonia eutropha is a facultatively chemolithoautotrophic bacterium able to grow with organic substrates or H2 and CO2 under aerobic conditions. Under conditions of nutrient imbalance, R. eutropha produces copious amounts of poly[(R)-3-hydroxybutyrate] (PHB). Its ability to utilize CO2 as a sole carbon source renders it an interesting new candidate host for the production of renewable liquid transportation fuels. We engineered R. eutropha for the production of fatty acid-derived, diesel-range methyl ketones. Modifications engineered in R. eutropha included overexpression of a cytoplasmic version of the TesA thioesterase, which led to a substantial (>150-fold) increase in fatty acid titer under certain conditions. In addition, deletion of two putative β-oxidation operons and heterologous expression of three genes (the acyl coenzyme A oxidase gene from Micrococcus luteus and fadB and fadM from Escherichia coli) led to the production of 50 to 65 mg/liter of diesel-range methyl ketones under heterotrophic growth conditions and 50 to 180 mg/liter under chemolithoautotrophic growth conditions (with CO2 and H2 as the sole carbon source and electron donor, respectively). Induction of the methyl ketone pathway diverted substantial carbon flux away from PHB biosynthesis and appeared to enhance carbon flux through the pathway for biosynthesis of fatty acids, which are the precursors of methyl ketones.
INTRODUCTION
Ralstonia eutropha is an industrially relevant, facultatively chemolithoautotrophic bacterium able to grow with organic substrates or H2 and CO2 under aerobic conditions. When experiencing nutrient limitation, R. eutropha directs most of its reduced carbon flux to the synthesis of poly[(R)-3-hydroxybutyrate] (PHB), a biopolymer stored in intracellular granules. When cultivated with H2 and CO2, R. eutropha is able to form up to 61 g/liter of PHB in 48 h, representing about 70% of total cell weight (1). Mutants defective in PHB production secrete large amounts of pyruvate into the growth medium when cultured chemolithoautotrophically, suggesting that these mutants maintain a comparable carbon flux in the presence or absence of PHB biosynthesis (2, 3).
Biosynthesis of PHB under nutrient-limiting conditions in R. eutropha has been industrially exploited in the production of biodegradable plastic for more than a decade (e.g., by ICI/Zeneca, Monsanto, and Metabolix). Only recently has R. eutropha been recognized as a candidate for biofuel production from CO2, potentially producing carbon-neutral biofuels from nonphotosynthetic sources. Li and coworkers engineered a R. eutropha strain for electromicrobial conversion of CO2 to C4 and C5 alcohols (4). In their system, electricity powered the electrochemical reduction of CO2 at a cathode to produce formate, which was then converted to isobutanol and 3-methyl-1-butanol by an engineered R. eutropha strain. To meet future transportation fuel demands, higher energy-density, diesel-compatible biofuels such as medium-chain hydrocarbons will also need to be developed.
Medium-chain methyl ketones are promising targets as diesel fuel blending agents (5). Recently, Goh and coworkers demonstrated the production of over 380 mg/liter of C11 to C15 methyl ketones from glucose (0.2%) in an engineered Escherichia coli DH1 strain, which represented a 700-fold increase in titer relative to a fatty acid-overproducing strain (5). The major features of this E. coli strain were a re-engineered β-oxidation pathway designed to overproduce β-ketoacyl coenzymes A (β-ketoacyl-CoAs) and overexpression of the native thioesterase FadM. Subsequently, Park and coworkers (6) described engineering of an E. coli MG1655 strain that was able to produce 14 mg/liter of methyl ketones (2% glucose) when overexpressing a β-ketoacyl-ACP (acyl carrier protein) thioesterase (shmks2) and a β-keto acid decarboxylase (shmks1) from Solanum habrochaites (wild tomato) and when adhE, ldhA, poxB, and pta were deleted from the chromosome. Batch incubations of the best strain under optimized growth conditions with 5% glucose resulted in a methyl ketone titer of ca. 500 mg/liter.
We report here for the first time the autotrophic production of methyl ketones by engineered bacteria (R. eutropha H16). Wild-type R. eutropha does not produce detectable levels of methyl ketones natively, and intracellular concentrations of fatty acids (methyl ketone precursors) are very low in this bacterium. In this study, methyl ketone-producing strains were initially developed under heterotrophic conditions, and the best-performing strain was then tested under autotrophic conditions. R. eutropha engineered with a modified β-oxidation pathway produced up to 180 mg/liter of methyl ketones from CO2 as the sole carbon source.
MATERIALS AND METHODS
Bacterial strains, media, and cultivation.
Strains used in this study are listed in Table 1. E. coli strains (S17-λpir, JM1, JM2, and JM3), R. eutropha strains (wild-type H16, JM7, JM8 [Re2303], JM9, and JM16), and plasmids (pJM3, pJM9, and pJM20), along with their associated information (annotated GenBank-format sequence files), have been deposited in the public version of the JBEI Registry (https://public-registry.jbei.org; entries JPUB_000923 to JPUB_000934) and are physically available from the authors and/or Addgene (http://www.addgene.org) upon request.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or descriptiona | Source or reference |
|---|---|---|
| E. coli | ||
| S17-λpir | thi pro recA hsdR [RP4-2Tc::Mu-Km::Tn7] λpir Tpr Smr | 9 |
| JM1 | S17 with pJM3 | This study |
| JM2 | S17 with pJM9 | This study |
| JM3 | S17 with pJM20 | This study |
| R. eutropha | ||
| H16 DSM28 | H16 wild type | DSM |
| JM4 | H16 with pJM3 | This study |
| JM5 | H16 with pJM9 | This study |
| JM6 | H16 with pJM20 | This study |
| JM7 | H16 ΔphaCAB (mutant is deficient in PHB biosynthesis) | This study |
| Re2303 (JM8) | H16 Δ(H16_A0459-0464, H16_A1526-1531); Δbeta ox; mutant is deficient in native β-oxidation | 10 |
| JM9 | H16 Δ(H16_A0459-0464, H16_A1526-1531)ΔphaCAB; (Δbeta ox ΔphaCAB); mutant is deficient in native β-oxidation and PHB biosynthesis | This study |
| JM10 | JM7 with pJM9 | This study |
| JM11 | JM7 with pJM20 | This study |
| JM12 | Re2303 with pJM9 | This study |
| JM13 | Re2303 with pJM20 | This study |
| JM14 | JM9 with pJM9 | This study |
| JM15 | JM9 with pJM20 | This study |
| JM16 | H16 Δ(H16_A0460, H16_A1530); mutant carries deletions in both copies of fadE | This study |
| JM17 | JM16 with pJM9 | This study |
| Plasmids | ||
| pJM3 | Kmr; broad host range vector pBBR1-MCS2 containing gfp under the control of BAD promoter | This study |
| pJM9 | Kmr; broad host range vector pBBR1-MCS2 containing ′tesA thioesterase under the control of BAD promoter | This study |
| pJM20 | Kmr; broad host range vector pBBR1-MCS2 containing entire MK pathway (′tesA, fadB, Mlut_11700, and fadM) under the control of BAD promoter | This study |
Numbers with the prefix “H16_A” are R. eutropha locus tags. For the sake of brevity, “Δbeta ox” is used to refer to host strain Re2303 and “Δbeta ox ΔphaCAB” is used to refer to host strain JM9.
E. coli strains were propagated at 37°C in lysogeny broth (LB). Where necessary, medium was solidified with 1.5% (wt/vol) agar and supplemented with 50 μg/ml kanamycin. R. eutropha H16 strains were grown at 30°C in LB medium supplemented with 300 μg/ml kanamycin and 15 μg/ml gentamicin, where required, or modified minimal medium (7) containing either 2% fructose and 0.1% NH4Cl (nitrogen-sufficient conditions) or 1% fructose and 0.01% NH4Cl (nitrogen-deficient growth conditions). For studies of heterologous gene expression, R. eutropha strains were grown in 15 ml minimal medium in 30-ml glass tubes with 200-rpm agitation at 30°C. Medium was supplemented as necessary with 300 μg/ml kanamycin and 15 μg/ml gentamicin. l-Arabinose was added at a final concentration of 0.2% (wt/vol) to cultures requiring gene induction (48 h postinoculation). All liquid cultures were inoculated with single colonies originating from freshly transformed strains and were grown for up to 120 h before being harvested for analysis. The first samples for methyl ketones were taken 48 h postinduction.
Plasmids and primers.
Plasmids and primers used in this study are summarized in Table 1 and in Table S1 in the supplemental material, respectively.
Plasmid construction for heterologous expression in R. eutropha.
Cloning of genes originating from E. coli and Micrococcus luteus was accomplished by ligase-dependent cloning techniques described elsewhere (5). All plasmids constructed in this study contain the backbone from the broad-host-range vector pBBR1-MCS2 (8), including the functions required for plasmid mobilization. Target genes were amplified using primers listed in Table S1 in the supplemental material. Proper plasmid construction was confirmed by sequencing before transformation into E. coli S17-λpir (9) (Table 1). Plasmids were subsequently transferred into the desired R. eutropha strain by conjugation. Following conjugation, plasmid DNA was extracted from R. eutropha and frozen. Before each expression experiment, R. eutropha strains were transformed by electroporation to preclude E. coli contamination. Notably, transformation efficiencies were at least 150-fold higher when the plasmid to be introduced into H16 wild-type by electroporation was prepared from H16 wild-type instead of from E. coli strains. For electroporation of R. eutropha, a single colony was inoculated into LB medium and grown to mid-exponential growth phase. One milliliter of the culture was pelleted and washed with 1 ml sorbitol (pH 7.4). The cell pellet was resuspended in 35 μl sorbitol (pH 7.4) and 1 to 2 μg of plasmid DNA was added. The cell suspension was transferred in a 1-mm cuvette, and electroporation was executed with the following parameters: 0.55 kV, 25 μF, and 200 Ω. Immediately following electroporation, 400 μl of SOC medium (Life Technologies) was added and the cells were transferred into a 5-ml culture tube. Electroporated cells were recovered for 2 h before being plated onto selective medium.
Strain construction for heterologous expression in R. eutropha.
Genome modifications in R. eutropha were performed by homologous recombination as described by Brigham and coworkers (10). Briefly, in-frame deletion constructs were created by amplifying the 500-bp regions flanking the target gene. Subsequently the flanking regions were fused via a 20-bp complementary tag that was added to the 5′ end of each inner primer. The fusion product was inserted into the BamHI-digested suicide vector pGY46. pGY46 was previously used for deletion of phaC1, so the phaC1 flanking regions had to be removed by BamHI restriction digestion. The mobilizing strain, E. coli S17-λpir (9), was transformed with this plasmid. Single-crossover events were selected for on LB plates containing kanamycin and confirmed by colony PCR using two primer combinations: (i) primer X-F and primer 3-O and (ii) primer X-R and primer 5-O, where primer X-F and primer X-R bind upstream and downstream of the flanking regions, respectively. Resolution of the integrated vector by a second crossover event was performed with positive strains. These strains were grown in LB medium without selection and plated onto solid LB medium containing 10% sucrose. Deletion events were verified by the PCR using primer X-F and primer X-R.
Quantification of fructose.
Culture medium of R. eutropha wild type and mutants was collected at specified time points. The supernatant was separated from cells by centrifugation, and cell debris was removed by filtration (0.45-μm pore size). An Agilent 1200 series binary-pump liquid chromatography system equipped with refractive index and diode array detectors was used to measure fructose in the supernatant. Isocratic separation of fructose was achieved with a Bio-Rad Aminex HPX-87H column (Richmond, CA) at 60°C using a mobile phase of 4 mM H2SO4 flowing at a rate of 0.6 ml per min.
Quantification of pyruvate.
Pyruvate in filtered culture medium was quantified using a commercially available fluorescence-based assay kit (K609-100; BioVision, Mountain View, CA) according to the manufacturer's instructions.
Extraction and quantification of PHB.
PHB was extracted and quantified according to a method described by Brandl and coworkers (11). One milliliter of chloroform was added to 5 to 20 mg of lyophilized cells, followed by the addition of 0.85 ml methanol and 0.15 ml concentrated sulfuric acid. The mixture was heated for 2.5 h at 100°C. After the solution had been allowed to cool down to room temperature, samples were stored on ice and 0.5 ml of water was added. Phases were separated by centrifugation at 2,000 × g for 5 min. Subsequently, the organic phase was removed, transferred to a new tube, and dried with 200 mg of anhydrous Na2SO4. Before analysis by gas chromatography/flame ionization detection (GC/FID), samples were filtered through a 0.2-μm polyvinylidene difluoride (PVDF) syringe filter. GC/FID analysis (1-μl injection) was performed with a Thermo Scientific Focus model GC, which was equipped with a DB-WAX column (15-m length, 0.32-mm inner diameter, 0.25-μm film thickness; J&W Scientific). The GC oven temperature was programmed to hold at 80°C for 5 min before increasing to 170°C at 20°C/min. The temperature of the injection port was 250°C. Ultra-high-purity helium served as the carrier gas, flowing at 1 ml/min. External standard quantification with 3-hydroxybutyrate methyl ester standards was performed.
Extraction of fatty acids from bacterial cultures.
Fatty acids were hexane extracted from cell pellets of R. eutropha using modifications of methods described previously (5). All solid materials that came in contact with the hexane extracts during the extraction process were solvent cleaned. Fifteen-ml cultures of R. eutropha wild-type and mutants were grown in 30-ml glass tubes with PTFE (polytetrafluorethylene)-lined screw caps and sacrificed at specified time points. Tubes were centrifuged for 20 min at 4,000 rpm and 20°C. Following centrifugation, the supernatant was decanted and the pellet was resuspended in 100 μl reagent-grade water by vortex mixing. Subsequently, 1 ml of high-purity methanol and 4 ml of high-purity hexane were added to the cells, and the mixture was homogenized by vortex mixing and sonication. To determine sample-specific recovery, the hexane was amended with perdeuterated alkane standards (C10D22 and C24D50). Sonication was performed for 15 min in an ice water bath. After sonication, samples were allowed to equilibrate for 10 min at 20°C before being subjected to another centrifugation step at 3,500 rpm for 15 min. After centrifugation, the hexane layer was transferred into a 10-ml conical vial and concentrated to 100 μl under a gentle stream of N2. Fifty microliters of the concentrated hexane extract were removed, derivatized with ethereal diazomethane to generate fatty acid methyl esters (FAME) (5) and concentrated to 50 μl for GC/mass spectrometry (GC/MS) analysis.
Extraction of methyl ketones from bacterial cultures.
Methyl ketones from R. eutropha cultures were extracted either using the above-described method for extraction of fatty acids or by means of a decane overlay (5). Fifty to 100 μl of the decane overlay was used for GC/MS analysis directly or was diluted in decane before injection.
Analysis of fatty acids and methyl ketones by GC/MS.
Fatty acids (derivatized as methyl esters) and methyl ketones extracted from R. eutropha were quantified using electron-ionization GC/MS as described previously (5).
Chemolithoautotrophic fermentation.
Precultures for chemolithoautotrophic fermentation were grown by inoculating a single colony of strain JM13 (Δbeta ox background; Table 1) in 5 ml of LB supplemented with 10 μg/ml gentamicin and 300 μg/ml kanamycin and incubated at 30°C for 24 h with shaking. The culture was diluted 1:100 into 50 ml of minimal medium (12) supplemented with 10 g/liter fructose, 2 g/liter ammonium sulfate, 10 μg/ml gentamicin, and 300 μg/ml kanamycin. Cultures were incubated in 250-ml baffled flasks at 30°C with shaking (250 rpm) for 72 h. Cells were harvested via centrifugation and washed once in minimal medium lacking any exogenous carbon source.
Chemoautotrophic cultures were grown in a 1-liter bioreactor. Reactors contained 700 ml of minimal medium lacking an organic carbon source and were supplemented with 5 g/liter ammonium sulfate and 2 g/liter arabinose (for induction of the PBAD promoter). Reactors were inoculated to a starting optical density of 1.0 (9.5 × 108 CFU/ml). Decane (66.67 ml) was added to the reactor following inoculation. A blend of hydrogen-oxygen-carbon dioxide (80:4:16) was introduced into the reactor at a flow rate of 0.1 l/min. The reactor was incubated at 30°C with constant stirring (200 rpm). Samples of the culture medium as well as the decane layer were removed periodically using sterile glass Pasteur pipettes and were analyzed as described above.
Detection of CO2 in batch cultures by the respiration activity monitoring system (RAMOS).
The general set-up and operation of the RAMOS system (Kuhner, Switzerland) have been described previously (13). Experiments were performed as follows. Eight 250-ml RAMOS flasks were filled with 40 ml of fructose minimal medium and equilibrated overnight. Following equilibration, a sample of the culture medium was taken for fructose analysis. Subsequently, four of the flasks were inoculated with 4 ml of either a preadapted JM13 (Δbeta ox background; Table 1) or JM15 (Δbeta ox ΔphaCAB background) culture and incubated at 30°C and 100 rpm. Sixty hours postinoculation, half of the RAMOS cultures were induced with 0.2% l-arabinose. All cultures were overlaid with 2.5 ml of decane at this point. Cultures were sampled for determination of optical density and fructose every 24 h postinoculation. Decane was sampled for methyl ketone analysis every 24 h postinduction. The experiment was terminated 156 h postinoculation and all culture cell pellets were collected for PHB analysis. During fermentation, a CO2 measuring cycle was continuously repeated; the measuring cycle consisted of a 240-min measuring phase and a 10-min purging phase.
RESULTS
Expression of ′tesA in R. eutropha led to enhanced production of fatty acids and methyl ketones under heterotrophic growth conditions.
Overproduction of fatty acids in E. coli was previously achieved by expressing a cytoplasmically directed version of the tesA thioesterase (′tesA) (14). Overexpression of ′tesA in a ΔfadE mutant, deficient in β-oxidation, was shown to produce 1.1 g/liter of free fatty acids in E. coli (14). Based on these findings, and the fact that methyl ketones derive from fatty acids, we attempted to build fatty acid-overproducing host strains for the production of methyl ketones in R. eutropha. Several strains heterologously expressing ′tesA were tested for fatty acid production: H16 wild-type (strain JM5; Table 1), a ΔfadE double mutant (strain JM17; Table 1), and Δbeta ox and Δbeta ox ΔphaCAB strains (JM12 and JM14, respectively; Table 1). These strains were cultivated for up to 96 h in either the presence or absence of a decane overlay.
Expression of ′tesA in R. eutropha wild-type (strain JM5) and a fadE double mutant host (JM17) led to the production of 10 mg/liter of fatty acids when the strains were cultivated in LB medium in the absence of a decane overlay. The fatty acids were detected in the cell pellets, and no fatty acids were found in the supernatant of these cultures. Trace amounts (55 μg/liter) of fatty acids were detected in a control strain expressing green fluorescent protein (GFP) (strain JM4). Thus, expression of ′tesA in wild-type H16 led to a 180-fold improvement in fatty acid production compared to the GFP control strain. Myristate and palmitate (14:0 and 16:0, respectively) were the major fatty acids produced by these strains (i.e., JM5 and JM17), with lesser amounts of palmitoleate and oleate (16:1 and 18:1, respectively). Notably, in-frame deletions of both fadE copies in R. eutropha did not increase fatty acid production, unlike results reported for E. coli (14). To ensure that β-oxidation was in fact eliminated, we tested ′tesA expression in a Δbeta ox mutant background (strain Re2303) that had been previously shown to be incapable of metabolizing oleic acid (10). Additionally, we created a Δbeta ox ΔphaCAB mutant strain (strain JM9) that was deficient in PHB synthesis, postulating that acetyl-CoA not being used for PHB biosynthesis might instead feed into fatty acid biosynthesis. Fatty acid production of ′tesA-overexpressing strains JM12 (Δbeta ox mutant) and JM14 (Δbeta ox ΔphaCAB mutant) was then compared to production under uninduced conditions and in wild-type H16. Under minimal medium conditions (2% fructose), Δbeta ox mutant strains expressing ′tesA (strain JM12) produced only 0.001 mg/liter of total fatty acids, which is comparable to fatty acid production when ′tesA expression was not induced (Fig. 1) and in wild-type H16 (data not shown). In contrast, expression of ′tesA in a Δbeta ox ΔphaCAB mutant strain (JM14) produced 0.017 mg/liter of total fatty acids. Thus, deletion of the PHB biosynthesis pathway contributed to a 17-fold increase in fatty acid titer compared to the uninduced strain or wild-type H16. Myristate and palmitate (14:0 and 16:0, respectively) were the only fatty acids produced under minimal medium conditions in strain JM14 (Δbeta ox ΔphaCAB mutant). Palmitate and stearate (16:0 and 18:0, respectively) were the two fatty acids detected in the cell pellets of strain JM12 (Δbeta ox mutant). While testing strains JM12 and JM14 for fatty acid overproduction, we observed that both strains also synthesized medium-chain methyl ketones (Fig. 1); ca. 0.5 mg/liter methyl ketones was detected in the cell pellets of these strains.
FIG 1.
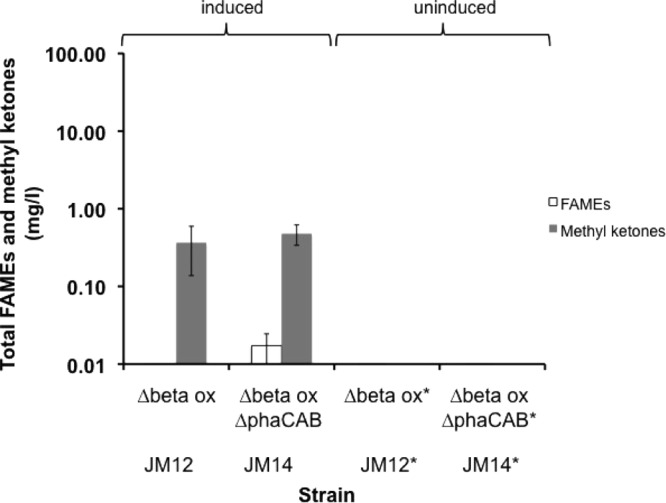
Fatty acid and methyl ketone production in R. eutropha strains expressing ′tesA. Titers of fatty acids (extracted from the cell pellet and derivatized as FAME) and methyl ketones (accumulated in the decane overlay) are shown for two fructose-grown R. eutropha mutant strains expressing ′tesA (mutants with β-oxidation and/or phaCAB chromosomal deletions; Table 1). Asterisks indicate strains that were not induced with arabinose. Values are averages for at least three independent biological replicates, and error bars represent one standard deviation at 96 h postinduction.
Re-engineering of β-oxidation in R. eutropha produced methyl ketones under heterotrophic growth conditions.
We observed that R. eutropha produced methyl ketones when ′tesA was overexpressed in both a β-oxidation-deficient host (strain JM12) and a β-oxidation- and PHB-deficient host (strain JM14). To further exploit this ability and increase methyl ketone titers, we modified R. eutropha based on a strategy described for the production of methyl ketones from glucose in E. coli (5). This strategy involved the following: (i) replacing the native β-oxidation pathway in R. eutropha with an overexpressed, heterologous, truncated version of β-oxidation in which the soluble and high-specific-activity acyl-CoA oxidase from M. luteus replaces FadE (acyl-CoA dehydrogenase), the E. coli version of FadB (which converts trans-2-enoyl-CoAs to β-ketoacyl-CoAs) is overexpressed, and fadA is deleted, all of which was designed to facilitate the overproduction of β-ketoacyl-CoAs, and (ii) overexpressing the E. coli thioesterase FadM, thereby converting β-ketoacyl-CoAs to β-keto acids, which can then spontaneously decarboxylate to form methyl ketones.
Of the four genetic backgrounds hosting the heterologous methyl ketone pathway, the highest methyl ketone titer was observed in strain JM13 (Δbeta ox mutant; Table 1) at 96 h postinduction (Fig. 2). This strain produced >50 mg/liter of total methyl ketones under heterotrophic growth conditions with fructose as the carbon source. The Δbeta ox ΔphaCAB mutant strain (strain JM15; Table 1) reached similar concentrations, whereas the wild-type (strain JM6) and the ΔphaCAB (strain JM11) hosts produced significantly lower concentrations of methyl ketones (<2 mg/liter). Thus, deletion of two putative β-oxidation operons in R. eutropha, both of which included fadA, was crucial for higher production of methyl ketones in this microorganism. Notably, almost all of the methyl ketone production occurred during a period of insignificant growth (48 to 96 h postinduction; Fig. 2). Methyl ketones were not detectable in any of these strains when the methyl ketone pathway was not induced (Fig. 2). In accordance with the fatty acid distributions observed in R. eutropha Δbeta ox ΔphaCAB mutants (JM14), the major methyl ketones detected in this host strain expressing the methyl ketone pathway (JM15) were 13:0 and 15:0 methyl ketones (derived, respectively, from 14:0 and 16:0 fatty acids) (Fig. 3). Minor amounts of 15:1 methyl ketones were observed (derived from 16:1 fatty acids). Unexpectedly, 13:0 and 15:0 methyl ketones were also the major methyl ketones and 15:1 the minor methyl ketone detected in Δbeta ox mutant strain JM13. This is at least partially in disagreement with the observed fatty acid distribution of this mutant strain (JM12), as the major fatty acids observed were palmitate and stearate (16:0 and 18:0 fatty acids). As shown in Fig. 3, wild-type (strain JM6) and ΔphaCAB (strain JM11) mutant backgrounds showed similar methyl ketone distributions. In these two low-titer strains, 35% of total methyl ketones were represented by 13:0 and 65% by 15:0 methyl ketones. The proportions of 13:0 and 15:1 methyl ketones were higher in the better producing strains (with β-oxidation knockouts).
FIG 2.

Heterotrophic methyl ketone production in R. eutropha. Methyl ketone titers are shown for four fructose-grown R. eutropha strains expressing the methyl ketone pathway (wild type and mutants with β-oxidation and/or phaCAB chromosomal deletions; Table 1). Asterisks indicate strains that were not induced with arabinose. For methyl ketones, values are averages for at least three independent biological replicates and error bars represent one standard deviation. For optical density, values are averages for at least three independent biological replicates.
FIG 3.

Distribution of 13:0, 15:0, and 15:1 methyl ketones produced by the R. eutropha strains represented in Fig. 2. Data are average methyl ketone distributions of three biological replicates at 96 h postinduction.
The methyl ketone pathway diverts carbon flux from the PHB pathway.
Biosynthesis of PHB from acetyl coenzyme A (acetyl-CoA) in R. eutropha is enabled by enzymes encoded by the phaCAB operon. R. eutropha produces PHB under conditions of nutrient imbalance when an excess of carbon is available and one or more essential growth factors, such as nitrogen or O2, are limited. We wanted to reroute carbon (specifically, acetyl-CoA) flux from PHB biosynthesis to fatty acid/methyl ketone biosynthesis by deleting the phaCAB operon. However, as reported earlier, deleting PHB biosynthesis in the wild-type H16 and Δbeta ox mutant strains expressing the methyl ketone pathway did not lead to increased methyl ketone production (Fig. 2). While deletion of PHB biosynthesis did not lead to improved methyl ketone production under the conditions tested, assessments of PHB production indicated that induction of the methyl ketone pathway did divert substantial carbon flux away from PHB biosynthesis (in strains with intact phaCAB). Specifically, we found that, when the methyl ketone pathway was induced, the percentage of fructose carbon allocated to PHB decreased from 9.4% to 2.1% in the wild-type background (strain JM6) and from 9.4% to 1.3% in the Δbeta ox mutant background (strain JM13) (Table 2). Under both induced and uninduced conditions, PHB production was negligible in ΔphaCAB control strains tested, namely, strains JM11 and JM15 (Table 2).
TABLE 2.
PHB production in R. eutropha strains bearing the methyl ketone pathway under induced and uninduced heterotrophic conditions (30-ml glass tube experiments)
| Straina | PHB yield (% fructose C consumed)b |
|
|---|---|---|
| Induced | Uninduced | |
| WT (JM6) | 2.1 | 9.4 |
| ΔphaCAB (JM11) | 0 | 0 |
| Δbeta ox (JM13) | 1.3 | 9.4 |
| Δbeta ox ΔphaCAB (JM15) | 0 | 0 |
All strains carried the MK pathway (Table 1).
Data represent a pooled sample of five independent cultures.
As the observed methyl ketone titers cannot account for the magnitude of the carbon flux diverted from PHB by induction of the methyl ketone pathway, we hypothesized that the carbon may be diverted to mineralization of fructose to CO2. To test this hypothesis, a RAMOS study was conducted with the Δbeta ox (strain JM13) and Δbeta ox ΔphaCAB (strain JM15) hosts bearing the methyl ketone pathway. Over the entire incubation period, CO2, fructose, and methyl ketone concentrations were determined in all samples and O2 was confirmed to be present. Methyl ketone concentrations in these shake flask cultures were comparable to those previously determined in 30-ml glass tubes (data not shown). As before, we observed a diversion of carbon flux from PHB biosynthesis under induced conditions (Table 3). Specifically, the Δbeta ox mutant (strain JM13) had 7% less fructose carbon utilized for PHB synthesis under induced conditions (Table 3). However, CO2 constituted less than 1% of fructose carbon consumed for both strains under induced conditions and thus could account for only a small portion of the carbon diverted from PHB. Similarly, methyl ketones accounted for a relatively small portion of fructose carbon (2.2%) (Table 3). Additionally, we have determined that pyruvate accounts for a negligible portion of the carbon diverted from PHB biosynthesis; no pyruvate was detected in the culture medium after incubation of strain JM13 (see Fig. S1 in the supplemental material). Also, acetate was not detected in these samples at concentrations greater than 5 μM. In summary, the fate of the diverted PHB carbon is not fully accounted for and merits further investigation.
TABLE 3.
PHB, CO2, and methyl ketone production in R. eutropha strains bearing the methyl ketone pathway under induced and uninduced heterotrophic conditions (RAMOS experiments)
| Conditions and straina | Carbon distribution (% of fructose C consumed)b |
||
|---|---|---|---|
| PHB | CO2 | MK | |
| Induced conditions | |||
| Δbeta ox (JM13) | 4 ± 0.73 | 0.1 | 2.2 ± 0.05 |
| Δbeta ox ΔphaCAB (JM15) | 0.3 ± 0.03 | 0.5 | 3.5 ± 0.66 |
| Uninduced conditions | |||
| Δbeta ox | 11 ± 0.77 | 0 | 0 |
| Δbeta ox ΔphaCAB | 0.1 | 0 | 0 |
All strains carried the MK pathway (Table 1).
Data are averages for two independent cultures ± standard deviations (standard deviations were not calculated for values near the detection limit).
Engineered R. eutropha produces methyl ketones autotrophically from hydrogen and carbon dioxide.
The ultimate goal of this study was to produce methyl ketones from CO2 as the sole carbon source. We chose the Δbeta ox mutant expressing the methyl ketone pathway (strain JM13) to investigate autotrophic production, as this strain had the best titer under heterotrophic conditions.
Chemolithoautotrophic cultures were inoculated with preadapted cells into minimal medium without an organic carbon source at an initial optical density of 1. l-Arabinose and methyl ketone concentrations as well as optical density were determined every 24 h postinduction. l-Arabinose concentrations remained constant over the entire fermentation period (<1.5% relative standard deviation for l-arabinose concentrations from the time of induction until the end of incubation in multiple experiments), confirming that R. eutropha was unable to utilize this sugar and, thus, that CO2 was the only available carbon source present in the culture. Figure 4 shows a time course of methyl ketone production under chemolithoautotrophic conditions. The methyl ketone concentration increased from undetectable levels at induction to ∼50 to 70 mg/liter at 48 h postinduction. In other experiments conducted under autotrophic conditions, methyl ketone titers were as high as 180 mg/liter (at OD values comparable to those shown in Fig. 4; see Fig. S2 in the supplemental material). The composition of methyl ketones produced under chemolithoautotrophic growth conditions was very similar to that observed under heterotrophic growth conditions (data not shown). Thus, R. eutropha engineered to overproduce methyl ketones is able to generate titers under chemolithoautotrophic conditions that are comparable to (or greater than) those observed under heterotrophic conditions.
FIG 4.

Autotrophic methyl ketone production in R. eutropha strain JM13 (β-oxidation mutant) under a H2-O2-CO2 atmosphere (80:4:16). Data for two biological replicates are shown.
DISCUSSION
Production of fatty acid-based methyl ketones from glucose was first shown in an engineered E. coli DH1 strain at titers of 380 mg/liter (5). Our data revealed that similar genetic modifications introduced into R. eutropha H16 led to autotrophic production of methyl ketones at concentrations of ∼50 to 180 mg/liter. Considering the relatively high energetic demands of carbon fixation, it is perhaps surprising that production of methyl ketones by R. eutropha strain JM13 under autotrophic conditions was comparable to that under heterotrophic conditions (Fig. 2 and 4). However, the lower titers of methyl ketones under heterotrophic growth conditions can be partially attributed to the low optical densities of the cultures (Fig. 2). Under heterotrophic growth conditions these cultures reached optical densities of ∼1, whereas the autotrophic cultures reached optical densities of ∼2.
A key feature of this engineered methyl ketone pathway in both R. eutropha and E. coli is the enhancement of early steps of β-oxidation (here catalyzed by acyl-CoA oxidase and FadB), allowing for production of β-ketoacyl-CoAs, and elimination of the final (FadA-catalyzed) reaction of β-oxidation, allowing β-ketoacyl-CoAs to be diverted (via FadM and spontaneous decarboxylation) to methyl ketones rather than acyl-CoAs that are two carbons shorter than the acyl-CoAs entering the cycle. In this context, one can rationalize the large improvement in methyl ketone titers in R. eutropha strains with native β-oxidation knocked out (strains JM13 and JM15 versus JM6 and JM11; Fig. 2): the deleted β-oxidation operons include fadA (5), which must be removed to truncate β-oxidation at β-ketoacyl-CoA.
Although fatty acids are precursors for methyl ketones, the relationship between fatty acid production and methyl ketone production is subtle. For example, deletion of the PHB biosynthesis pathway resulted in increased fatty acid production (Fig. 1) but comparable methyl ketone production, either with or without the heterologous methyl ketone pathway (Fig. 2 and 1, respectively). Nonetheless, our data strongly suggest that expression of the heterologous methyl ketone pathway greatly increases flux through the fatty acid biosynthesis pathway. To illustrate, in Δβ-oxidation (JM13) and Δβ-oxidation and ΔphaCAB (JM15) strains expressing the methyl ketone pathway, methyl ketones accounted for 2 to 4% of the fructose carbon consumed. However, in the analogous strains expressing ′tesA but lacking the rest of the heterologous methyl ketone pathway (JM12 and JM14, respectively), we found that free fatty acids accounted for only ∼0.00016% of the fructose carbon consumed. As methyl ketones derive from fatty acids, it appears that expression of the methyl ketone pathway substantially increases flux through the fatty acid biosynthetic pathway (assuming that the amount of fatty acids converted to phospholipids in strains with and without the methyl ketone pathway does not differ by several orders of magnitude).
Contrary to our expectations, deletion of PHB biosynthesis in R. eutropha did not improve methyl ketone production. Although we were unsuccessful in diverting acetyl-CoA toward methyl ketone production in PHB-negative strains, induction of the methyl ketone pathway in PHB-positive strains led to substantial diversion of carbon flux away from PHB biosynthesis (Tables 2 and 3). While we demonstrated that the fate of the diverted carbon is primarily not methyl ketones, CO2, acetate, or pyruvate, the disposition of this carbon is not currently known. A better understanding of metabolic flux in methyl ketone-producing R. eutropha strains, and use of that information for engineering refinements, should allow us to substantially increase methyl ketone titers and enhance the viability of chemolithoautotrophic biofuel production.
Supplementary Material
ACKNOWLEDGMENTS
We thank Chris J. Brigham for providing us with the Re2303 mutant strain. We also thank the A. J. Sinskey laboratory (MIT) for general technical advice. We are grateful to Ee-Been Goh for technical advice and helpful discussions, and we thank Veronica Teixeira Benites for technical assistance and strain maintenance.
This work was supported by funding from DOE's ARPA-E Electrofuels Program to Lawrence Berkeley National Laboratory under contract DE-AC02-05CH11231 and performed at the Joint BioEnergy Institute.
Footnotes
Published ahead of print 17 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00973-13.
REFERENCES
- 1.Ishizaki A, Tanaka K, Taga N. 2001. Microbial production of poly-d-3-hydroxybutyrate from CO2. Appl. Microbiol. Biotechnol. 57:6–12 [DOI] [PubMed] [Google Scholar]
- 2.Cook A, Schlegel H. 1978. Metabolite concentrations in Alcaligenes eutrophus H16 and a mutant defective in poly-β-hydroxybutyrate synthesis. Arch. Microbiol. 119:231–235 [Google Scholar]
- 3.Steinbüchel A, Schlegel HG. 1989. Excretion of pyruvate by mutants of Alcaligenes eutrophus, which are impaired in the accumulation of poly(β-hydroxybutyric acid) (PHB), under conditions permitting synthesis of PHB. Appl. Microbiol. Biotechnol. 31:168–175 [Google Scholar]
- 4.Li H, Opgenorth PH, Wernick DG, Rogers S, Wu TY, Higashide W, Malati P, Huo YX, Cho KM, Liao JC. 2012. Integrated electromicrobial conversion of CO2 to higher alcohols. Science 335:1596. [DOI] [PubMed] [Google Scholar]
- 5.Goh EB, Baidoo EE, Keasling JD, Beller HR. 2012. Engineering of bacterial methyl ketone synthesis for biofuels. Appl. Environ. Microbiol. 78:70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park J, Rodriguez-Moya M, Li M, Pichersky E, San KY, Gonzalez R. 2012. Synthesis of methyl ketones by metabolically engineered Escherichia coli. J. Ind. Microbiol. Biotechnol. 39:1703–1712 [DOI] [PubMed] [Google Scholar]
- 7.Peoples OP, Sinskey AJ. 1989. Poly-beta-hydroxybutyrate (PHB) biosynthesis in Alcaligenes eutrophus H16. Identification and characterization of the PHB polymerase gene (phbC). J. Biol. Chem. 264:15298–15303 [PubMed] [Google Scholar]
- 8.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, 2nd, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176 [DOI] [PubMed] [Google Scholar]
- 9.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1:784–791 [Google Scholar]
- 10.Brigham CJ, Budde CF, Holder JW, Zeng Q, Mahan AE, Rha C, Sinskey AJ. 2010. Elucidation of beta-oxidation pathways in Ralstonia eutropha H16 by examination of global gene expression. J. Bacteriol. 192:5454–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandl H, Gross RA, Lenz RW, Fuller RC. 1988. Pseudomonas oleovorans as a source of poly(beta-hydroxyalkanoates) for potential applications as biodegradable polyesters. Appl. Environ. Microbiol. 54:1977–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuster E, Schlegel HG. 1967. Chemolithotrophic growth of Hydrogenomonas H16 in a chemostat with electrolytic production of oxygen and hydrogen. Arch. Mikrobiol. 58:380–409 [PubMed] [Google Scholar]
- 13.Anderlei T, Zang W, Papaspyrou M, Büchs J. 2004. Online respiration activity measurement (OTR, CTR, RQ) in shake flasks. Biochem. Eng. J. 17:187–194 [Google Scholar]
- 14.Steen EJ, Kang Y, Bokinsky G, Hu Z, Schirmer A, McClure A, Del Cardayre SB, Keasling JD. 2010. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463:559–562 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.