

Abstract
Lactobacillus plantarum is a lactic acid bacterium able to degrade tannins by the subsequent action of tannase and gallate decarboxylase enzymes. The gene encoding tannase had previously been identified, whereas the gene encoding gallate decarboxylase is unknown. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of gallic-acid induced L. plantarum extracts showed a 54-kDa protein which was absent in the uninduced cells. This protein was identified as Lp_2945, putatively annotated UbiD. Homology searches identified ubiD-like genes located within three-gene operons which encoded the three subunits of nonoxidative aromatic acid decarboxylases. L. plantarum is the only bacterium in which the lpdC (lp_2945) gene and the lpdB and lpdD (lp_0271 and lp_0272) genes are separated in the chromosome. Combination of extracts from recombinant Escherichia coli cells expressing the lpdB, lpdC, and lpdC genes demonstrated that LpdC is the only protein required to yield gallate decarboxylase activity. However, the disruption of these genes in L. plantarum revealed that the lpdB and lpdC gene products are essential for gallate decarboxylase activity. Similar to L. plantarum tannase, which exhibited activity only in esters derived from gallic and protocatechuic acids, purified His6-LpdC protein from E. coli showed decarboxylase activity against gallic and protocatechuic acids. In contrast to the tannase activity, gallate decarboxylase activity is widely present among lactic acid bacteria. This study constitutes the first genetic characterization of a gallate decarboxylase enzyme and provides new insights into the role of the different subunits of bacterial nonoxidative aromatic acid decarboxylases.
INTRODUCTION
Vegetable tannins are present in a variety of plants utilized as food and feed. High tannin concentrations are found in nearly every part of the plant, such as the bark, wood, leaf, fruit, root, and seed. Tannins also widely occur in common foodstuffs, such as pomegranate, banana, strawberry, grape, cashew nut, and hazelnut. Drinks like wine and tea also contain these phenolic compounds (1). Tannins have been described to exhibit opposing health effects (2). They are beneficial to health due to their chemopreventive activities against carcinogenesis and mutagenesis. However, tannins are considered nutritionally undesirable because of their ability to bind to proteins to form indigestible complexes and to chelate heavy metals, and they may be involved in cancer formation and hepatotoxicity (2).
Tannase (tannin acyl hydrolase, EC 3.1.1.20) catalyzes the hydrolysis of ester linkages in hydrolyzable tannins. The products of hydrolysis are glucose and gallic acid. In addition to esters, gallic acid can be found in plants in the free state or in the form of ethers (e.g., syringic acid and other lignins constituents), being a major pollutant present in the wastewater generated in processes involving the boiling of cork and in food manufacturing industries. Gallic acid and its derivatives are used in industry as antioxidants (3).
Although gallic acid is widely distributed in nature, it is easily oxidized at neutral or alkaline pH, at which point it becomes a product difficult for bacteria to use as a carbon source for growth. In fact, only bacteria of the genus Pseudomonas have been reported to be able to utilize free gallic acid as the sole carbon and energy source under aerobic conditions (4). The aerobic metabolism of gallic acid usually starts with a direct ring-cleavage reaction and formation of the central intermediate 4-oxalomesaconic acid, which then undergoes hydration to 4-carboxy-4-hydroxy-2-oxoadipic acid and aldol cleavage to oxaloacetic and pyruvic acids (5). In addition to microorganisms that use gallic acid as the sole carbon and energy source, there are also microorganisms that nonoxidatively decarboxylate gallic acid but do not possess appropriate mechanisms to further degrade the pyrogallol produced by this dead-end pathway. Strains of the species Pantoea agglomerans (6), Enterococcus faecalis (7), Klebsiella pneumoniae (7), Streptococcus gallolyticus (8), and Lactobacillus plantarum (9–11) were described to decarboxylate gallic acid to pyrogallol, without further metabolism. Even though several gallate decarboxylases, mainly from anaerobic sources, have been described, most of these enzymes have not been purified due to their instability. Gallate decarboxylases from E. faecalis and P. agglomerans are inducible enzymes which, due to their oxygen sensitivity, were extremely unstable when they were purified (6, 7). So far, none of these gallate decarboxylase enzymes has been genetically identified or characterized.
L. plantarum is a lactic acid bacterial species that is most frequently encountered in the fermentation of plant materials where tannins are abundant. These plant fermentations include several food and feed products, e.g., olives, grape must, and a variety of vegetable fermentation products. Among food lactic acid bacteria, strains from the L. plantarum group are the only ones which possess tannase activity (12–14). The proposed biochemical pathway for the degradation of tannins by L. plantarum implies the action of a tannase and a gallate decarboxylase to decarboxylate the gallic acid formed by tannase action (9, 10). Of the genes involved in tannin degradation in L. plantarum, only the gene encoding tannase has been identified so far (15), and the gene encoding the gallate decarboxylase enzyme involved in this degradation remains unknown. In this work, the genes involved in L. plantarum gallate decarboxylation have been identified. For the first time, a gallate decarboxylase enzyme has been molecularly identified and characterized. In addition, our results provide new important insights into bacterial nonoxidative aromatic acid decarboxylases.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
L. plantarum WCFS1, used throughout this study, was kindly provided by Michiel Kleerebezem (NIZO Food Research, The Netherlands). This strain is a colony isolate of L. plantarum NCIMB 8826, which was isolated from human saliva. L. plantarum WCFS1 derivative strains, lactic acid bacteria, and the Escherichia coli strains used in this study are described in Table 1. Escherichia coli JW2308-4 (CGSC 9853), which has a deletion in the ubiX gene, was generously provided by the E. coli Genetic Stock Center.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description/relevant marker(s)a | Source or referenceb |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| DH5α | F− ϕ80dlacZΔM15 Δ(lacIZYA-argF) recA1 gyrA endA1 relA1 supE44 hsdR17 | Clontech |
| BL21(DE3) | E. coli B F− dcm ompT hsdSB(rB− mB−), gal λ (DE3) | Novagen |
| JW2308-4 (CGSC 9853) | Δ(araD-araB)567 ΔlacZ4787 (::rrnB-3) rph-1 λ− Δ(rhaD-rhaB)568 ΔubiX732(del)::kan | 31 |
| Lactobacillus plantarum | ||
| ATCC 14917T | Wild-type strain | CECT |
| WCFS1 | Wild-type strain | M. Kleerebezem |
| WCFS1ΔlpdB | WCFS1 derivative, ΔlpdB | This study |
| WCFS1ΔlpdC | WCFS1 derivative, ΔlpdC | This study |
| WCFS1ΔlpdD | WCFS1 derivative, ΔlpdD | This study |
| Other | ||
| Enterococcus faecium CECT 410T | Wild-type strain | CECT |
| Enterococcus faecium CECT 4102 | Wild-type strain | CECT |
| Lactobacillus brevis CECT 4121T | Wild-type strain | CECT |
| Lactobacillus brevis CECT 5354 | Wild-type strain | CECT |
| Lactobacillus hilgardii RM62 | Wild-type strain | 42 |
| Lactobacillus hilgardii RM63 | Wild-type strain | 42 |
| Lactobacillus pentosus DSM 20314T | Wild-type strain | DSMZ |
| Lactobacillus sakei DSM 15831T | Wild-type strain | DSMZ |
| Leuconostoc mesenteroides CECT 219T | Wild-type strain | CECT |
| Oenococcus oeni CECT 4100T | Wild-type strain | CECT |
| Oenococcus oeni RM1 | Wild-type strain | 42 |
| Pediococcus pentosaceus CECT 4695T | Wild-type strain | CECT |
| Streptococcus gallolyticus UCN34 | Wild-type strain | P. Glaser |
| Plasmids | ||
| pIN-III-A3 | Expression vector for producing proteins in E. coli | 30 |
| pURI3 | Expression vector for producing His-tagged proteins in E. coli; pT7-7 derivative, Ampr | 42 |
| pIN-lpdB | pIN-III-A3 carrying lpdB | This study |
| pIN-lpdC | pIN-III-A3 carrying lpdC | This study |
| pIN-lpdD | pIN-III-A3 carrying lpdD | This study |
| pURI3-lpdB | pURI3-carrying lpdB | This study |
| pURI3-lpdC | pURI3-carrying lpdC | This study |
| pUCE191 | L. plantarum integrative vector, pUC19 derivative, Ampr Emr Lmr | 18 |
| pUCE191-lpdB | pUCE191 carrying an internal fragment of lpdB | This study |
| pUCE191-lpdC | pUCE191 carrying an internal fragment of lpdC | This study |
| pUCE191-lpdD | pUCE191 carrying an internal fragment of lpdD | This study |
Ampr, ampicillin resistance; Emr, erythromycin resistance; Cmr, chloramphenicol resistance.
CECT, Spanish Type Culture Collection; DSMZ, German Collection of Microorganisms and Cell Cultures.
Lactic acid bacteria were routinely grown on MRS broth. When gallate activity was assayed and in order to avoid the presence of phenolic compounds included in nondefined medium, bacteria were cultivated in a modified basal medium, RPM, described previously (16). The sterilized modified basal medium was supplemented with filter-sterilized gallic or protocatechuic acid at a 3 mM final concentration. Where appropriate, erythromycin was added to the culture medium at 10 μg/ml. The inoculated media were incubated at 30°C in the dark, without shaking, for 7 to 10 days. Incubated media with cells and without phenolic compound were used as controls. The phenolic compounds present in the supernatants were extracted by a standard protocol involving two extraction steps with one-third of the reaction mixture volume of ethyl acetate.
E. coli cells were routinely grown in LB medium (17) at 37°C with agitation. The E. coli JW2308-4 strain was grown in medium containing kanamycin at 30 μg/ml. E. coli transformant cells harboring recombinant plasmids were selected on LB medium plates supplemented with 100 μg of ampicillin or 200 μg of erythromycin per ml.
Molecular biology techniques.
Standard molecular biology techniques were performed as previously described (17). Plasmid DNA was extracted by a High Pure plasmid isolation kit (Roche). PCR products were purified with a QIAquick gel extraction kit (Qiagen). All cloned inserts and DNA fragments were confirmed by DNA sequencing with fluorescently labeled dideoxynucleotide terminators and AmpliTaq FS DNA polymerase (Applied Biosystems) in an ABI Prism 377 automated DNA sequencer (Applied Biosystems). Transformation of E. coli cells was carried out by using the RbCl method (17). Oligonucleotides were purchased from Eurofins MWG Operon (Ebersberg, Germany) (see Table S1 in the supplemental material). Proteins were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Coomassie stained according to standard protocols (17). The protein concentration was determined by using a Bio-Rad protein assay.
L. plantarum cell extract preparations.
To identify the protein involved in gallate decarboxylation, cell extracts containing all soluble proteins were prepared. L. plantarum WCFS1 was grown in MRS medium at 30°C until exponential phase (optical density at 600 nm [OD600], 0.6). The culture was induced by adding 3 mM gallic acid and incubated for 1 h at 30°C. Uninduced culture was prepared as a control. In the experiments with L. plantarum lpd knockout mutants, the mutants were grown in MRS medium containing 10 μg/ml erythromycin until the OD600 was 0.6. In all experiments, after incubation the cells were harvested by centrifugation, washed three times with phosphate buffer (50 mM, pH 6.5), and subsequently resuspended in the same buffer for cell rupture. Bacterial cells were disintegrated three times by using a French press at a 1,100-lb/in2 pressure (Amicon French pressure cell; SLM Instruments). The cell disruption steps were carried out on ice to ensure the low-temperature conditions required for most enzymes. The disintegrated cell suspension was centrifuged at 12,000 × g for 20 min at 4°C in order to sediment the cell debris. The supernatant containing the soluble proteins was filtered aseptically using sterile filters with a pore size of 0.2 μm (Sarstedt, Germany).
Gallate decarboxylase assay.
The gallate decarboxylase activity of the bacterial cultures was assayed by growing the strains in RPM or LB medium supplemented with 3 mM gallic acid in the dark for several days. The phenolic compounds present in the supernatant were extracted with ethyl acetate as described above. In E. coli cultures expressing recombinant L. plantarum lpd proteins, gallate decarboxylase activity was assayed in cell extracts incubated at 37°C for 1 h in the presence of 3 mM gallic acid. Similarly, cell extracts were used to assay the gallate decarboxylase activity of L. plantarum lpd knockout mutants incubated for 18 h in 3 mM gallic acid. Recombinant His-tagged proteins purified in 50 mM phosphate buffer, pH 6.5, containing 300 mM NaCl, 150 mM imidazole, 1 mM dithiothreitol (DTT), and 50 mM Na2S2O5 were assayed for gallate decarboxylase activity (6). Eluted purified proteins (100 μg) were incubated at 37°C in the presence of 3 mM gallic acid and 50 mM l-ascorbate for 1 h.
The reaction products were extracted with ethyl acetate and analyzed by high-pressure liquid chromatography (HPLC) with a diode array detector. A Thermo chromatograph (Thermo Electron Corporation, Waltham, MA) equipped with a P400 SpectraSystem pump, an AS3000 autosampler, and a UV6000LP photodiode array detector was used. A gradient of solvent A (water and acetic acid, 98:2, vol/vol) and solvent B (water, acetonitrile, and acetic acid, 78:20:2, vol/vol/vol) was applied to a reversed-phase Nova-pack C18 cartridge (25 cm by 4.0 mm [inner diameter]; particle size, 4.6 μm) at room temperature as follows: 0 to 55 min, 0 to 80% solvent B, linear, 1.1 ml/min; 55 to 57 min, 80 to 90% solvent B, linear, 1.2 ml/min; 57 to 70 min, 90 to 95% solvent B, isocratic, 1.2 ml/min; 70 to 80 min, 95 to 100% solvent B, linear, 1.2 ml/min; 80 to 90 min, 100% linear, 1.2 ml/min; 100 to 120 min, washing with methanol 1.0 ml/min; and reequilibration of the column under initial gradient conditions. Detection was performed by scanning from 220 to 380 nm. Samples were injected onto the cartridge in duplicate, after being filtered through a 0.45-μm-pore-size polyvinylidene difluoride filter. The identification of degradation compounds was carried out by comparing the retention times and spectral data of each peak with those of standards from commercial suppliers.
Construction of L. plantarum lpd knockout mutant strains.
To ascertain the participation of particular lpd genes in gallate decarboxylase activity, insertion-duplication mutagenesis was employed. Internal fragments from the lpdB (lp_0271), lpdC (lp_2945), and lpdD (lp_0272) genes were cloned into the pUCE191 suicide vector. Plasmid pUCE191 is a pUCE19 derivative that does not replicate in L. plantarum but expresses lincomycin resistance upon integration into the lactobacillus genome. Plasmid pUCE191 was constructed by introducing the LnR gene from plasmid pFB9 into pUC19 (18). When pUCE191 and its derivatives were used as donor DNA, L. plantarum transformants were selected by plating with 10 μg/ml erythromycin and E. coli transformants were selected by plating with ampicillin at 100 μg/ml. Plasmids pUCE191-lpdB, pUCE191-lpdC, and pUCE191-lpdD (Table 1), constructed in E. coli, were used to transform L. plantarum WCFS1 competent cells by electroporation (19). Knockout mutants were selected by plating in MRS medium plates containing erythromycin. The correct insertion of the donor pUCE191 derivative plasmid into the L. plantarum WCFS1 chromosome was checked by PCR analysis using primers flanking the target region combined with vector-specific primers (primers 1131 and 1233 for lpdB, 388 and 1224 for lpdC, and 1109 and 1224 for lpdD) (see Table S1 in the supplemental material).
Expression of the lpd genes in E. coli and purification of the His6-tagged lpdB and lpdC recombinant enzymes.
The lpdB, lpdC, and lpdD genes from L. plantarum WCFS1 were PCR amplified by using HS Prime Start DNA polymerase (TaKaRa) and primer pairs 455 and 390 (lpdB), 454 and 388 (lpdC), and 1141 and 1142 (lpdD). The purified PCR products were inserted into the pIN-III-A3 vector following the restriction enzyme- and ligation-free cloning strategy described previously (20, 21). The procedure used to clone lpdB and lpdC containing an amino-terminal His6 tag into the pURI3 vector was essentially the same as that described above for the native protein, but primers 389 and 390 and primers 387 and 388, respectively, were used. The pURI3 vector is a pT7-7 derivative that was used to overproduce recombinant proteins containing a six-histidine tag at their N termini (20).
Cells carrying the recombinant plasmids were grown at 37°C in LB medium containing ampicillin (100 μg/ml), until they reached an optical density at 600 nm of 0.4, and induced by adding IPTG (isopropyl-β-d-thiogalactopyranoside; final concentration, 0.4 mM). After induction, the cells were grown at 22°C for 20 h and collected by centrifugation. Cells were resuspended in phosphate buffer (50 mM, pH 6.5). Crude extracts were prepared by French press lysis of the cell suspension (three times at 1,100 lb/in2). The insoluble fraction of the lysate was removed by centrifugation at 47,000 × g for 30 min at 4°C, and the supernatant was filtered through a 0.2-μm-pore-size filter.
For purification of the recombinant His-tagged LpdB and LpdC proteins, the cultures were similarly prepared but the cells were resuspended in 50 mM phosphate buffer, pH 6.0, containing 30 mg/ml FeSO4, 1 mM DTT, 1 mM l-ascorbate, and 50 mM Na2S2O5 (6). The supernatants were applied to a Talon Superflow resin (Clontech) equilibrated with the buffer described above containing 0.3 M NaCl and 10 mM imidazole to improve the interaction specificity in the affinity chromatography step. The bound enzymes were eluted using 150 mM imidazole in the same buffer. The purity of the enzymes was determined by SDS-PAGE in Tris-glycine buffer. Fractions containing the His6-tagged protein were pooled and analyzed for gallate decarboxylase activity. Eluted purified LpdB and LpdC proteins (100 μg) were incubated at 37°C in the presence of 3 mM gallic acid for 1 h.
In vitro protein-protein cross-linking experiments.
The LpdB-LpdC interaction was assayed by glutaraldehyde cross-linking. For glutaraldehyde treatment, LpdB and LpdC proteins, at concentrations ranging from 2 and 10 μM, in 50 mM sodium phosphate, 300 mM NaCl, and 150 mM imidazole buffer (pH 7) were treated with glutaraldehyde solution (0.1, 0.2, and 0.5 μM) for 20 min at room temperature. As a control, a similar glutaraldehyde treatment was applied to monomeric lysozyme. The reactions were stopped by adding Laemmli sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, β-mercaptoethanol, 10% glycerol) containing 0.005% bromophenol blue. Samples were separated by SDS-PAGE and revealed by Coomassie blue staining.
PCR detection of gallate decarboxylase.
Genes encoding LpdB and LpdC were amplified by PCR using chromosomal DNA from several lactic acid bacterial strains. Amplifications were performed by using degenerate primers 450 and 451 and degenerate primers 448 and 449 to amplify lpdB and lpdC, respectively. These degenerate primers were based on the well-conserved domains of the B and C proteins. The reactions were performed in a Personal thermocycler (Eppendorf), using 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 1 min, and extension at 72°C for 30 s. The expected sizes of the amplicons were 158 bp (subunit B) and 300 bp (subunit C). Amplified fragments were resolved on 2% agarose gels.
Protein identification via MS.
The protein band was excised manually and then digested automatically using a Proteineer DP protein digestion station (Bruker-Daltonics, Bremen, Germany). The protocol described by Shevchenko et al. (22) was used for trypsin digestion. The digestion was analyzed in an Ultraflex time-of-flight mass spectrometer (MS; Bruker-Daltonics) equipped with a LIFT-MS/MS device. Spectra were acquired in the positive-ion mode at a 50-Hz laser frequency, and 100 to 1000 individual spectra were averaged. Automated analysis of mass data was performed using flexAnalysis software (Bruker-Daltonics). Matrix-assisted laser desorption ionization MS and MS/MS data were combined through the BioTools program (Bruker-Daltonics) to search a nonredundant protein database (NCBInr Swiss-Prot) using Mascot software (Matrix Science, London, United Kingdom).
Sequence data analyses.
A homology search with finished and unfinished microbial genome databases was performed with the TBLAST algorithm at the National Center for Biotechnology Information server (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi). Multiple alignments were made using the Clustal Omega program (http://www.ebi.ac.uk/Tools/msa/clustalo/) on the EBI site, after retrieval of sequences from the GenBank and Swiss-Prot databases.
RESULTS
Identification of the enzyme responsible for gallate decarboxylation in L. plantarum WCFS1.
In order to know if gallate decarboxylase is an inducible enzyme, mid-exponential-phase L. plantarum WCFS1 cultures were incubated in MRS medium containing glucose as the carbon source, with or without the addition of 3 mM gallic acid for 1 h at 30°C. Cell extracts prepared from these cultures were tested for activity on gallic acid. The extract from the control culture (grown in the absence of gallic acid) did not show decarboxylase activity. However, the extract from the culture grown in the presence of gallic acid for 1 h was able to decarboxylate the gallic acid present in the reaction mixture (data not shown). Similar to the result for p-coumaric acid decarboxylase previously described in L. plantarum (23, 24), this result might indicate that gallate decarboxylase is an inducible enzyme.
Cell extracts were resolved by SDS-PAGE in order to find proteins overproduced from the induced culture (Fig. 1). The only difference clearly detected was in the gallate-induced culture and consisted of a protein band of approximately 50 kDa which was absent in the uninduced sample. The overproduced protein was excised from the gel, and its identification was done by in-gel trypsin and chymotrypsin digestion and subsequent mass spectrometry analyses. The result obtained unambiguously identified the protein as 3-octaprenyl-4-hydroxybenzoate carboxy-lyase or UbiD (NP_786283), encoded by the lp_2945 locus.
Fig 1.
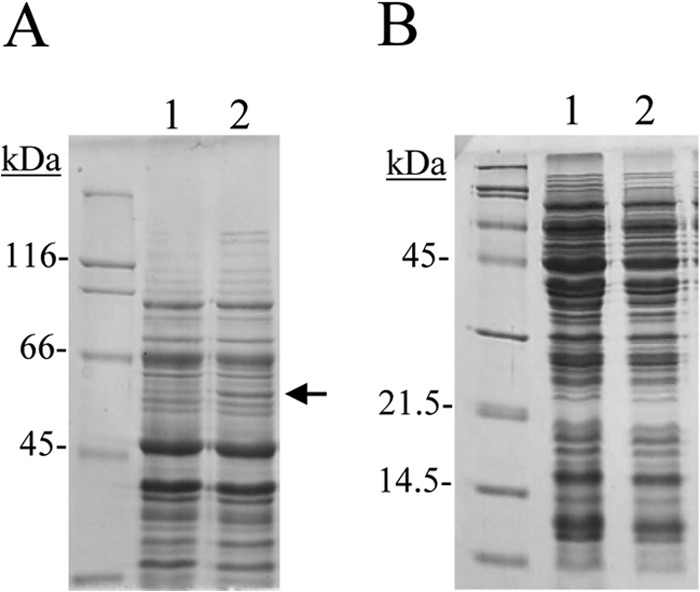
SDS-PAGE analysis of protein extracts from L. plantarum WCFS1 grown in the presence of 3 mM gallic acid. Lanes 1, uninduced cell extracts; lanes 2, extracts induced with gallic acid for 1 h. The arrow indicates the induced protein. The 8% (A) and 15% (B) gels were stained with Coomassie blue. Molecular mass markers are located on the left (SDS-PAGE standards; Bio-Rad).
In E. coli, the UbiD protein is involved in the biosynthesis of ubiquinone (or coenzyme Q) (25). Ubiquinone plays an essential role in aerobic respiratory electron transfer for energy generation. The biosynthesis of ubiquinone involved at least nine reactions. In one of these reactions, the 3-octaprenyl-4-hydroxybenzoate is decarboxylated to 2-octaprenylphenol by the enzyme 3-octaprenyl-4-hydroxybenzoate decarboxylase. There are two isofunctional enzymes in E. coli K-12, UbiD and UbiX, which catalyze this reaction (26). Their amino acid sequences share no similarity. UbiX, a 21-kDa protein, may require a flavin nucleotide as a cofactor, whereas UbiD is a 55-kDa protein requiring divalent metal for activity. Of the two enzymes, UbiD accounts for almost 80% of the total activity (27). However, it has been described that L. plantarum does not produce mena- or ubiquinones, as it needs the exogenous addition of at least menaquinones for heme-assisted respiration (28). Therefore, it is unlikely that UbiD is involved in the self-production of ubiquinones by L. plantarum. The precise biochemical function of the L. plantarum protein annotated UbiD (Lp_2945) is likely to be a gallate decarboxylase.
Genes encoding L. plantarum gallate decarboxylase.
Recent studies indicated that ubiD-like genes in many prokaryotes are located within operons that encode partner proteins, including proteins homologous to UbiX, which are required to decarboxylate a variety of hydroxyarylic or aromatic acids. Bacterial nonoxidative, reversible multisubunit hydroxyarylic acid decarboxylases/phenol carboxylases are encoded by three clustered genes (genes encoding the B, C, and D subunits) (29). The corresponding genes from Sedimentibacter hydroxybenzoicus, Bacillus subtilis, Streptomyces sp. strain D7, E. coli O157:H7, K. pneumoniae, and Salmonella enterica serovar Typhimurium were cloned and expressed in E. coli and shown to code for proteins exhibiting nonoxidative aromatic acid decarboxylase activity (29). Database searches revealed the existence of different gene organizations among these decarboxylases: the BCD gene arrangement (such as in Enterobacter cloacae, Pantoea ananatis, B. subtilis, and a Streptomyces sp.), CDB (S. hydroxybenzoicus, Clostridium sp. strain D5, and Olsenella uli DSM 7084), CDB in several lactic acid bacteria (Oenococcus oeni, Lactobacillus brevis, and Enterococcus faecium), and DBC in S. gallolyticus (Fig. 2). Surprisingly, the genes that putatively encoded gallate decarboxylase, the lpd (from Lactobacillus plantarum decarboxylase, to be consistent with the existing nomenclature of genes encoding aromatic acid decarboxylases) genes, are not close only in the genome of L. plantarum. The gene encoding the C subunit, lpdC or lp_2945, which was overproduced in the presence of gallate, is located close to the gene encoding tannase (tanLp1 or lp_2956). However, the genes encoding the B (lp_0271) and D (lp_0272) subunits are located more than 1 Mb apart in the L. plantarum genome. This unusual gene organization could indicate a different catalytic mechanism of L. plantarum gallate decarboxylase.
Fig 2.

Genetic organization of the L. plantarum WCFS1 chromosomal region containing the genes encoding gallate decarboxylase (GenBank accession no. NC_004567, positions 243093 to 252815 and 2618290 to 2635122). The genetic organization from several bacterial nonoxidative aromatic acid decarboxylases (B, C, and D subunits) is also represented, such as those of Enterobacter cloacae subsp. cloacae ATCC 13047 (GenBank accession no. NC_014121, positions c/4181733 to 4189439 [where “c/” indicates positions on a complementary strand]), Pantoea ananatis LMG20103 (GenBank accession no. NC_013956, positions 1814209 to 1820127), Bacillus subtilis BSn5 (GenBank accession no. NC_014976, positions 2624421 to 2630650), Streptomyces sp. strain e14 (GenBank accession no. NZ_ACUR00000000, positions c/7010912 to 7018020), Sedimentibacter hydroxybenzoicus (GenBank accession no. AF128880, positions 1 to 5796), Clostridium sp. D5 (GenBank accession no. NZ_ADBG00000000, positions 5032836 to 5040967), Olsenella uli DSM 7084 (GenBank accession no. NC_014363, positions c/1673164 to 1680578), Oenococcus oeni PSU-1 (GenBank accession no. NC_008528, positions c/1104544 to 1114618), Lactobacillus brevis ATCC 367 (GenBank accession no. NC_008497, positions c/1942529 to 1951428), Enterococcus faecium DO (GenBank accession no. NC_017960, positions 1147858 to 1155656), Lactobacillus rhamnosus HN001 (GenBank accession no. NZ_ABWJ00000000, positions 1905984 to 1914300), and Streptococcus gallolyticus UCN34 (GenBank accession no. NC_013798, positions 1699210 to 1708634). Arrows indicate genes. Genes having putative identical functions are represented by identical shading. The genes having brick-like shading encode putative LysR-type transcriptional regulators. Genes coding for putative tannase proteins are represented by black arrows.
Pyrogallol production by recombinant E. coli cells harboring lpdB, lpdC, and lpdD genes.
In order to know the catalytic subunits involved in gallate decarboxylation, the lpd genes were individually cloned into the expression vector pIN-III-A3 under the control of the lppp-5 and lacPO promoters, which can be induced to high levels with IPTG (30). The correct sequence of the recombinant plasmids pIN-lpdB, pIN-lpdC, and pIN-lpdD was verified by DNA sequencing.
Cell extracts were prepared from E. coli DH5α cells harboring the recombinant plasmids. The extracts were used to detect the presence of hyperproduced proteins. Control cells containing the pIN-III-A3 vector plasmid alone did not show expression over the time course analyzed (overnight), whereas expression of additional 54-, 21-, and 15-kDa proteins was apparent in DH5α cells harboring pIN-lpdC, pIN-lpdB, and pIN-lpdD, respectively (Fig. 3A and B). These molecular masses are in good agreement with the relative molecular masses deduced from the nucleotide sequences of the lpdC, lpdB, and lpdD genes.
Fig 3.

Pyrogallol production by cell extracts from recombinant E. coli cells harboring lpdB, lpdC, and lpdD genes. (A and B) SDS-PAGE analysis of cell extracts of IPTG-induced cultures of E. coli DH5α bearing recombinant pIN-III-A3 plasmids for LpdB, LpdC, and LpdD protein production. (A) LpdC, LpdB, and LpdD production (15% gel). Lane 1, E. coli DH5α(pIN-III-A3); lane 2, E. coli DH5α(pIN-lpdC); lane 3, E. coli DH5α(pIN-lpdB); lane 4, E. coli DH5α(pIN-lpdD). (B) LpdC production (12% gel). Lane 1, E. coli DH5α(pIN-III-A3); lane 2, E. coli DH5α(pIN-lpdC). The arrows indicate the overproduced proteins. The gels were stained with Coomassie blue. Molecular mass markers are located on the left. (C) Gallate decarboxylase activity of E. coli DH5α cell extracts harboring lpdB, lpdC, and lpdD genes. HPLC chromatograms of E. coli cell extracts, at the same total protein concentration, incubated in 3 mM gallic acid for 1 h: pIN-III-A3 (control), pIN-III-A3 plus pIN-lpdB (LpdB), pIN-III-A3 plus pIN-lpdD (LpdD), pIN-III-A3 plus pIN-lpdB and pIN-lpdD (LpdB LpdD), pIN-III-A3 plus pIN-lpdC (LpdC), pIN-III-A3 plus pIN-lpdB and pIN-lpdC (LpdB LpdC), pIN-III-A3 plus pIN-lpdC and pIN-lpdD (LpdC LpdD), and pIN-lpdB plus pIN-lpdC and pIN-lpdD (LpdB LpdC LpdD). The gallic acid (GA) and pyrogallol (P) detected are indicated. Chromatograms were recorded at 280 nm. mAU, milli-absorbance units.
Extracts from E. coli cells carrying the pIN-III-A3, pIN-lpdC, pIN-lpdB, and pIN-lpdD plasmids adjusted to the same protein concentration were assayed for gallate decarboxylase activity. Reactions with mixtures containing the same total protein concentration were done by mixing these extracts in all possible combinations, e.g., in mixtures containing the B, C, or D subunit individually; mixtures containing subunits B and C, B and D, and C and D; and finally, mixtures simultaneously containing the three different lpd subunits. Reactions were carried out for 1 h, and after that, the phenolic compounds present in the reaction mixtures were extracted by ethyl acetate and analyzed by HPLC.
Figure 3C shows that all the reactions with mixtures containing LpdC were able to partially decarboxylate gallic acid to a similar extent. In contrast, in reactions in which LpdC was absent from the reaction mixture, gallic acid was not metabolized. These results indicate the involvement of LpdC in the catalysis of decarboxylation, and in addition, they suggest that LpdC is the only subunit required to yield gallate decarboxylase activity.
From the results obtained using E. coli extracts, the possibility that in E. coli the missing subunits can be replaced by other E. coli proteins, e.g., UbiX, to yield enzyme activity could not be excluded. As explained before, UbiX is involved in ubiquinone biosynthesis and catalyzes the reaction of 3-octaprenyl-4-hydroxybenzoate to 2-octaprenylphenol. In order to avoid the presence of a functional E. coli UbiX protein in the extracts, plasmid pIN-lpdC was introduced into E. coli JW2308-4 (CGSC 9853), a UbiX-defective mutant (31). Gallate decarboxylase activity was assayed in cell extracts prepared from E. coli JW2308-4 harboring pIN-lpdC. The results indicated that, similar to E. coli DH5α extracts, pyrogallol was produced from gallic acid in the presence of LpdC even in the absence of a functional E. coli UbiX protein (see Fig. S1 in the supplemental material). Therefore, the possibility of the involvement of E. coli UbiX in the gallate decarboxylase activity observed in E. coli extracts could be discarded.
Enzymatic activity of purified of His6-LpdC.
To further investigate the decarboxylase activity of LpdC and the possible involvement of lpdB or lpdB-like genes, His-tagged proteins LpdB and LpdC were constructed for expression and purification from E. coli. His6-tagged proteins were cloned into E. coli BL21(DE3), overproduced, and purified by a one-step affinity procedure as described in Materials and Methods. Only the His6-LpdC protein showed gallate decarboxylase activity. However, even though His6-LpdC was produced at a high yield, it presented low gallate decarboxylase activity, as only degradation similar to that of the cell extracts was observed (Fig. 4).
Fig 4.

Purification and enzymatic activity of recombinant L. plantarum LpdC protein. (A) SDS-PAGE analysis of the expression and purification of the His6-tagged LpdC. Results of analysis of soluble cell extracts of IPTG-induced E. coli BL21(DE3)(pURI3) (lane 1) or E. coli BL21(DE3)(pURI3-lpdC) (lane 2), flowthrough from the affinity resin (lane 3), or fractions eluted after His affinity resin (lanes 4 to 6) are shown. The 8% gel was stained with Coomassie blue. Molecular mass markers are located on the left (SDS-PAGE standards; Bio-Rad). (B) Gallate decarboxylase activity of purified His6-LpdC protein. An HPLC chromatogram of LpdC (100 μg) incubated in 3 mM gallic acid for 1 h (LpdC) is shown. A chromatogram without protein (control) is also shown. The gallic acid (GA) and pyrogallol (P) detected are indicated. Chromatograms were recorded at 280 nm.
In order to know the involvement of LpdB on activity, reactions were performed by using both purified His6-LpdB and His6-LpdC proteins. Decarboxylase activity did not increase due to the presence of the LpdB subunit (see Fig. S2 in the supplemental material). Moreover, in vitro protein-protein cross-linking experiments using glutaraldehyde did not show physical interaction between the two proteins (see Fig. S3 in the supplemental material).
Effects of disruption of lpdB, lpdC, and lpdD on gallate decarboxylation by L. plantarum.
To corroborate previous results, insertion-duplication mutagenesis was employed to construct L. plantarum mutants with knockouts in the lpdB, lpdC, and lpdD genes. The correct insertion of the donor plasmids into the L. plantarum WCFS1 chromosome was verified by PCR. Unexpectedly, when these mutants were grown in the presence of gallic acid, the lpdB and lpdC mutants were unable to decarboxylate it to pyrogallol (Fig. 5), suggesting the participation of both proteins, LpdB and LpdC, in the decarboxylation of this hydroxybenzoic acid. Taking into account the probable operonic organization of the lpdBD genes (Fig. 2), the lpdB mutant could, in fact, be a double-knockout mutant. The lpdD mutant was the only mutant able to decarboxylate gallic acid. Similar to gallic acid, protocatechuic acid was decarboxylated in the wild type and disrupted L. plantarum cells, except cells in which the lpdB and lpdC genes were interrupted (Fig. 5). The results obtained from the L. plantarum knockout mutants indicate that the B and C subunits of the decarboxylase seem to be essential for gallate and protocatechuate decarboxylase activity in L. plantarum WCFS1, whereas the D subunit is not involved.
Fig 5.

Effects of disruption of lpdB, lpdC, and lpdD on gallate and protocatechuate decarboxylation in L. plantarum WCFS1. HPLC chromatograms of L. plantarum cultures incubated in 3 mM gallic acid (A) or protocatechuic acid (B) are shown for L. plantarum WCFS1 (wild type [wt]), L. plantarum WCFS1(pUCE191-lpdB) (lpdB mutant), L. plantarum WCFS1(pUCE191-lpdC) (lpdC mutant), and L. plantarum WCFS1(pUCE191-lpdD) (lpdD mutant). Results for uninoculated medium are also shown (control). The gallic acid (GA), protocatechuic acid (PA), pyrogallol (P), and catechol (C) detected are indicated. Chromatograms were recorded at 280 nm.
To ascertain the participation of lpd genes in gallate decarboxylase activity, cell extracts were prepared from L. plantarum wild type and knockout mutants. The extracts were adjusted to the same protein concentration, and, similar to the reactions with the E. coli extracts described above, the reactions were done with mixtures in which these extracts were mixed in all possible combinations in the presence of gallic acid. Reactions were carried out for 18 h, and after that, the phenolic compounds present in the reaction mixtures were extracted with ethyl acetate and analyzed by HPLC.
Figure 6 shows that only the reaction with a mixture which contained a functional copy of the lpdB and lpdC genes from the same strain was able to decarboxylate gallic acid. Surprisingly, gallate decarboxylase activity was not observed when functional LpdB and LpdC proteins came from different extracts. The only explanation for this result could be that LpdB has a possible role during the maturation (e.g., folding) or activation of LpdC, the main catalytic subunit.
Fig 6.

Pyrogallol production by cell extracts from L. plantarum knockout mutants. HPLC chromatograms of L. plantarum cell extracts, at the same total protein concentration, incubated in 3 mM gallic acid for 18 h are shown for L. plantarum WCFS1 (wild type), L. plantarum WCFS1(pUCE191-lpdB) (lpdB mutant), L. plantarum WCFS1(pUCE191-lpdC) (lpdC mutant), and L. plantarum WCFS1(pUCE191-lpdB) plus L. plantarum WCFS1 (pUCE191-lpdC) (lpdB and lpdC mutants). The gallic acid (GA) and pyrogallol (P) detected are indicated. Chromatograms were recorded at 280 nm.
Gallate decarboxylase activity in lactic acid bacteria.
Once the direct involvement of lpd genes in gallate/protocatechuate decarboxylation was demonstrated, as shown in Fig. 2, it seems probable that other species of lactic acid bacteria could also decarboxylate these aromatic acids. The sequences of LpdB and LpdC proteins from nine lactic acid bacteria were aligned. The degree of identity among these LpdB proteins ranged from 56 to 80% (see Fig. S4 in the supplemental material). The identity shown among the LpdC proteins was higher, ranging from 73 to 90% (see Fig. S5 in the supplemental material). In both cases, proteins from L. plantarum and Lactobacillus pentosus presented a 98% identity. These alignments allowed us to identify conserved amino acid domains to design degenerate oligonucleotides to detect the presence of both genes by PCR. Oligonucleotides 450 and 451 amplify a 158-bp internal fragment of the subunit B gene in lactic acid bacteria; similarly, oligonucleotides 448 and 449 amplify a 300-bp fragment of the gene encoding subunit C.
In order to associate the presence of the lpdB and lpdC genes and the ability to degrade gallic and protocatechuic acids, selected strains of lactic acid bacteria (Table 1) were grown in culture medium containing these hydroxybenzoic acids, and their supernatants were analyzed for the production of pyrogallol or catechol. In addition, their DNAs were used as the templates in PCRs using oligonucleotides 450 and 451 and oligonucleotides 448 and 449 to detect the presence of the genes encoding subunits B and C, respectively.
Strains belonging to the species E. faecium, L. brevis, L. pentosus, L. plantarum, O. oeni, and S. gallolyticus amplified fragments from both genes (Fig. 7A and B); Lactobacillus sakei DSM 15831T amplified only the gene encoding subunit C; and finally, strains of the species Lactobacillus hilgardii, Leuconostoc mesenteroides, and Pediococcus pentosaceus did not amplify either of the two genes. These results are mostly in agreement with the information obtained from the complete genome sequences of representative strains of these lactic acid bacterial species. However, unexpected results were obtained with L. sakei and P. pentosaceus. The genome sequence of L. sakei subsp. sakei 23K revealed the presence of a copy of the gene encoding subunit B which was absent from the L. sakei subsp. carnosus DSM 15831T strain used in this study. Similarly, the genome sequence of P. pentosaceus ATCC 25745 revealed the presence of both genes; however, these genes were absent from Pediococcus claussenii ATCC BAA-344, as revealed from its sequenced genome, and from P. pentosaceus CECT 4695, used in this study. These results might indicate that, at least in these species, the ability to decarboxylate gallic and protocatechuic acids is strain specific.
Fig 7.

Gallate and protocatechuate decarboxylase activity in lactic acid bacteria. (A and B) PCR amplification of the B and C subunits of putative gallate decarboxylases. Chromosomal DNA from the following strains was used for PCR amplification with oligonucleotides 450 and 451 (A) or oligonucleotides 448 and 449 (B) to amplify 158 bp or 300 bp of the B or C subunit, respectively: E. faecium CECT 410T (lane 1), E. faecium CECT 4102 (lane 2), L. brevis CECT 4121T (lane 3), L. brevis CECT 5354 (lane 4), L. hilgardii RM62 (lane 5), L. hilgardii RM63 (lane 6), L. mesenteroides CECT 219T (lane 7), L. pentosus DSM 20314T (lane 8), L. plantarum ATCC 14917T (lane 9), L. plantarum WCFS1 (lane 10), L. sakei DSM 15831T (lane 11), O. oeni CECT 4100T (lane 12), O. oeni RM1 (lane 13), P. pentosaceus CECT 4695T (lane 14), and S. gallolyticus UCN34 (lane 15). PCR products were subjected to gel electrophoresis and stained with Gel Red. Left lane, 100-bp molecular size ladder. Numbers indicate some of the molecular sizes. (C) HPLC chromatograms of supernatants from lactic acid bacteria. The L. brevis CECT 5354 and P. pentosaceus CECT 4695T strains were grown in RMP medium containing 3 mM gallic acid (A) or protocatechuic acid (B) for 10 days. The gallic acid (GA), protocatechuic acid (PA), pyrogallol (P), and catechol (C) detected are indicated. Chromatograms were recorded at 280 nm.
HPLC analysis of the supernatants from cultures of these bacteria in the presence of gallic or protocatechuic acid indicated that only the bacteria which possess the genes encoding subunits B and C are able to decarboxylate gallic acid to pyrogallol and protocatechuic acid to catechol (Fig. 7C). Strains from the species E. faecium, L. brevis, L. pentosus, L. plantarum, and O. oeni were able to decarboxylate gallic and protocatechuic acids; however, L. hilgardii, L. mesenteroides, L. sakei, and P. pentosaceus strains were not. Therefore, the results obtained seem to indicate that the ability to decarboxylate some hydroxybenzoic acids (gallic and protocatechuic acids) is widely extended among lactic acid bacterial strains. Moreover, the ability to decarboxylate these acids is related to the presence of the B and C subunits of a putative aromatic acid decarboxylase found in these bacteria.
DISCUSSION
Bacterial nonoxidative, reversible multisubunit aromatic acid decarboxylases are encoded by three clustered genes, B, C, and D. The functions of these proteins remain unknown, and the question that arises is, which genes encode the catalytic protein? Initially, when these decarboxylases were purified from cell extracts in an active form, the results indicated that only one multimeric protein, composed of identical subunits, was involved. Purified 4-hydroxybenzoate decarboxylase from S. hydroxybenzoicus showed a single band on both native gradient PAGE and denatured SDS-PAGE, having an apparent molecular mass of 350 kDa and consisted of six identical subunits of 57 kDa (32). Similarly, in P. agglomerans, SDS-PAGE analysis indicated that the purified gallate decarboxylase was homogeneous and consisted of six identical subunits of 57 kDa in molecular mass (6). In addition, purified 4-hydroxybenzoate decarboxylase from E. cloacae was a homohexamer of identical 60-kDa subunits (33). The molecular masses of these proteins are in accordance with the masses of their respective C subunits deduced from in silico sequence analysis.
However, when the genetic organization of these proteins was known, results involving the activity of the additional B and D subunits were obtained. Contradictory results about the involvement of the B, C, and D subunits in the activity of these decarboxylases were obtained (29, 32, 34, 35). It seems that purification from cell extracts indicated that, on the basis of the size of the purified protein, only the C subunit is involved in enzymatic activity; however, experiments of heterologous expression indicated that the three genes, or at least subunits C and D, are needed for activity (29, 36). It was also speculated that during heterologous expression in E. coli, the B subunit can be at least partially replaced by another gene product from E. coli, such as UbiX (36).
As shown in Fig. 2, genes encoding nonoxidative decarboxylase are clustered in different organizations in bacteria. However, L. plantarum is the only bacterial species in which the genes are separated in the chromosome by more than 1 Mb. This unusual configuration could indicate a different enzymatic organization. The first result suggesting the involvement of LpdC in gallate decarboxylase activity in L. plantarum was that, in induced cell extracts, only this protein was significantly overproduced, as observed in the 8 and 15% SDS-PAGE analysis (Fig. 1). In addition, only the LpdC protein was overproduced (more than 7-fold) in L. plantarum when it was exposed to a tannic acid challenge, as revealed by a proteomic analysis (37). Similarly, only the proteins equivalent to LpdC were overproduced in response to other phenolic acids, such as protein 3717 (VdcC) in Streptomyces sp. D7 upon exposure to vanillic acid (34) and BsdC in B. subtilis in response to salicylic acid (38). However, in Streptomyces sp. D7 as well as in B. subtilis, it has been described that at least subunits C and D are required to confer decarboxylase activity (29, 36).
The only involvement of LpdC in L. plantarum decarboxylation also arose from E. coli extracts producing LpdB, LpdC, and LpdD proteins. The three proteins were independently overproduced in E. coli, and the expression of lpdC was enough to confer gallate decarboxylase activity to E. coli, even in an E. coli UbiX-defective mutant. Finally, to ascertain the exclusive role of LpdC in gallate decarboxylase activity, the recombinant LpdC protein was purified and gallate decarboxylase activity was demonstrated in vitro. However, even though LpdC was produced in a high yield, it presented low gallate decarboxylase activity. It is noteworthy that addition of pure LpdB protein did not increase the activity of LpdC. It could be argued that the presence of a His tag could result in differences in activity. More likely, the low activity observed could be because these enzymes are extremely unstable in usual buffer solutions due to their oxygen sensitivity (6, 7). In addition, the batch purification protocol followed for protein purification and the presence of immobilized metal ions (cobalt) in the resin could contribute to the inactivation of the enzyme.
Unexpected results were obtained by the use of L. plantarum mutants with knockouts in the three lpd genes, as it was demonstrated that the disruption of subunit B and subunit C avoids gallate decarboxylase activity in L. plantarum. Decarboxylase activity was restored only when extracts containing functional B and C proteins were present. These results could be compatible with those obtained from E. coli extracts only if a protein from E. coli could assume the function of LpdB. In spite of the similarity of UbiX and LpdB, the expression of lpdC in a ubiX-negative E. coli mutant (E. coli JW2308-4) indicates that UbiX is not assuming the LpdB function. An unknown E. coli protein different from UbiX could be involved. The involvement of the B subunit in the decarboxylation reaction has also been clearly demonstrated in B. subtilis since antisense mRNA inactivation of the B subunit highly reduces the enzyme activity to below 2% of that of the wild type (29, 36).
The biochemical activities of the three different protein subunits have not been assigned. So far, it has not been possible to unequivocally correlate genes coding for aromatic acid decarboxylase and their function. In this study, interesting results came from the use of L. plantarum knockout mutants. In L. plantarum, the LpdD protein did not seem to be necessary for gallate decarboxylase activity, while the LpdC protein seemed to be the main catalytic subunit. However, the function of LpdB is unknown. To achieve gallate decarboxylase activity fully, it is not enough to have functional copies of the LpdB and LpdC proteins, since it seems that both proteins need to be synthesized in the same strain. Both mature proteins do not seem to interact, as revealed by the in vitro cross-linking experiments. It is tempting to speculate that LpdB could have a possible role during the maturation (e.g., folding) or activation of LpdC, and therefore, LpdB and LpdC need to be synthesized simultaneously in the same host. The mechanism of decarboxylation followed by these aromatic acid decarboxylases is a paradigm for a new type of biological decarboxylation reaction. As far as we know, this study in L. plantarum constitutes the first description of the involvement of only subunits B and C in the nonoxidative decarboxylation of an aromatic acid.
Apart from E. faecalis, among lactic acid bacteria, decarboxylation of aromatic acids has been described only in L. plantarum (11), L. brevis (39), and S. gallolyticus (8, 40). These bacteria decarboxylate the same hydroxybenzoic acids, gallic acid and protocatechuic acid, and all possess genes similar to the gallate decarboxylase genes described in this work (Fig. 2). Such decarboxylase activity has never been described in E. faecium and O. oeni species; however, strains from these species also decarboxylate both acids and possess both genes (Fig. 2 and 7). From the data obtained in this study, it could be assumed that, at least in some bacterial species, the ability to decarboxylate gallic and protocatechuic acids might be strain dependent, similar to the ability of some specific E. coli strains (e.g., strains EDL933 and VT2-Sakai from E. coli O157:H7) to decarboxylate 4-hydroxybenzoate (29).
The identification of the L. plantarum gallate decarboxylase involved in tannin degradation completes the analysis of the first route of degradation of a phenolic compound in lactic acid bacteria. The proposed biochemical pathway for the degradation of tannins by L. plantarum implies that tannins are hydrolyzed to gallic acid and glucose by a tannase action, and the gallic acid formed is decarboxylated to pyrogallol by the action of a gallate decarboxylase (9, 10). When purified L. plantarum tannase was assayed against 18 phenolic acid esters, only esters derived from gallic and protocatechuic acids were hydrolyzed (41), with these esters apparently sharing the same substrate specificity as the decarboxylase enzyme. This substrate specificity suggests a concomitant activity of tannase and gallate decarboxylase on specific phenolic substrates. This is more obvious when the chromosomal location of these genes is considered. The genes encoding gallate decarboxylase (lp_2945) and tannase (lp_2956) are only 6.5 kb apart on the L. plantarum WCFS1 chromosome. Interestingly, in S. gallolyticus, another tannin-degrading lactic acid bacterium, the gene encoding tannase (GALLO_1609) is separated by only one open reading frame from the genes encoding decarboxylase (GALLO_1611, GALLO_1612, and GALLO_1613) (Fig. 2). More interestingly, S. gallolyticus strains showed metabolism of these phenolic compounds identical to that of L. plantarum, as they hydrolyzed hydrolyzable tannins to release gallic acid, which was subsequently decarboxylated to pyrogallol, and protocatechuic acid, which was decarboxylated to catechol (8, 40). Neither the S. gallolyticus nor L. plantarum bacterial species possesses appropriate mechanisms to further degrade the compounds produced by these dead-end pathways. The physiological relevance of these reactions is unknown, but in natural ecosystems, it could be imagined that other organisms in a consortium mineralize and remove these dead-end metabolites. These enzymatic activities have ecological advantages for L. plantarum, as it is often associated with fermentation of plant materials. Therefore, L. plantarum plays an important role when tannins are present in food and the intestine, having the capability of degrading and detoxifying harmful and antinutritional constituents into simpler and innocuous compounds.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants AGL2008-01052, AGL2011-22745, Consolider INGENIO 2010 CSD2007-0063 FUN-C-FOOD (Comisión Interministerial de Ciencia y Tecnología), S2009/AGR-1469 (ALIBIRD; Comunidad de Madrid), and RM2008-00002 (Instituto Nacional de Investigación Agraria y Alimentaria). N. Jiménez is the recipient of an FPI fellowship from MINECO.
We are grateful to J. L. Ruíz-Barba for his help with the L. plantarum electroporation experiments and to M. V. Santamaría and J. M. Barcenilla for their assistance.
Footnotes
Published ahead of print 3 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00840-13.
REFERENCES
- 1. Shahidi F, Naczk M. 2003. Phenolics in food and nutraceuticals. CRC Press, London, United Kingdom [Google Scholar]
- 2. Chung K-T, Wei C-I, Johnson MG. 1998. Are tannins a double-edged sword in biology and health? Trends Food Sci. Technol. 9:168–175 [Google Scholar]
- 3. Ow YY, Stupans I. 2003. Gallic acid and gallic acid derivatives: effects on drug metabolizing enzymes. Curr. Drug Metab. 3:241–248 [DOI] [PubMed] [Google Scholar]
- 4. Chowdhury SP, Khanna S, Verma SC, Tripathi AK. 2004. Molecular diversity of tannic acid degrading bacteria isolated from tannery soil. J. Appl. Microbiol. 97:1210–1219 [DOI] [PubMed] [Google Scholar]
- 5. Nogales J, Canales A, Jiménez-Barbero J, Serra B, Pingarrón JM, García JL, Díaz E. 2011. Unravelling the gallic acid degradation pathway in bacteria: the gal cluster from Pseudomonas putida. Mol. Microbiol. 79:359–374 [DOI] [PubMed] [Google Scholar]
- 6. Zeida M, Wieser M, Yoshida T, Sugio T, Nagasawa T. 1998. Purification and characterization of gallic acid decarboxylase from Pantoea agglomerans T71. Appl. Environ. Microbiol. 64:4743–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakajima H, Otani C, Niimura T. 1992. Decarboxylation of gallate by cell-free extracts of Streptococcus faecalis and Klebsiella pneumoniae isolated from rat feces. J. Food Hyg. Soc. Jpn. 33:371–377 [Google Scholar]
- 8. Chamkha M, Patel BKC, Traore A, Garcia Labat J-LM. 2002. Isolation from a shea cake digester of a tannin-degrading Streptococcus gallolyticus strain that decarboxylates protocatechuic acid hydroxycinnamic acids, and emendation of the species. Int. J. Syst. Evol. Microbiol. 52:939–944 [DOI] [PubMed] [Google Scholar]
- 9. Osawa R, Kuroiso K, Goto S, Shimizu A. 2000. Isolation of tannin-degrading lactobacilli from humans and fermented foods. Appl. Environ. Microbiol. 66:3093–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodríguez H, de las Rivas B, Gómez-Cordovés MC, Muñoz R. 2008. Degradation of tannic acid by cell-free extracts of Lactobacillus plantarum. Food Chem. 107:664–670 [DOI] [PubMed] [Google Scholar]
- 11. Rodríguez H, Landete JM, de las Rivas B, Muñoz R. 2008. Metabolism of food phenolic acids by Lactobacillus plantarum CECT 748T. Food Chem. 107:1393–1398 [Google Scholar]
- 12. Nishitani Y, Osawa R. 2003. A novel colorimetric method to quantify tannase activity of viable bacteria. J. Microbiol. Methods 54:281–284 [DOI] [PubMed] [Google Scholar]
- 13. Nishitani Y, Sasaki E, Fujisawa T, Osawa R. 2004. Genotypic analyses of lactobacilli with a range of tannase activities isolated from human feces and fermented foods. Syst. Appl. Microbiol. 27:109–117 [DOI] [PubMed] [Google Scholar]
- 14. Vaquero I, Marcobal A, Muñoz R. 2004. Tannase activity by lactic acid bacteria isolated from grape must and wine. Int. J. Food Microbiol. 96:199–204 [DOI] [PubMed] [Google Scholar]
- 15. Iwamoto K, Tsuruta H, Nishitani Y, Osawa R. 2008. Identification and cloning of a gene encoding tannase (tannin acylhydrolase) from Lactobacillus plantarum ATCC 14917T. Syst. Appl. Microbiol. 31:269–277 [DOI] [PubMed] [Google Scholar]
- 16. Rozès N, Peres C. 1998. Effects of phenolic compounds on the growth and the fatty acid composition of Lactobacillus plantarum. Appl. Microbiol. Biotechnol. 49:108–111 [Google Scholar]
- 17. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 18. Arrecubieta C, García E, López R. 1995. Sequence and transcriptional analysis of a DNA region involved in the production of capsular polysaccharide in Streptococcus pneumoniae type 3. Gene 167:1–7 [DOI] [PubMed] [Google Scholar]
- 19. Aukrust T, Blom H. 1992. Transformation of Lactobacillus strains used in meat and vegetable fermentations. Food Res. Int. 25:253–261 [Google Scholar]
- 20. de las Rivas B, Curiel JA, Mancheño JM, Muñoz R. 2007. Expression vectors for enzyme restriction- and ligation-independent cloning for producing recombinant His-fusion proteins. Biotechnol. Prog. 23:680–686 [DOI] [PubMed] [Google Scholar]
- 21. Curiel JA, de las Rivas B, Mancheño JM, Muñoz R. 2011. The pURI family of expression vectors: a versatile set of ligation independent cloning plasmids for producing recombinant His-fusion proteins. Protein Expr. Purif. 76:44–53 [DOI] [PubMed] [Google Scholar]
- 22. Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 68:850–858 [DOI] [PubMed] [Google Scholar]
- 23. Cavin J-F, Barthelmebs L, Guzzo J, van Beeumen J, Samyn B, Travers J-F, Divies C. 1997. Purification and characterization of an inducible p-coumaric acid decarboxylase from Lactobacillus plantarum. FEMS Microbiol. Lett. 147:291–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cavin J-F, Barthelmebs L, Divies C. 1997. Molecular characterization of an inducible p-coumaric acid decarboxylase from Lactobacillus plantarum: gene cloning, transcriptional analysis, overexpression in Escherichia coli, purification, and characterization. Appl. Environ. Microbiol. 63:1939–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bentinger M, Tekle M, Dallner G. 2010. Coenzyme Q—biosynthesis and functions. Biochem. Biophys. Res. Commun. 396:74–79 [DOI] [PubMed] [Google Scholar]
- 26. Gulmezian M, Hyman KR, Marbois BN, Clarke CF, Javor GT. 2007. The role of UbiX in Escherichia coli coenzyme Q biosynthesis. Arch. Biochem. Biophys. 467:144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rangarajan ES, Yunge L, Iannuzzi P, Tocilj A, Hung L-W, Matte A, Cygler M. 2004. Crystal structure of a dodecameric FMN-dependent UbiX-like decarboxylase (Pad1) from Escherichia coli O157:H7. Protein Sci. 13:3006–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brooijmans R, Smit B, Santos F, van Riel J, de Vos WM, Hugenholtz J. 2009. Heme and menaquinone induced electron transport in lactic acid bacteria. Microb. Cell Fact. 8:28. 10.1186/1475-2859-8-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lupa B, Lyon D, Gibbs MD, Reeves RA, Wiegel J. 2005. Distribution of genes encoding the microbial non-oxidative reversible hydroxyarylic acid decarboxylases/phenol carboxylases. Genomics 86:342–351 [DOI] [PubMed] [Google Scholar]
- 30. Masui Y, Mizuno T, Inouye M. 1984. Novel high level expression cloning vehicles: 104-fold amplification of Escherichia coli minor protein. Biotechnology (NY) 2:81–85 [Google Scholar]
- 31. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio Collection. Mol. Syst. Biol. 2:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He Z, Wiegel J. 1995. Purification and characterization of an oxygen-sensitive reversible 4-hydroxybenzoate decarboxylase from Clostridium hydroxybenzoicum. Eur. J. Biochem. 229:77–82 [DOI] [PubMed] [Google Scholar]
- 33. Matsui T, Yoshida T, Hayashi T, Nagaswa T. 2006. Purification, characterization, and gene cloning of 4-hydroxybenzoate decarboxylase of Enterobacter cloacae P240. Arch. Microbiol. 186:21–29 [DOI] [PubMed] [Google Scholar]
- 34. Chow KT, Pope MK, Davies J. 1999. Characterization of a vanillic acid non-oxidative decarboxylation gene cluster from Streptomyces sp. D7. Microbiology 145:2393–2403 [DOI] [PubMed] [Google Scholar]
- 35. Huang J, He Z, Wiegel J. 1999. Cloning, characterization, and expression of a novel gene encoding a reversible 4-hydroxybenzoate decarboxylase from Clostridium hydroxybenzoicum. J. Bacteriol. 181:5119–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lupa B, Lyon D, Shaw LN, Sieprawska-Lupa M, Wiegel J. 2008. Properties of the reversible nonoxidative vanillate/4-hydroxybenzoate decarboxylase from Bacillus subtilis. Can. J. Microbiol. 54:75–81 [DOI] [PubMed] [Google Scholar]
- 37. Curiel JA, Rodríguez H, de las Rivas B, Anglade P, Baraige F, Zagorec M, Champomier-Vergès M, Muñoz R, López de Felipe F. 2011. Response of a Lactobacillus plantarum human isolate to tannic acid challenge assessed by proteomic analyses. Mol. Nutr. Food Res. 55:1454–1465 [DOI] [PubMed] [Google Scholar]
- 38. Duy NV, Mäder U, Tran NP, Cavin J-F, Tam LT, Alberecht D, Hecker M, Anterlmann H. 2007. The proteome and transcriptome analysis of Bacillus subtilis in response to salicylic acid. Proteomics 7:698–710 [DOI] [PubMed] [Google Scholar]
- 39. Curiel JA, Rodríguez H, Landete JM, de las Rivas B, Muñoz R. 2010. Ability of Lactobacillus brevis strains to degrade food phenolic acids. Food Chem. 120:225–229 [Google Scholar]
- 40. Osawa R, Fujisawa T, Sly LI. 1995. Streptococcus gallolyticus sp. nov.; gallate degrading organisms formerly assigned to Streptococcus bovis. Syst. Appl. Microbiol. 18:74–78 [Google Scholar]
- 41. Curiel JA, Rodríguez H, Acebron I, Mancheño JM, de las Rivas B, Muñoz R. 2009. Production and physicochemical properties of recombinant Lactobacillus plantarum tannase. J. Agric. Food Chem. 57:6224–6230 [DOI] [PubMed] [Google Scholar]
- 42. de las Rivas B, Rodríguez H, Curiel JA, Landete JM, Muñoz R. 2009. Molecular screening of wine lactic acid bacteria degrading hydroxycinnamic acids. J. Agric. Food Chem. 57:490–494 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.