

Abstract
Strains of Bifidobacterium animalis subsp. lactis are extensively exploited by the food industry as health-promoting bacteria, although the genetic variability of members belonging to this taxon has so far not received much scientific attention. In this article, we describe the complete genetic makeup of the B. animalis subsp. lactis Bl12 genome and discuss the genetic relatedness of this strain with other sequenced strains belonging to this taxon. Moreover, a detailed comparative genomic analysis of B. animalis subsp. lactis genomes was performed, which revealed a closely related and isogenic nature of all currently available B. animalis subsp. lactis strains, thus strongly suggesting a closed pan-genome structure of this bacterial group.
INTRODUCTION
Bifidobacteria are intensively exploited by the food industry due to the presumed health beneficial effects they exert on the human host (1–5). However, the molecular mechanisms underlying these proclaimed health-promoting activities are still largely unknown. Recently, significant efforts have been made to decode and analyze bifidobacterial genome sequences, which is part of a novel discipline called probiogenomics, aimed at the discovery of genetic determinants responsible for the adaptation of these microorganisms to the gastrointestinal tract of their host (6–11). In the context of probiogenomics attempts involving bifidobacterial strains, members of the Bifidobacterium animalis subsp. lactis taxon are worth mentioning, as they have been the target of several genome sequencing projects which have resulted in the complete genomic decoding of nine B. animalis subsp. lactis strains (12–18). The availability of such a large set of genome sequences of strains belonging to the same species allows the identification of the pan-genome structure of this taxon, as well as the determination of the extent of genetic variability occurring among members of this species. So far, several attempts have been carried out to delineate the evolutionary development of B. animalis subsp. lactis, as well as the genetic relatedness with other bifidobacterial species (14, 19–21). However, these studies drew their conclusions from a limited set of genomic information, as opposed to the much larger data set of B. animalis subsp. lactis genomes that is currently available. Here, we describe the sequence analysis of the B. animalis subsp. lactis Bl12 genome. Furthermore, we analyzed all complete and publicly available genome sequences of B. animalis subsp. lactis strains and highlight the strictly monomorphic nature as well as the closed pan-genomic structure of this taxon.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study and their origins are listed in Table S1 in the supplemental material. All Bifidobacterium strains were cultivated in an anaerobic atmosphere (2.99% H2, 17.01% CO2, and 80% N2) in a chamber (Concept 400; Ruskin) on de Man-Rogosa-Sharpe (MRS) medium (Scharlau Chemie, Barcelona, Spain) supplemented with 0.05% (wt/vol) l-cysteine hydrochloride and incubated at 37°C. Cell growth on semisynthetic MRS medium supplemented with 1% (wt/vol) of a particular sugar was monitored by optical density at 600 nm using a plate reader (Biotek, VT). The plate reader was run in discontinuous mode, with absorbance readings performed at 60-min intervals and preceded by 30 s of shaking at medium speed. Cultures were grown in biologically independent triplicates, and the resulting growth data were expressed as the mean of these replicates. Carbohydrates were purchased from Sigma (Milan, Italy) and Carbosynth (Berkshire, United Kingdom).
Susceptibility to tetracycline.
Susceptibility to antibiotics, in terms of MIC to tetracycline (Sigma-Aldrich), was determined using the broth microdilution method. MIC, considered the lowest biocide concentration that prevents detectable growth of a particular bacterium, was determined using the standardized bifidobacterium susceptibility test medium (LSM-Cys) broth formulation as indicated in ISO 10932:2010 for antibiotic sensitivity assessment of bifidobacteria (22), which is expected to ensure adequate growth of the test organism (Bifidobacterium longum ATCC 15707). LSM-Cys consists of a mixture of Iso-Sensitest broth medium (Oxoid) (90%) and MRS broth medium (10%) added with 0.3 g of l-cysteine per liter of LSM, adjusted to pH 6.7. MIC testing was performed in a 5-ml final volume. Each strain included in this study was inoculated in triplicate for each antibiotic concentration tested at a final inoculum density of 105 bacteria/ml, starting from cultures incubated for 48 h under anaerobic conditions. The bacterial cell concentration of the overnight culture was determined microscopically by use of an improved Neubauer counting chamber (Marienfeld GmbH, Lauda-Königshofen, Germany). Cultures were incubated at 37°C under anaerobic conditions for 48 h. After incubation, culture cell density was measured spectrophotometrically (optical density [OD] at 600 nm).
Genome sequencing and bioinformatics analyses.
The genome sequence of Bl12 was determined by GenProbio srl using the Ion Torrent Personal Genome Machine (Life Technologies, Germany). A genomic library was generated using 1 μg of genomic DNA and an Ion Xpress Plus fragment library kit and employing the Ion Shear chemistry according to the user guide. After dilution to 2.66 × 107 molecules/μl, 4.5 × 108 molecules were used as the templates for clonal amplification on Ion Sphere particles during the emulsion PCR according to the Ion Xpress Template 200 kit manual. The quality of the amplification was estimated, and the sample was loaded onto an Ion 316 chip and subsequently sequenced using 125 sequencing cycles according to the Ion Sequencing 200 kit user guide. This number of sequencing cycles resulted in an average reading length of approximately 200 nucleotides. The MIRA program (version 3.4.0) was used for de novo assembly of the Bl12 genome sequence (23). The generated sequencing output consisted of 600 Mb of DNA sequences, corresponding to about 300× coverage of the Bl12 genome. Quality improvement of the genome sequence involved sequencing of more than 50 PCR products across the entire genome to ensure correct assembly, double stranding, and the resolution of any remaining base conflicts.
Sequence annotation.
The genomes analyzed consisted of nine complete and publicly available B. animalis subsp. lactis genome sequences plus the B. animalis subsp. lactis Bl12 genome, which was sequenced as part of this study and is described in Table 1. In order to ensure the identical sequence quality standards for all investigated genomes, the publicly available nucleotide sequences corresponding to these genomes were reanalyzed using common software and parameters (see below). Overall DNA similarity analyses between the B. animalis subsp. lactis genomes were carried out using BLASTN (24) and Artemis (25).
Table 1.
Alignment to Bl12 and general genome features of the nine publicly available B. animalis subsp. lactis, B. animalis subsp. lactis Bl12, and B. animalis subsp. animalis complete genome sequences
| Organism | Strain | Accession no. | Alignment against Bl12 (%) | Genome size (bp) | No. of ORFs | GC content (%) | No. of tRNAs | rRNA locusa | No. of transposases | R/M systemsb | No. of EPS genes | No. of prophages |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B. animalis subsp. lactis | Bl12 | CP004053 | 100.000 | 1,938,605 | 1,518 | 60.5 | 52 | 4 | 9 | 3 (incomplete) | 1 | 1 |
| BLC1 | CP003039 | 99.84 | 1,943,983 | 1,518 | 60.5 | 52 | 4 | 9 | 3 (incomplete) | 1 | 1 | |
| DSM10140 | CP001606 | 99.99 | 1,938,483 | 1,518 | 60.5 | 51 | 4 | 9 | 3 (incomplete) | 1 | 1 | |
| V9 | CP001892 | 99.84 | 1,944,050 | 1,521 | 60.5 | 52 | 4 | 10 | 3 (incomplete) | 1 | 1 | |
| Bl-04 | CP001515 | 99.99 | 1,938,709 | 1,518 | 60.5 | 52 | 4 | 9 | 3 (incomplete) | 1 | 1 | |
| Bi-07 | NC_017867 | 99.99 | 1,938,822 | 1,518 | 60.5 | 52 | 4 | 9 | 3 (incomplete) | 1 | 1 | |
| B420 | NC_017866 | 99.99 | 1,938,595 | 1,518 | 60.5 | 52 | 4 | 9 | 3 (incomplete) | 1 | 1 | |
| BB12 | CP001853 | 99.82 | 1,942,198 | 1,521 | 60.5 | 52 | 4 | 10 | 3 (incomplete) | 1 | 1 | |
| CNCM I-2494 | CP002915 | 99.82 | 1,943,113 | 1,521 | 60.5 | 52 | 4 | 10 | 3 (incomplete) | 1 | 1 | |
| AD011 | CP001213 | 99.83 | 1,933,695 | 1,520 | 60.5 | 52 | 2 + single 5S rRNA | 10 | 3 (incomplete) | 1 | 1 | |
| B. animalis subsp. animalis | ATCC 25527 | CP002567 | 91.77 | 1,932,693 | 1,538 | 60.5 | 52 | 3 + single 16S rRNA and 23S rRNA | 8 | 1 | 1 | 1 |
These numbers refer to a complete locus encompassing 16S rRNA, 23S rRNA, and 5S rRNA genes.
Determined by REBASE (29).
Protein-encoding open reading frames (ORFs) were predicted using a combination of Prodigal (26) and BLASTX (24) for comparative analysis. Results of the gene-finder program were combined manually with data from BLASTP (27) analysis against a nonredundant protein database provided by the National Center for Biotechnology Information. The combined results were inspected by Artemis (25), which was used for a manual editing effort to verify and, if necessary, to redefine the start of each predicted coding region or to remove or add coding regions.
Assignment of protein function to predicted coding regions of the B. animalis subsp. lactis genomes was performed manually. Moreover, the revised gene-protein set was searched against the Swiss-Prot (http://www.expasy.ch/sprot//TrEMBL), PRIAM (http://priam.prabi.fr/), protein family (Pfam; http://pfam.sanger.ac.uk/), TIGRFAMs (http://www.jcvi.org/cms/research/projects/tigrfams/overview/), Interpro (INTERPROSCAN; http://www.ebi.ac.uk/Tools/InterProScan/), Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/), and COGs (http://www.ncbi.nlm.nih.gov/COG/) databases, in addition to BLASTP (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Functional assignments were defined by manual processing of the combined results. Manual corrections to automated functional assignments were completed on an individual gene-by-gene basis as needed.
Additional bioinformatic analyses included tRNA gene identification using tRNAscan-SE (28) and rRNA gene detection using RNAmmer (http://www.cbs.dtu.dk/services/RNAmmer/), followed by manual annotation on the basis of BLASTN searches.
Insertion sequence (IS) families were assigned using ISFinder (http://www-is.biotoul.fr/is.html), restriction/modification (R/M) systems were searched on the basis of the REBASE database (29), transporter classification was performed according to the Transporter Classification Database scheme (30), and ORF attribution to a specific COG family was made by searching against the COGs database (http://www.ncbi.nlm.nih.gov/COG/).
Proteome comparison and extraction of shared and unique genes.
Each predicted proteome of the 10 analyzed B. animalis subsp. lactis strains (Table 1), B. animalis subsp. animalis ATCC 25527 (31), Bifidobacterium longum subsp. longum NCC2705 (32), Bifidobacterium longum subsp. infantis ATCC 15697 (33), Bifidobacterium bifidum PRL2010 (34), Bifidobacterium dentium Bd1 (35), Bifidobacterium breve UCC2003 (36), and Bifidobacterium adolescentis ATCC 15703 (NCBI source) was searched for orthologues against the total proteome, where orthology between two proteins was defined as the best bidirectional FASTA hits (37). Identification of orthologues, paralogues, and unique genes was performed following a preliminary step consisting of the comparison of each protein against all other proteins using BLAST analysis (27) (cutoff: E value of 1 × 10-4 and 30% identity over at least 80% of both protein sequences), and then all proteins were clustered into protein families using MCL (graph theory-based Markov clustering algorithm) (38). Following this, the unique protein families for each of the 17 bifidobacterial genomes were classified. Protein families shared between all genomes, named core gene families, were defined by selecting the families that contained at least one single protein member for each genome.
Each set of orthologous proteins was aligned using CLUSTAL_W (39), and phylogenetic trees were constructed using the maximum-likelihood in PhyML (40). The supertree was built using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Whole-genome alignments, single nucleotide polymorphism (SNP) or indel detection, and in silico optical map reconstruction.
Whole-genome sequence alignments for similarity and dot plot analysis were performed at DNA level using LAST (http://last.cbrc.jp/), while Mauve (41) was used for whole-genome sequence alignments for SNPs and indel identification and manual editing of genome sequences. Clusters based on the heat maps of SNPs and indels were constructed using TIGR MultiExperiment Viewer (TMeV) software (42). For each B. animalis subsp. lactis genome, nucleotide sequences corresponding to 20-bp regions spanning the verified SNPs listed by Barrangou et al. (14) were used to build a supertree according to the procedure described above. An in silico optical map was constructed by in silico digestion with NotI and visualized through Geneious software (43).
Pan-genome.
For all 10 B. animalis subsp. lactis genomes used in this study, a pan-genome calculation was performed using the PGAP pipeline (44); the ORF content of each genome was organized in functional gene clusters using the gene family (GF) method. A pan-genome profile was built using all possible BLAST combinations for each genome being sequentially added. Finally, the PGAP pipeline (44) performed also a power law regression in order to extrapolate the best function fitting, according to the Heaps law pan-genome model (45).
PCR validation of the indels.
After identification of putative indels between the 10 genomes analyzed, primers were designed to amplify regions spanning indels from all these genomes. PCR amplicons were purified using a Qiaquick kit (Qiagen) and then submitted to DNA sequencing (Macrogen, South Korea).
Nucleotide sequence accession numbers.
The sequence reported in this article has been deposited in the GenBank database under accession number CP004053.
RESULTS AND DISCUSSION
Identification of B. animalis subsp. lactis Bl12 strain.
Seventy-six strains belonging to B. animalis subsp. lactis originally identified from different ecological niches, including fecal as well as colonoscopic samples (see Table S1 in the supplemental material), were investigated for their resistance to tetracycline using the MIC assay. It has previously been established that the B. animalis subsp. lactis genomes sequenced so far encompass a putative conjugative transposon carrying a tet(W) gene (46–48), as well as a miaA gene (see below), which confers a high level (32 μg/ml) of tetracycline resistance. Thus, we decided to analyze this metabolic feature in order to characterize all 76 B. animalis subsp. lactis strains and to reveal possible genetic differences existing within this bifidobacterial taxon (see Table S1). Notably, only two B. animalis subsp. lactis strains, named 646 and Bl12, were shown to display MICs of 24 μg/ml and 16 μg/ml, respectively, which are significantly lower than those identified for several commercially exploited B. animalis subsp. lactis strains (e.g., BB12 and BLC1 strains possess a MIC of 32 μg/ml). Furthermore, the growth profile of these two B. animalis subsp. lactis strains on different carbohydrates was evaluated and compared to those known for other B. animalis subsp. lactis strains. As displayed in Table S2 in the supplemental material, no evident differences, except for fucose, were noticed in carbohydrate utilization profiles between these strains, suggestive of a high level of conservation of the genetic arsenal of carbohydrate-degrading enzymes and sugar transporters in the B. animalis subsp. lactis taxon.
To further assess the genetic variability of the B. animalis subsp. lactis taxon, we decided to sequence the genome of strain Bl12 based on its deviating MIC for tetracycline.
General genome features of B. animalis subsp. lactis Bl12 strain.
The chromosome of B. animalis subsp. lactis Bl12 was fully decoded using the Ion Torrent Personal Genome Machine, and the achieved reads were assembled using MIRA (see Materials and Methods). The determined genome sequence consists of 1,938,605 bp, with a G+C content of 60.5%, which is (nearly) identical to that of other sequenced B. animalis subsp. lactis genomes (Table 1). Furthermore, the genome sequence of B. animalis subsp. lactis Bl12 encompasses a collection of tRNA and rRNA genes which is highly similar to that identified in other B. animalis subsp. lactis chromosomes (Table 1).
Analysis of clusters of orthologous groups (COGs) of the Bl12 predicted proteome allowed a functional assignment for 78.9% of the total number of predicted ORFs. Interestingly, about 9.53% and 7.89% of the genes identified in the B. animalis subsp. lactis Bl12 genome encode proteins that are predicted to be involved in amino acid and carbohydrate transport and metabolism, respectively (Fig. 1). Such an extensive genetic adaptation to amino acid and carbohydrate metabolism is similar to that identified in the genomes of other intestinal bifidobacteria (32–35, 49) and thus appears to represent a key genetic adaptation of members of the genus Bifidobacterium.
Fig 1.

Comparative genomic analysis of B. animalis subsp. lactis Bl12 with other fully sequenced B. animalis subsp. lactis strains. Panel a represents a circular genome atlas of B. animalis subsp. lactis Bl12 (circle 1) with mapped orthologues (defined as reciprocal best BLASTp hits with more than 30% identity over at least 80% of both protein lengths) in nine publicly available B. animalis subsp. lactis genomes (circles 2 through 10). Circle 11 illustrates B. animalis subsp. lactis DSM10140 G+C% deviation, followed by circle 12, which highlights B. animalis subsp. lactis DSM10140 GC skew (G-C/G+C). Panel b shows a graphical representation of the COG families of B. animalis subsp. lactis Bl12 and other Bifidobacterium species. Each COG family is identified by a one-letter abbreviation: A, RNA processing and modification; B, chromatin structure and dynamics; C, energy production and conversion; D, cell cycle control and mitosis; E, amino acid metabolism and transport; F, nucleotide metabolism and transport; G, carbohydrate metabolism and transport; H, coenzyme metabolism; I, lipid metabolism; J, translation; K, transcription; L, replication and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, and chaperone functions; P, inorganic ion transport and metabolism; Q, secondary structure; T, signal transduction; U, intracellular trafficking and secretion; Y, nuclear structure; V, defense mechanisms; Z, cytoskeleton; R, general functional prediction only; S, function unknown.
Homologues from other bacterial species with known function were identified for 83% of the B. animalis subsp. lactis Bl12 ORFs, while the remaining 17% do not possess a predicted function.
Genetics of the tetracycline susceptibility of B. animalis subsp. lactis Bl12.
All strains of B. animalis subsp. lactis described so far display a medium level of resistance to the antibiotic tetracycline. Even though the genetic locus responsible for this tetracycline resistance is not fully characterized in B. animalis subsp. lactis, it is likely to be linked to the presence of the tet(W) gene in their genomes (46, 47). The tet(W) gene encodes an alternative elongation factor that belongs to the GTP-binding elongation factor family, also referred to as the Tet(M)/Tet(O) subfamily, which protects ribosomes from the translation inhibition of tetracycline (50). However, alignment of the relevant DNA sequences of the tet(W) gene in all of the available B. animalis subsp. lactis genomes, including that of the B. animalis subsp. lactis 646 strain, revealed that these are identical to that of the homologue present in the genome of Bl12 (data not shown). In contrast, we identified a genetic difference in the DNA sequence of the miaA gene (Bl12_1042). The miaA gene product has been reported to affect the efficiency of Tet(W) and ultimately the susceptibility to tetracycline (51). The nucleotide alignment of the DNA spanning the miaA loci of the nine currently available B. animalis subsp. lactis strains, including B. animalis subsp. lactis 646, displayed identical sequences, while the miaA gene sequence of B. animalis subsp. lactis Bl12 showed a single nucleotide polymorphism (SNP), consisting of a thymine instead of a guanosine, located at nucleotide position 1154 in the gene sequence (Fig. 2). The identified SNP was further validated experimentally by PCR followed by direct DNA sequencing of the obtained amplicon. This causes a nonsynonymous mutation (from a glutamine residue to a lysine residue) in the Mia protein sequence of Bl12 strain at position 52 (Fig. 2). This finding may thus explain the higher level of sensitivity of the Bl12 strain to tetracycline than that of other tested B. animalis subsp. lactis strains (see above).
Fig 2.

(a) Comparison of the miaA locus in B. animalis subsp. lactis Bl12 with corresponding loci in various other B. animalis subsp. lactis strains. Each arrow indicates an ORF. The length of the arrow is proportional to the length of the predicted ORF. Corresponding genes are marked with the same color. The putative function of the protein is indicated above each arrow. The percent amino acid identity is indicated. (b) Nucleotide alignment of the portion of the miaA encompassing the identified SNP. (c) Amino acid alignment of the portion of the Mia protein around the identified nonsynonymous mutation.
Phylogenomic analyses of B. animalis subsp. lactis.
The availability of whole-genome sequences allows a more robust reconstruction of the phylogeny occurring within a particular bacterial taxon (52 –55). A comparative study was undertaken to determine putative orthology between completed bifidobacterial genome sequences of strains of B. longum subsp. longum, B. longum subsp. infantis, B. breve, B. bifidum, B. adolescentis, B. dentium, B. animalis subsp. animalis, and B. animalis subsp. lactis, which resulted in the identification of 886 orthologues that were shared between all these genomes (Fig. 3). This orthologue collection represents the most updated, at the time of this writing, of core genome sequences of the genus Bifidobacterium. A concatenated protein sequence that includes the product of each of these core genes, as described above, was used to build a Bifidobacterium supertree (Fig. 3). This phylogenomic analysis produced a highly reliable evolutionary positioning of B. animalis subsp. lactis within the genus Bifidobacterium, by placing all strains of B. animalis subsp. lactis on the same cluster of B. animalis subsp. animalis (Fig. 3). Remarkably, all investigated B. animalis subsp. lactis strains were placed on the same branch of the tree, indicating the absence of substantial amino acid sequence differences between the individual core proteins of these strains.
Fig 3.

Genomic diversity of the Bifidobacterium animalis subsp. lactis species. Panel a displays a Venn diagram of homologues shared between type strain B. animalis subsp. lactis Bl12 and other fully sequenced bifidobacterial species. Panel b shows a Venn diagram representation of shared homologues between B. animalis subsp. lactis Bl12 and the nine publicly available B. animalis subsp. lactis genomes. Panel c depicts a phylogenetic supertree based on the sequences of identified core proteins shared by the analyzed Bifidobacterium genomes.
Comparative analyses of B. animalis subsp. lactis genomes.
The nine publicly available B. animalis subsp. lactis genomes and B. animalis subsp. lactis Bl12 were analyzed so as to identify shared orthologues. In silico analyses show that 1,518 ORFs are shared between these strains, while 3 ORFs appear to be present only in strains BB12, V9, AD011, and CNCM I-2494. PCR attempts targeting these ORFs did not provide any experimental evidence for their existence, thus suggesting that they represent assembly and/or annotation mistakes of these genomic sequences (Fig. 3).
The genomic structure of B. animalis subsp. lactis is highly syntenic between the 10 strains investigated here, with a nucleotide identity (using B. animalis subsp. lactis strain Bl12 as a reference) of more than 99.82%, as obtained from a LAST alignment (Table 1). Dot plot comparison involving the investigated B. animalis subsp. lactis genomes revealed a perfect alignment of their chromosomes with the exception of alignments involving B. animalis subsp. lactis AD011 (Fig. 4), which may have been caused by sequencing and/or assembly mistakes. No major disruption of gene conservation between the 10 B. animalis subsp. lactis genome sequences was identified (Fig. 4). In contrast, dot plot analyses involving B. animalis subsp. animalis ATCC 25527 displayed less colinearity and highlighted differences, including small rearrangements and insertions or deletions (Fig. 4). The genome sequences of the 10 B. animalis subsp. lactis strains were further employed to reconstruct theoretical NotI restriction profiles. The generated optical maps were shown to be highly similar for the investigated strain set, thereby substantiating the notion of a high degree of genome conservation in terms of size, organization, and sequence (Fig. 5).
Fig 4.

Dot plot comparison based on genomic sequence alignments of B. animalis subsp. lactis Bl12, the nine publicly available B. animalis subsp. lactis strains, and B. animalis subsp. animalis ATCC 25527.
Fig 5.
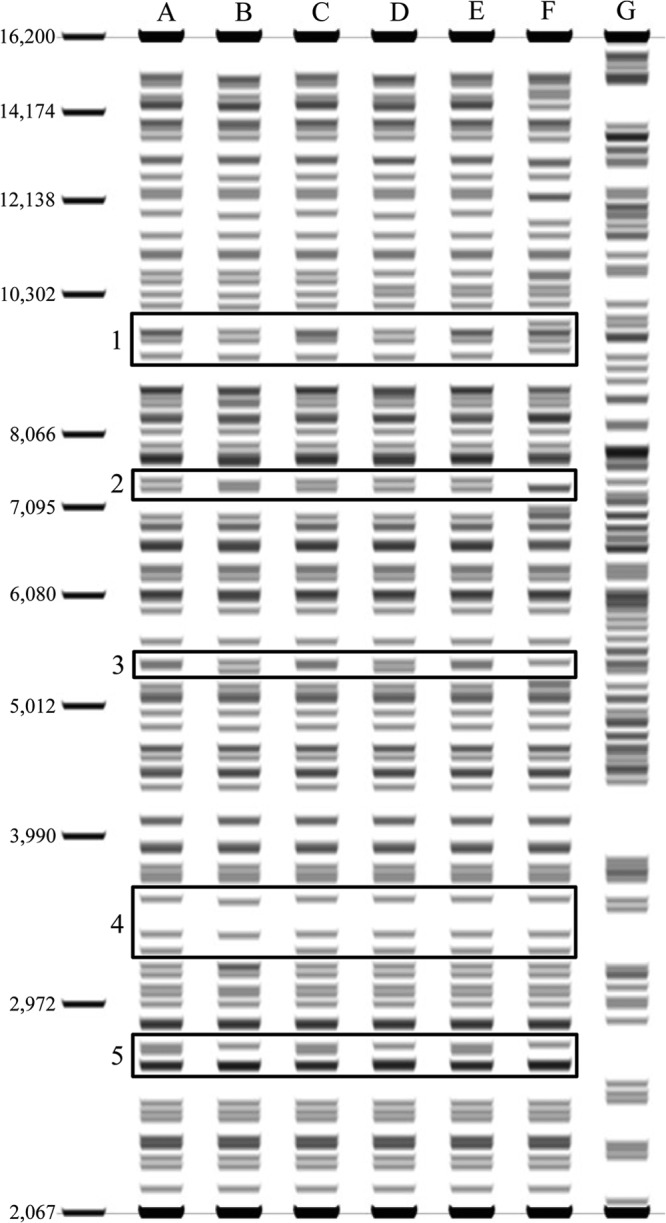
Restriction profiles of B. animalis subsp. lactis strains by in silico prediction. The optical map was generated by in silico digestion with NotI and visualized through Geneious software. The strains analyzed are Bl12 (lane A), BB12 (lane B), BLC1 (lane C), CNCM I-2494 (lane D), DSM10140 (lane E), AD011 (lane F), and B. animalis subsp. animalis ATCC 25527 (lane G); regions of variability are highlighted.
To further explore the level of similarity among the different B. animalis subsp. lactis genomes, we performed a comparative genome analysis using Mauve software, which highlighted a very similar genome sequence for all strains analyzed, with the exception of seven regions where differences were observed (named indel 1 to indel 7). These include the four previously identified B. animalis subsp. lactis insertion/deletion sites (indels), named indel 1 to indel 4 (14), and possibly three additional indels (indel 5 to indel 7) encompassing DNA regions ranging from 22 to 5,422 bp (Table 2). However, PCR efforts together with direct DNA sequencing of the resulting amplicons obtained using PCR primers spanning these seven indel sequences revealed no differences between the analyzed strains except for indel 3 (Table 2), which encompasses the CRISPR locus (see below), and further support the high isogenic nature of the B. animalis subsp. lactis taxon. These findings therefore suggest that indel 1, indel 2, and indel 4 to indel 7 are a consequence of sequencing or assembly mistakes.
Table 2.
Indels identified between B. animalis subsp. lactis Bl12 and the nine publicly available B. animalis subsp. lactis genome sequences
| Strain | Indel positionsa |
||||||
|---|---|---|---|---|---|---|---|
| Indel 1b | Indel 2b | Indel 3c | Indel 4b | Indel 5b | Indel 6b | Indel 7b | |
| Bl12 | 881333–881454 | 902870–902924 | 1715409–1715463 | 1459078–1459101 | 1814984–1815078 | ||
| AD011 | 1607778–1607899 | 1629316–1629370 | 1173764–914478 | 1067909–1067932 | |||
| B420 | 881338–881459 | 902875–902929 | 1715488–1715541 | 1459086–1459109 | |||
| BB12 | 880707–880828 | 902230–902284 | 1304338–1309729 | 1818740–1818832 | |||
| Bi-07 | 881341–881462 | 902878–902932 | 1512259–1512473 | 1715707–17115761 | 1459092–1459114 | ||
| Bl-04 | 881340–881461 | 1512204–1512418 | 1459035–1459058 | ||||
| BLC1 | 881311–881432 | 902848–902902 | 1715408–1715462 | 1459026–1459049 | 1814988–1815081 | ||
| CNCM I-2494 | 881074–881195 | 902611–902665 | 1720186–1720219 | 1304961–1310351 | 1463854–1463877 | ||
| DSM10140 | 902759–902813 | 1715369–1715423 | 1458967–1458990 | ||||
| V9 | 881343–881464 | 902880–902934 | 1720938–1720992 | 1305555–1311003 | 1464537–1464560 | ||
Positions refers to the corresponding strain genome.
The indel was absent from all strains.
The indel was real in all strains, and the PCR validation gave a positive result.
SNP analyses of the B. animalis subsp. lactis genomes.
SNP analysis has recently been developed to compare the genomes of B. animalis subsp. lactis Bl-04 and B. animalis subsp. lactis DSM10140 (14), resulting in the identification of 47 validated SNPs upon comparison of these two complete genome sequences. These 47 validated SNPs may represent a valid reference database for analyzing the genomic variability or polymorphism within the B. animalis subsp. lactis taxon. Thus, we analyzed all genome sequences of B. animalis subsp. lactis strains and B. animalis subsp. animalis ATCC 25527 for the presence or absence of these SNPs. Furthermore, we decided to infer the phylogeny among these strains by analyzing the phylogenetic tree based on a 20-bp sequence region that surrounds each of these SNPs (Fig. 6). This analysis highlighted an evolutionary development of B. animalis subsp. lactis consisting of four phylogenetic clusters encompassing DSM10140 (group 1); CNCM I-2494 (group 2); strains Bl12, BLC1, BB12, V9, AD011, and B420 (group 3); and strains Bl-04 and Bi-07 (group 4).
Fig 6.

Putative SNPs in B. animalis subsp. lactis Bl12 and the nine publicly available B. animalis subsp. lactis genomes. Panel a shows a heat map of the 47 SNPs listed by Barangou et al. (14) mapped on B. animalis subsp. lactis Bl12 and the nine publicly available B. animalis subsp. lactis genomes. Each color represents a base as indicated. The dendrogram shows genome clustering produced by hierarchical clustering based on the heat map data. Panel b depicts a phylogenetic supertree based on B. animalis subsp. lactis Bl12 and the nine publicly available B. animalis subsp. lactis nucleotide sequences corresponding to 20-bp regions spanning the verified SNPs listed by Barrangou et al. (14). The different B. animalis subsp. lactis groups are highlighted.
Furthermore, optical mapping was used to analyze the genome layout of Bl12 as well as of other publicly available B. animalis subsp. lactis strains, such as the BLC1, BB12, CNCM I-2494, AD011, and DSM10140 strains. The resulting optical maps of Bl12, BLC1, and DSM10140 showed very similar patterns, thus suggesting a high degree of genome conservation in terms of size, organization, and sequence within these strains (Fig. 5). In contrast, optical mapping of strains BB12, AD011, and CNCM I-2494 resulted in different restriction profiles compared to those obtained for strains Bl12, DSM10140, and BLC1. Notably, the discrepancies noticed in the optical maps of BB12 and CNCM I-2494 compared to those of strains Bl12, BLC1, and DSM10140 were not confirmed by the DNA sequences of the amplicons spanning the five presumed DNA regions of differences (named 1 to 5) as outlined in the in silico optical map (Fig. 5). Such findings corroborate the apparent quality issues of some of the DNA genome sequences retrieved from public databases, including those of the BB12 strain, which is one of the most intensely commercially exploited bifidobacterial strains.
Evaluation of B. animalis subsp. lactis intraspecific variable genome regions.
In order to investigate the genetic variability occurring between B. animalis subsp. lactis strains, we focused our analyses on those genomic regions that are considered to be highly variable in bifidobacterial chromosomes, representing the so-called mobilome of bifidobacterial genomes (55). These include, for example, (remnants of) prophages, putative pilus biosynthesis genes, genes encoding restriction/modification (R/M) systems, and exopolysaccharide (EPS) biosynthesis gene clusters (55). Some of the regions that specify extracellular structures (e.g., pili or exopolysaccharides) are believed to be involved in the interaction with the host, which may be a strong selective driver for specialization in this specific ecological niche (for a review, see reference 56). In addition, genetic diversity is observed for bifidobacterial genes encoding R/M systems, which protect bacterial cells against acquisition of alien DNA such as bacteriophages (36, 57). Thus, the genome sequences of strains Bl12, BLC1, AD011, BB12, DSM10140, Bi-07, B420, Bl-04, CNCM I-2494, and V9 were analyzed for the presence of these putative mobile or diversity elements. Notably, alignments between homologous regions displayed a high level of identity, ranging from 99% to 100% (see Fig. S1 in the supplemental material). Other DNA sequences considered to represent key components of the bifidobacterial mobilome are transposase-encoding genes (55). The dissection of Bl12 and all publicly available genome sequences of B. animalis subsp. lactis for transposases revealed an identical data set. Another genetic locus, which is known to be highly variable at the intraspecies level in bifidobacteria, is encompassed by the cluster of regularly interspersed short palindromic repeats (CRISPR) loci (55), which includes DNA repeats and the cas genes (CRISPR-associated genes) (58). CRISPR loci represent the most widely distributed family of repeats among prokaryotic genomes (59, 60), acting as a defense system against the invasion of foreign genetic material, in particular phages (58, 61). As previously described for B. animalis subsp. lactis Bl-04 and B. animalis subsp. lactis DSM10140 (14), CRISPR loci represent genetic regions of the B. animalis subsp. lactis genome where polymorphisms have been identified. Interestingly, a genetic survey of all 10 B. animalis subsp. lactis genome sequences revealed the presence of a 36-bp CRISPR repeat, 5′-ATCTCCGAAGTCTCGGCTTCGGAGCTTCATTGAGGG-3′. Furthermore, the CRISPR locus of the Bl12 genome was shown to encompass 19 repeats instead of the previously identified 20 copies of this repeat (as present in the genomes of strains DSM10140, BB12, V9, B420, V9, CNCM I-2494, and BLC1 [13, 14]) or 23 copies (as present in the genomes of strains Bl-04 and Bi-07 [14, 18]).
The B. animalis subsp. lactis pan-genome.
In order to evaluate the total gene repertoire of the B. animalis subsp. lactis taxon, i.e., the B. animalis subsp. lactis pan-genome, we used a previously described methodology (45), which calculates both the overall number of genes discovered and the expected number of new genes contributed by each additional genomic sequence, using the same permutation scheme as employed in the analysis of core genes. The total number of different genes identified when all 10 genomes are compared is 1,518 (Fig. 7). The pan-genome size, when plotted on a log-log scale versus the number of genomes, shows a clear asymptotic behavior, and a data regression analysis, based on the Heaps law pan-genome model (45), found a robust fit for α = 3.37 ± 0.014, in accordance with a closed pan-genome state, which clearly supports the idea that B. animalis subsp. lactis has a closed pan-genome.
Fig 7.

The distribution of the number of total genes (a) and core genes (b) found upon sequential addition of n genomes. In panel a, power law fit to the pan-genome size is shown as solid curve. In panel b, an exponential regression to core genome data is shown as a solid curve.
The number of new genes discovered by sequential addition of genome sequences of B. animalis subsp. lactis is shown in Fig. 7. Notably, the number of specific genes added to the pan-genome dramatically decreases after the addition of the third strain. This result probably reflects the fact that B. animalis subsp. lactis is a highly clonal, recently evolved taxon of the Bifidobacterium animalis species in which genome variability is associated only with SNPs or indels (see above). These findings highlight that further efforts toward genomic sequencing of other B. animalis subsp. lactis strains are unlikely to result in the discovery of new genes within this taxon, since all its genetic variability seems to have been resolved and detected with the currently available genome sequences.
Recently, the analysis of publicly available genomes from bifidobacterial species, including Bifidobacterium longum subsp. longum, Bifidobacterium longum subsp. infantis, Bifidobacterium breve, Bifidobacterium adolescentis, Bifidobacterium dentium, Bifidobacterium bifidum, and B. animalis subsp. lactis, showed that such bifidobacterial genomes display an open pan-genome structure (55). Mathematical extrapolation of the data indicates that the genome reservoir available to the bifidobacterial pan-genome consists of more than 5,000 genes (55). The pan-genomic structure of microbial genomes is also influenced by the ecological niche where bacteria reside. An open pan-genome is commonly found for those species that colonize multiple habitats and possess diverse ways of exchanging genetic material. Microorganisms such as streptococci, meningococci, Helicobacter pylori, Salmonella species, and Escherichia coli possess these features and display an open-pangenomic structure (62). In contrast, it is known that bacterial species residing in restricted environments and lacking mechanisms of gene exchange may have evolved with considerably less genome variation. Bacteria such as Buchnera aphidicola or Bacillus anthracis possess a closed pan-genome, where no or very limited chromosome rearrangements or gene acquisitions have occurred during the course of evolution (63). A closer look at the structures of the genetic trees of open pan-genomic taxa and closed pan-genomic species (e.g., Buchnera aphidicola or Bacillus anthracis) indicates that the latter species resemble a clone organization rather than being a true independent species. Thus, the identification of a closed pan-genomic structure of B. animalis subsp. lactis might provide further genetic evidence of the clonal origin of this taxon.
In addition, the closed pan-genomic structure of B. animalis subsp. lactis subspecies might be a consequence of the worldwide distribution of this taxon as a health-promoting bacterium and to its limited ability to colonize and persist within the human host. This might reduce the possibility that alien DNA is acquired by members of the B. animalis subsp. lactis taxon.
Conclusions.
In this report, we describe the complete genome sequence of the recently identified B. animalis subsp. lactis Bl12 strain and its use in establishing the genetic variability among known members of the B. animalis subsp. lactis taxon. Bl12 represents the first strain of B. animalis subsp. lactis possessing a clear human ecological origin (being isolated from a human colonoscopic sample) and a phenotype (higher susceptibility to tetracycline) which is different from that displayed by other characterized strains of this species. This strain was isolated from a healthy individual that had not consumed probiotic products. In silico analyses of the Bl12 strain revealed limited genetic diversity which is restricted to SNPs, one of which corresponds to miaA and which may be responsible for the reduced tetracycline resistance compared to those of other tested B. animalis subsp. lactis strains. Overall, the very high genome sequence similarity observed within members of B. animalis subsp. lactis as well as the close evolutionary distances of this investigated strain collection revealed a high degree of genome conservation in terms of size, organization, and sequence. This lack of polymorphism is indicative of a genomically monomorphic subspecies and an isogenic nature of all B. animalis subsp. lactis strains. These findings are also supported by the closed pan-genome structure of the B. animalis subsp. lactis taxon, which clearly suggests that no novel genes will be discovered by further genomic attempts. This result probably reflects the fact that B. animalis subsp. lactis is a highly clonal, recently evolved taxon from the B. animalis species. Alternatively, the sequenced strains may belong to the same evolutionary clade and may not adequately represent the diversity that exists within strains belonging to B. animalis subsp. lactis.
The low level of genetic variability displayed by this taxon of bacterium as revealed in this study has important implications in terms of the use of various B. animalis subsp. lactis strains as health-promoting bacteria. The apparent lack of major genomic differences among the 10 analyzed B. animalis subsp. lactis strains suggests that these strains exert similar, if not identical, health-promoting activities. Furthermore, this study revealed the existence of microvariability of the miaA gene within members of the B. animalis subsp. lactis taxon, while also identifying the first B. animalis subsp. lactis strain that appears to be an authochtonous member of the human gut microbiota.
Supplementary Material
ACKNOWLEDGMENTS
We thank GenProbio srl for the financial support of the Laboratory of Probiogenomics. This work was financially supported by Fondazione Cariparma to M.V. and by a FEMS Advanced Fellowship 2011 and an IRCSET Embark postdoctoral fellowship to F.T. and to Spinner, Emilia Romagna, to S.D. D.V.S. is a member of The Alimentary Pharmabiotic Centre, which is a Centre for Science and Technology (CSET) funded by the Science Foundation Ireland (SFI) through the Irish government's National Development Plan (grant no. 07/CE/B1368).
Footnotes
Published ahead of print 3 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00984-13.
REFERENCES
- 1. Chenoll E, Casinos B, Bataller E, Astals P, Echevarria J, Iglesias JR, Balbarie P, Ramon D, Genoves S. 2011. Novel probiotic Bifidobacterium bifidum CECT 7366 strain active against the pathogenic bacterium Helicobacter pylori. Appl. Environ. Microbiol. 77:1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shirasawa Y, Shibahara-Sone H, Iino T, Ishikawa F. 2010. Bifidobacterium bifidum BF-1 suppresses Helicobacter pylori-induced genes in human epithelial cells. J. Dairy Sci. 93:4526–4534 [DOI] [PubMed] [Google Scholar]
- 3. Khailova L, Mount Patrick SK, Arganbright KM, Halpern MD, Kinouchi T, Dvorak B. 2010. Bifidobacterium bifidum reduces apoptosis in the intestinal epithelium in necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 299:G1118–G1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guglielmetti S, Mora D, Gschwender M, Popp K. 2011. Randomised clinical trial: Bifidobacterium bifidum MIMBb75 significantly alleviates irritable bowel syndrome and improves quality of life—a double-blind, placebo-controlled study. Aliment. Pharmacol. Ther. 33:1123–1132 [DOI] [PubMed] [Google Scholar]
- 5. Malago JJ, Tooten PC, Koninkx JF. 2010. Anti-inflammatory properties of probiotic bacteria on Salmonella-induced IL-8 synthesis in enterocyte-like Caco-2 cells. Benef. Microbes 1:121–130 [DOI] [PubMed] [Google Scholar]
- 6. Ventura M, Turroni F, Motherway MO, MacSharry J, van Sinderen D. 2012. Host-microbe interactions that facilitate gut colonization by commensal bifidobacteria. Trends Microbiol. 20:467–476 [DOI] [PubMed] [Google Scholar]
- 7. Ventura M, O'Flaherty S, Claesson MJ, Turroni F, Klaenhammer TR, van Sinderen D, O'Toole PW. 2009. Genome-scale analyses of health-promoting bacteria: probiogenomics. Nat. Rev. Microbiol. 7:61–71 [DOI] [PubMed] [Google Scholar]
- 8. Turroni F, van Sinderen D, Ventura M. 2011. Genomics and ecological overview of the genus Bifidobacterium. Int. J. Food Microbiol. 149:37–44 [DOI] [PubMed] [Google Scholar]
- 9. Ventura M, Canchaya C, Zhang Z, Bernini V, Fitzgerald GF, van Sinderen D. 2006. How high G+C Gram-positive bacteria and in particular bifidobacteria cope with heat stress: protein players and regulators. FEMS Microbiol. Rev. 30:734–759 [DOI] [PubMed] [Google Scholar]
- 10. Ventura M, Turroni F, van Sinderen D. 2012. Probiogenomics as a tool to obtain genetic insights into adaptation of probiotic bacteria to the human gut. Bioeng. Bugs 3:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee JH, O'Sullivan DJ. 2010. Genomic insights into bifidobacteria. Microbiol. Mol. Biol. Rev. 74:378–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garrigues C, Johansen E, Pedersen MB. 2010. Complete genome sequence of Bifidobacterium animalis subsp. lactis BB-12, a widely consumed probiotic strain. J. Bacteriol. 192:2467–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bottacini F, Dal Bello F, Turroni F, Milani C, Duranti S, Foroni E, Viappiani A, Strati F, Mora D, van Sinderen D, Ventura M. 2011. Complete genome sequence of Bifidobacterium animalis subsp. lactis BLC1. J. Bacteriol. 193:6387–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barrangou R, Briczinski EP, Traeger LL, Loquasto JR, Richards M, Horvath P, Coute-Monvoisin AC, Leyer G, Rendulic S, Steele JL, Broadbent JR, Oberg T, Dudley EG, Schuster S, Romero DA, Roberts RF. 2009. Comparison of the complete genome sequences of Bifidobacterium animalis subsp. lactis DSM 10140 and Bl-04. J. Bacteriol. 191:4144–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim JF, Jeong H, Yu DS, Choi SH, Hur CG, Park MS, Yoon SH, Kim DW, Ji GE, Park HS, Oh TK. 2009. Genome sequence of the probiotic bacterium Bifidobacterium animalis subsp. lactis AD011. J. Bacteriol. 191:678–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chervaux C, Grimaldi C, Bolotin A, Quinquis B, Legrain-Raspaud S, van Hylckama Vlieg JE, Denariaz G, Smokvina T. 2011. Genome sequence of the probiotic strain Bifidobacterium animalis subsp. lactis CNCM I-2494. J. Bacteriol. 193:5560–5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun Z, Chen X, Wang J, Gao P, Zhou Z, Ren Y, Sun T, Wang L, Meng H, Chen W, Zhang H. 2010. Complete genome sequence of probiotic Bifidobacterium animalis subsp. lactis strain V9. J. Bacteriol. 192:4080–4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stahl B, Barrangou R. 2012. Complete genome sequences of probiotic strains Bifidobacterium animalis subsp. lactis B420 and Bi-07. J. Bacteriol. 194:4131–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ventura M, Reniero R, Zink R. 2001. Specific identification and targeted characterization of Bifidobacterium lactis from different environmental isolates by a combined multiplex-PCR approach. Appl. Environ. Microbiol. 67:2760–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ventura M, Zink R. 2003. Comparative sequence analysis of the tuf and recA genes and restriction fragment length polymorphism of the internal transcribed spacer region sequences supply additional tools for discriminating Bifidobacterium lactis from Bifidobacterium animalis. Appl. Environ. Microbiol. 69:7517–7522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Briczinski EP, Loquasto JR, Barrangou R, Dudley EG, Roberts AM, Roberts RF. 2009. Strain-specific genotyping of Bifidobacterium animalis subsp. lactis by using single-nucleotide polymorphisms, insertions, and deletions. Appl. Environ. Microbiol. 75:7501–7508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. International Organization for Standardization 2010. ISO 10932:2010 (IDF 223:2010). Milk and milk products—determination of the minimal inhibitory concentration (MIC) of antibiotics applicable to bifidobacteria and non-enterococcal lactic acid bacteria (LAB). International Organization for Standardization, Geneva, Switzerland [Google Scholar]
- 23. Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WE, Wetter T, Suhai S. 2004. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14:1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gish W, States DJ. 1993. Identification of protein coding regions by database similarity search. Nat. Genet. 3:266–272 [DOI] [PubMed] [Google Scholar]
- 25. Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945 [DOI] [PubMed] [Google Scholar]
- 26. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 28. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roberts RJ, Vincze T, Posfai J, Macelis D. 2010. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 38:D234–D236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Busch W, Saier MH., Jr 2002. The transporter classification (TC) system, 2002. Crit. Rev. Biochem. Mol. Biol. 37:287–337 [DOI] [PubMed] [Google Scholar]
- 31. Loquasto JR, Barrangou R, Dudley EG, Roberts RF. 2011. Short communication: the complete genome sequence of Bifidobacterium animalis subspecies animalis ATCC 25527(T) and comparative analysis of growth in milk with B. animalis subspecies lactis DSM 10140(T). J. Dairy Sci. 94:5864–5870 [DOI] [PubMed] [Google Scholar]
- 32. Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, Zwahlen MC, Desiere F, Bork P, Delley M, Pridmore RD, Arigoni F. 2002. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl. Acad. Sci. U. S. A. 99:14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, Lapidus A, Rokhsar DS, Lebrilla CB, German JB, Price NP, Richardson PM, Mills DA. 2008. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. U. S. A. 105:18964–18969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turroni F, Bottacini F, Foroni E, Mulder I, Kim JH, Zomer A, Sanchez B, Bidossi A, Ferrarini A, Giubellini V, Delledonne M, Henrissat B, Coutinho P, Oggioni M, Fitzgerald GF, Mills D, Margolles A, Kelly D, van Sinderen D, Ventura M. 2010. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc. Natl. Acad. Sci. U. S. A. 107:19514–19519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ventura M, Turroni F, Zomer A, Foroni E, Giubellini V, Bottacini F, Canchaya C, Claesson MJ, He F, Mantzourani M, Mulas L, Ferrarini A, Gao B, Delledonne M, Henrissat B, Coutinho P, Oggioni M, Gupta RS, Zhang Z, Beighton D, Fitzgerald GF, O'Toole PW, van Sinderen D. 2009. The Bifidobacterium dentium Bd1 genome sequence reflects its genetic adaptation to the human oral cavity. PLoS Genet. 5:e1000785. 10.1371/journal.pgen.1000785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Connell Motherway M, Zomer A, Leahy SC, Reunanen J, Bottacini F, Claesson MJ, O'Brien F, Flynn K, Casey PG, Munoz JA, Kearney B, Houston AM, O'Mahony C, Higgins DG, Shanahan F, Palva A, de Vos WM, Fitzgerald GF, Ventura M, O'Toole PW, van Sinderen D. 2011. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proc. Natl. Acad. Sci. U. S. A. 108:11217–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pearson WR. 2000. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol. Biol. 132:185–219 [DOI] [PubMed] [Google Scholar]
- 38. van Dongen S. 2000. Graph clustering by flow simulation. Ph.D. thesis. University of Utrecht, Utrecht, The Netherlands [Google Scholar]
- 39. Thompson JD, Gibson TJ, Higgins DG. 2002. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics 2002:2.3.1–2.3.22. 10.1002/0471250953.bi0203s00 [DOI] [PubMed] [Google Scholar]
- 40. Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704 [DOI] [PubMed] [Google Scholar]
- 41. Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378 [DOI] [PubMed] [Google Scholar]
- 43. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. 2012. PGAP: pan-genomes analysis pipeline. Bioinformatics 28:416–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tettelin H, Riley D, Cattuto C, Medini D. 2008. Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11:472–477 [DOI] [PubMed] [Google Scholar]
- 46. Gueimonde M, Florez AB, van Hoek AH, Stuer-Lauridsen B, Stroman P, de los Reyes-Gavilan CG, Margolles A. 2010. Genetic basis of tetracycline resistance in Bifidobacterium animalis subsp. lactis. Appl. Environ. Microbiol. 76:3364–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ammor MS, Florez AB, Alvarez-Martin P, Margolles A, Mayo B. 2008. Analysis of tetracycline resistance tet(W) genes and their flanking sequences in intestinal Bifidobacterium species. J. Antimicrob. Chemother. 62:688–693 [DOI] [PubMed] [Google Scholar]
- 48. Flórez AB, Ammor MS, Alvarez-Martin P, Margolles A, Mayo B. 2006. Molecular analysis of tet(W) gene-mediated tetracycline resistance in dominant intestinal Bifidobacterium species from healthy humans. Appl. Environ. Microbiol. 72:7377–7379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee JH, Karamychev VN, Kozyavkin SA, Mills D, Pavlov AR, Pavlova NV, Polouchine NN, Richardson PM, Shakhova VV, Slesarev AI, Weimer B, O'Sullivan DJ. 2008. Comparative genomic analysis of the gut bacterium Bifidobacterium longum reveals loci susceptible to deletion during pure culture growth. BMC Genomics 9:247. 10.1186/1471-2164-9-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Scott KP, Barbosa TM, Forbes KJ, Flint HJ. 1997. High-frequency transfer of a naturally occurring chromosomal tetracycline resistance element in the ruminal anaerobe Butyrivibrio fibrisolvens. Appl. Environ. Microbiol. 63:3405–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Burdett V. 1993. tRNA modification activity is necessary for Tet(M)-mediated tetracycline resistance. J. Bacteriol. 175:7209–7215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Henz SR, Huson DH, Auch AF, Nieselt-Struwe K, Schuster SC. 2005. Whole-genome prokaryotic phylogeny. Bioinformatics 21:2329–2335 [DOI] [PubMed] [Google Scholar]
- 53. Chan CX, Beiko RG, Ragan MA. 2006. Detecting recombination in evolving nucleotide sequences. BMC Bioinformatics 7:412. 10.1186/1471-2105-7-412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, van Sinderen D. 2007. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 71:495–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bottacini F, Medini D, Pavesi A, Turroni F, Foroni E, Riley D, Giubellini V, Tettelin H, van Sinderen D, Ventura M. 2010. Comparative genomics of the genus Bifidobacterium. Microbiology 156:3243–3254 [DOI] [PubMed] [Google Scholar]
- 56. Ventura M, Canchaya C, Fitzgerald GF, Gupta RS, van Sinderen D. 2007. Genomics as a means to understand bacterial phylogeny and ecological adaptation: the case of bifidobacteria. Antonie Van Leeuwenhoek 91:351–372 [DOI] [PubMed] [Google Scholar]
- 57. O'Connell Motherway M, Fitzgerald GF, van Sinderen D. 2011. Metabolism of a plant derived galactose-containing polysaccharide by Bifidobacterium breve UCC2003. Microb. Biotechnol. 4:403–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712 [DOI] [PubMed] [Google Scholar]
- 59. Jansen R, Embden JD, Gaastra W, Schouls LM. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 43:1565–1575 [DOI] [PubMed] [Google Scholar]
- 60. Mojica FJ, Diez-Villasenor C, Soria E, Juez G. 2000. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 36:244–246 [DOI] [PubMed] [Google Scholar]
- 61. Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 1:7. 10.1186/1745-6150-1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. 2005. The microbial pan-genome. Curr. Opin. Genet. Dev. 15:589–594 [DOI] [PubMed] [Google Scholar]
- 63. Tamas I, Klasson L, Canback B, Naslund AK, Eriksson AS, Wernegreen JJ, Sandstrom JP, Moran NA, Andersson SG. 2002. 50 million years of genomic stasis in endosymbiotic bacteria. Science 296:2376–2379 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.