

Abstract
Mutations in the ribosomal protein L22 that impair peptide-mediated translation arrest in Escherichia coli have been shown to reduce the expression of several genes, including secA, which encodes an ATPase that drives protein export via the Sec pathway. Here, we used a comparative proteomic approach to obtain insight into the global effects of the L22(Δ82-84) mutation on gene expression and protein synthesis. While the mutation did not affect or modestly affected the level of most soluble proteins, it dramatically reduced the level of antigen 43 (Ag43), a secreted virulence factor that promotes autoaggregation. The reduced protein concentration correlated with a sharp decrease in the abundance and stability of Ag43 mRNA. We found that the overexpression of secA or the inactivation of genes that encode presecretory and membrane proteins restored Ag43 production in the L22 mutant strain. Furthermore, impairment of the Sec pathway in a wild-type strain reduced Ag43 production but did not significantly affect the synthesis of other presecretory proteins. Taken together, these results indicate that Ag43 gene expression is exquisitely sensitive to the status of the Sec machinery and strongly suggest that the L22 mutation decreases the Ag43 concentration indirectly by reducing secA expression. Our results imply the existence of a novel regulatory mechanism in which the efficiency of protein export is coupled to gene expression and help to explain the modulation of SecA synthesis that has been observed in response to secretion stress.
INTRODUCTION
Nascent polypeptides travel through a long water-filled tunnel in the large ribosomal subunit that extends from the peptidyltransferase center (PTC) to an exit site ∼100 Å away before entering the cytoplasm. The ribosome tunnel is, on average, only ∼15 Å in diameter and highly irregular in shape (1). The surface of the tunnel is comprised primarily of rRNA, but in bacteria it also contains the nonglobular segments of three ribosomal proteins. Two of the proteins, called L4 and L22, are located relatively close to the tunnel entrance, and the third protein, L23, resides near the exit. A conserved β hairpin loop of L22 is situated in close proximity to L4 at the narrowest part of the tunnel (the constriction point) about 30 Å from the PTC.
Despite over 40 years of intensive research on the ribosome, the function of the ribosome tunnel has remained obscure. Although it is conceivable that the tunnel evolved simply to direct nascent chains away from the PTC and to insulate them from the cytosolic environment, there is growing evidence that it plays a significant role both in protein biogenesis and the regulation of protein synthesis. The rate of translation, for example, can be influenced by the considerable variation in electrostatic potential that exists at different sites in the tunnel (2). Moreover, although early studies showed that ribosomes protect ∼30 to 40 amino acids of typical nascent chains and suggested that polypeptides traverse the tunnel in an extended conformation (3, 4), recent work has shown that at least some nascent chains adopt specific, more compact conformations (5–8). In at least one instance, the conformation of a peptide inside the ribosome tunnel appears to affect protein folding (9). Furthermore, the detection of specific polypeptides inside the tunnel has been shown to promote the binding of protein targeting and translocation factors or affect ribosome activity. The presence of a membrane spanning segment inside the tunnel, for example, stimulates the recruitment of the signal recognition particle and a protein associated with the Sec complex in the endoplasmic reticulum (10, 11). Remarkably, the recognition of specific peptide sequence motifs can also arrest translation elongation or termination (12). This cis-acting translational attenuation serves to regulate the expression of a downstream gene and frequently requires a threshold concentration of a small molecule. In Escherichia coli, a 24-amino-acid leader peptide encoded by the tnaC gene inhibits its own termination in the presence of tryptophan and thereby activates the transcription of a downstream operon (13). Likewise, the expression of several different antibiotic resistance genes is regulated by the production of a leader peptide (14). In the presence of low levels of the antibiotic, elongation of the nascent peptide stalls at a specific position, and a resulting alteration of the structure of the mRNA activates translation of the downstream gene. Intriguingly, the ribosome stalling peptides identified to date vary in length and share no sequence homology.
Small molecules are not the only physiological signals that regulate peptide-mediated translation arrest. In E. coli, the synthesis of SecA, an essential ATPase that drives protein translocation through the SecYEG complex and facilitates the insertion of many inner membrane (IM) proteins, increases as much as 5- to 10-fold under secretion-defective conditions in which the Sec machinery is compromised experimentally (15). secA expression is regulated at the translational level by secM, a gene encoding a presecretory protein that resides immediately upstream of secA in the same operon. SecM contains a C-terminal sequence motif whose recognition inside the ribosome tunnel induces translation arrest (16). Under normal conditions, translation is arrested transiently and is rapidly released by a physical force exerted by the Sec machinery during translocation (17, 18). During the transient translation arrest, ribosomes briefly denature a hairpin in the secM-secA mRNA that masks the secA Shine-Dalgarno sequence and thereby facilitate the synthesis of a basal level of SecA (19). Experimental manipulations that impair SecM export dramatically prolong the translation arrest and lead to an increased production of SecA by increasing the exposure of the Shine-Dalgarno sequence (20–22). The physiological benefits of the regulation of SecA synthesis, however, are not entirely clear. While it is tempting to speculate that this regulatory process evolved to enable cells to adapt to changes in the level of secreted proteins, conditions that increase the secretory burden have not been reported. The ability to increase secA expression so far has only been shown to be required to compensate for an inherent cold sensitivity of Sec pathway function and to maintain cell growth at 20°C (19).
In this study, we sought to gain insight into the function of the ribosome tunnel at a global level by examining the effect of a deletion in the L22 β hairpin loop [L22 Δ82-84] on the composition of the E. coli proteome. The L22(Δ82-84) mutation increases the diameter of the ribosome tunnel and confers erythromycin resistance (23, 24). More significantly, this and other mutations in the L22 β hairpin loop have been shown to impair the recognition of several different translation arrest-inducing peptides and to affect protein folding (9, 16, 25–29). We hypothesized that the mutation would significantly alter the level of any protein whose synthesis or stability is sensitive to perturbation of the tunnel environment. In principle, because the L22 mutation impairs the recognition of the SecM arrest motif and thereby suppresses SecA synthesis (16, 19, 28), it might reduce the secretion of specific proteins and lead to their degradation in the cytoplasm. The mutation might also affect the level of other proteins whose synthesis depends on the recognition of an uncharacterized upstream translation arrest motif or whose stability depends on early folding events that occur inside the tunnel. Surprisingly, we found that the L22(Δ82-84) mutation did not affect or only modestly affected the levels of most soluble proteins. The level of a secreted protein called antigen 43 (Ag43), however, was drastically reduced in the mutant strain. Ag43 is a member of the autotransporter family of virulence factors that promotes autoaggregation, biofilm formation, and long-term colonization in a mouse model of urinary tract infections (30, 31). The transcription of the Ag43 gene is regulated by a phase variation mechanism that involves a competition between two global regulatory proteins, OxyR and the Dam methylase. Our results strongly suggest that the reduced level of SecA in the L22(Δ82-84) mutant strain impairs secretion and thereby activates a previously unrecognized feedback mechanism that leads to the destabilization of Ag43 mRNA. Our findings not only reveal a novel connection between secretion and gene expression but also help to explain why the level of SecA is so finely regulated.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
E. coli strain MG1655 (F− λ− rfb-50 rph-1 ilvG) and its derivatives, MNY15 [rplV(Δ82-84)], MNY16 (rplV ins105/2), MNY34 (Ag43::cat), MNY37 (secY39cs zhd::tet), MNY40 (Ag43::His6), and HDB148 [rplW(Δ65-74) rpsL150(strR)], were used in all experiments. Mutations in the gene that encodes L22 (rplV) were introduced into the MG1655 chromosome by homologous recombination using the Lambda Red method (32) to create MNY15 and MNY16. To generate MNY34 and MNY40, a 1.1-kb DNA fragment encoding the cat gene was amplified by PCR using oligonucleotide pairs P558/P559 and P686/P687 and pKD3 (32) as a template (all oligonucleotide sequences are listed in Table S1 in the supplemental material). The cat gene was removed from MNY40 by FLP recombination target (FRT)-mediated excision (32). To construct HDB148, the rplW(Δ65-74) allele [encoding L23(loopΔ10)] was moved from strain HDB143 (9) into MG1655 by P1 transduction. Cells were grown at 37°C in LB supplemented with ampicillin (100 μg/ml) as needed.
Plasmid construction.
Plasmid pMF8 is a pBR322 derivative that contains the secM-secA locus (33). The SecA(D133N) mutation was introduced into pMF8 using the oligonucleotide pair P714/P715 and the QuikChange mutagenesis kit (Stratagene). Plasmid pMY28, which encodes the hemagglutinin (HA)-tagged passenger domain of Ag43 (Ag43PD-HA), and pMY29, which encodes the HA-tagged passenger domain lacking its signal peptide (ΔSP-Ag43PD-HA), were constructed by first amplifying portions of the Ag43 gene by PCR using the oligonucleotide pairs P600/P690 and P604/P690 and genomic DNA from MG1655 as a template. The DNA fragments were then digested with EcoRI and HindIII and cloned into the cognate sites of pTrc99A (34).
2D-DIGE.
For two-dimensional difference gel electrophoresis (2D-DIGE), MG1655 and MNY15 cultures (500 ml) were grown to an optical density at 600 nm (OD600) of ∼0.8. Cells were poured over ice and pelleted (7,500 × g, 15 min, 4°C). Pellets were washed twice with buffer A [10 mM Tris, pH 8, 5 mM Mg(OAc)2, 0.5 mM phenylmethylsulfonyl fluoride (PMSF)] and resuspended in buffer A (0.5 ml per gram of wet weight). The cell suspension was passed through a French press three times at 8,000 lb/in2, and cell debris was removed by centrifuging the samples twice (23,420 × g, 30 min, 4°C). Membranes were then removed by ultracentrifugation (125,000 × g, 30 min, 4°C). Total soluble proteins (300 μg) were labeled with Cy3 (MG1655) and Cy5 (MNY15) using the CyDye DIGE Fluors kit (GE Life Sciences) according to the manufacturer's instructions.
2D-DIGE (35), quantitative analysis, and mass spectrometry were performed by Applied Biomics (Hayward, CA). Gels were scanned following SDS-PAGE using a Typhoon Trio imager (GE Healthcare). The images were then analyzed by ImageQuant software (version 6.0), and the fold difference in protein levels was determined using DeCyder software (version 6.5). Spots of interest were excised using an Ettan spot picker (Amersham BioSciences), and proteins were subjected to in-gel digestion with modified porcine trypsin (Trypsin Gold; Promega). The tryptic peptides were desalted using C18 ZipTips (Millipore) and eluted in 0.5 μl matrix solution (5 mg/ml α-cyano-4-hydroxycinnamic acid in 50% acetonitrile, 0.1% trifluoroacetic acid, 25 mM ammonium bicarbonate). Samples were then placed in an AB SCIEX MALDI Opti-TOF 384-well insert plate, and matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI-TOF MS) and tandem TOF (TOF/TOF) MS were performed using an AB SCIEX TOF/TOF 5800 system. MALDI-TOF mass spectra were acquired in reflectron-positive ion mode, averaging 4,000 laser shots per spectrum. TOF/TOF tandem MS fragmentation spectra were acquired for the 10 most abundant ions in each sample (excluding trypsin autolytic peptide ions and other known background ions), averaging 4,000 laser shots per spectrum. Both peptide mass and the associated fragmentation spectra were submitted to a Mascot search engine (Matrix Science) to search the National Center for Biotechnology Information nonredundant protein database (NCBInr) for possible matches. Searches were performed without constraining the protein molecular weight or isoelectric point. The search parameters were set to allow variable carbamidomethylation of cysteine residues, variable oxidation of methionine residues, and one missed cleavage. Confidence intervals of >95% were considered significant.
Autoaggregation assay.
Overnight cultures were diluted 1:100 into 4 ml of fresh media and grown to an OD600 of ∼0.8. Culture tubes were then placed in a rack at room temperature overnight. Cell clumps that settle at the bottom of the tubes are indicative of an autoaggregative (flu+) phenotype. Phenotypic tests were repeated at least three times with 10 parallel cultures seeded with randomly chosen colonies to ensure that the results were not skewed by extremes in the phase variation of Ag43 gene expression.
Tn5 mutagenesis and insertion mapping.
Random Tn5 insertions were introduced into the chromosome of MNY15 using the EZ-TN5<R6Kγori/Kan-2> Tnp transposome kit (Epicentre) by following the manufacturer's protocol. Kanamycin-resistant colonies were selected and screened for the restoration of a flu+ phenotype. To identify the Tn5 insertion sites on the chromosome of the flu+ clones, total genomic DNA was isolated using the Wizard genomic DNA purification kit (Promega) and was digested with one or more restriction enzymes. Digested DNA fragments were self ligated and propagated in E. coli DH5αλpir on LB agar plates containing kanamycin. Bidirectional DNA sequencing was conducted using the outward-facing, Tn5-specific oligonucleotides R6KAN-2 RP-1 and KAN-2 FP-1 supplied in the kit to map the Tn5-disrupted genes. Finally, genes disrupted by Tn5 insertions were introduced into a clean MNY15 background by P1 transduction.
Quantitative RT-PCR.
Total RNA was isolated from cultures grown to an OD600 of ∼0.7 to 0.8 using a modified hot phenol-SDS extraction method (36) and the RNeasy kit (Qiagen). DNA contaminants were then removed using two successive digestions with Turbo DNase I (Ambion), and the integrity of the RNA was verified using the Bioanalyzer RNA 6000 Nano kit (Agilent). cDNA was then synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosystem) with 1 μg of total RNA in a 20-μl reaction mixture. Reverse transcription (RT) was performed at 25°C for 10 min, 37°C for 120 min, and 85°C for 5 min. Primers were designed using Primer Express 3.0 software (Applied Biosystems) and were specific to the Ag43 gene (P486/P487, passenger domain; P699/P700, β domain), secA (P551/P552), tnaA (P435/P436), and the internal reference gene, gapA (P405/P406). The gapA transcript is often used as an internal standard in quantitative RT-PCR experiments because it is especially stable (based on the geNorm algorithm; see reference 37 and http://medgen.ugent.be/∼jvdesomp/genorm) and because its level remains constant in different strain backgrounds and under different growth conditions. Quantitative PCRs were performed in triplicate using an Applied Biosystems 7200 real-time PCR detection system. Each 20-μl reaction mixture contained the Power SYBR green master mix, primers (100 nM), and 1 μl of cDNA. PCRs consisted of a denaturation cycle (50°C, 2 min; 95°C, 10 min), 40 amplification and quantification cycles (95°C, 15 s; 60°C, 1 min), and a dissociation step (95°C, 15 s; 60°C, 1 min; 95°C, 15 s). The threshold cycle (CT) was determined using the manufacturer's software with default settings. To ensure that the RNA was free of DNA contamination, a parallel set of quantitative PCRs was performed using RNA that was not subject to reverse transcription. Differences in mRNA levels were calculated using a published formula (38).
Rifampin chase.
MNY40 cultures harboring appropriate plasmids were divided in half at an OD600 of 0.4, and 500 μM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to one half. After 30 min, rifampin (200 μg/ml) was added to both halves and allowed to equilibrate for 2 min. At that time point (which was defined as t0) and at subsequent time points, 3-ml aliquots of cells were withdrawn for RNA extraction and quantitative RT-PCR. mRNA decay relative to t0 was calculated as described previously (38, 39).
Analysis of steady-state protein levels.
Overnight cultures were washed and diluted into fresh medium at an OD600 of 0.2. When cultures reached an OD600 of 0.8, 1-ml aliquots were removed from each culture and subjected to trichloroacetic acid (TCA) precipitation. TCA precipitates were then solubilized in SDS-PAGE sample buffer, and proteins were resolved on 8 to 16% Tris-glycine minigels (Invitrogen) or Mini-PROTEAN AnykD TGX gels (Bio-Rad) and transferred to nitrocellulose using an iBlot system (Invitrogen). Polyclonal rabbit antisera against Ffh, BamA, and YidC have been described previously (40–42). A polyclonal antiserum against the OmpC peptide (NH2-QSKGKNLGRGYDDGDILKYVD-COOH) was generated by Wanyoike Kang'ethe. Additional polyclonal antisera were obtained from Don Oliver (SecA), Peter Owens (Ag43 β domain), Gigi Storz (RpoD), P. C. Tai (OmpA), Jon Beckwith (DegP), Loan Lascu (Ndk), Gary Jacobson (MtlA), New England BioLabs (MalE), Stressgen (DsbA), Santa Cruz Biotechnology (HA epitope), and Qiagen (Penta-His). Horseradish peroxidase (HRP)-linked protein A (Amersham) was used in conjunction with the SuperSignal West Dura chemiluminescence kit (Pierce) to detect antibody-antigen complexes.
RESULTS
An L22 mutation causes a dramatic decline in the level of Ag43 mRNA and its protein product.
We initially measured the relative abundance of soluble proteins in E. coli strain MG1655 and an isogenic strain that harbors the L22(Δ82-84) mutation (MNY15) using two-dimensional difference gel electrophoresis (2D-DIGE). Surprisingly, we found that the mutation had no effect or a modest effect on the level of most proteins. We isolated spots corresponding to 32 proteins that appeared to increase or decrease 2- to 3-fold in the mutant strain and were able to confidently identify 25 of them by mass spectrometry (see Fig. S1A in the supplemental material). The vast majority of these proteins are cytoplasmic, although three are cell envelope proteins (OmpX, TonA, and Ag43). Western blot analysis confirmed that the levels of NuoE, Ndk, and Crl decreased and OmpX increased ∼2- to 3-fold, while the level of many other proteins did not change significantly in MNY15 (see Fig. S1B). The same changes in protein abundance were detected in MNY16, a strain harboring a small insertion in the L22 β hairpin loop [L22(ins.105/2) (43)] (see Fig. S1B). This finding provided further evidence that the effects we observed were due to the alteration of the ribosome tunnel near the constriction point. The only protein whose level appeared to change more than 3-fold was tryptophanase (TnaA), which decreased 4.6-fold. Previous studies have shown that a defect in the recognition of the upstream TnaC peptide inside the ribosome tunnel in L22 mutant strains causes transcriptional attenuation of tnaA (25), so this observation validated the results of our 2D-DIGE experiments.
Further examination revealed that 2D-DIGE underestimated the magnitude of the decrease in the level of one protein, Ag43. Like all autotransporters, Ag43 has an N-terminal extracellular (passenger) domain and a C-terminal β barrel (β) domain that anchors the protein to the outer membrane (OM) (Fig. 1A). Following its translocation across the OM, the Ag43 passenger domain is released by proteolytic cleavage (44). A 60-kDa polypeptide corresponding to the cleaved passenger domain of Ag43 was identified on 2D-DIGE as a protein that was reduced 2.7-fold in MNY15 (Fig. 1B and C; also see Fig. S1A in the supplemental material). Western blot analysis using an antiserum raised against the Ag43 β domain that recognizes the full-length Ag43 precursor as well as the cleaved β domain, however, showed that the level of the protein was actually reduced >10-fold in the mutant strain (Fig. 2A, lane 2). Consistent with the dramatic reduction in the level of Ag43, MNY15 cultures exhibited a flu phenotype; that is, they did not form large aggregates and settle under laboratory conditions (31) (Fig. 2B). The discrepancy between the 2D-DIGE and Western blot results is almost certainly due to the release of the cleaved passenger domain from the cell surface and its loss during sample preparation. MNY16 also produced almost no Ag43 and exhibited a flu phenotype, but a strain that harbored a 10-residue deletion in a conserved L23 loop situated close to the distal end of the ribosome tunnel [L23(loopΔ10)] produced a wild-type level of Ag43 (Fig. 2A, lanes 3 and 4, and B). Thus, like the more modest changes described above, the sharp drop in the level of Ag43 appeared to be due to the alteration of a specific region of the ribosome tunnel. Furthermore, the fact that we did not see a marked change in the level of Ag43 when we introduced L22 mutations into some strain backgrounds (data not shown) suggested that the production of the protein is highly sensitive to differences in cell physiology.
Fig 1.
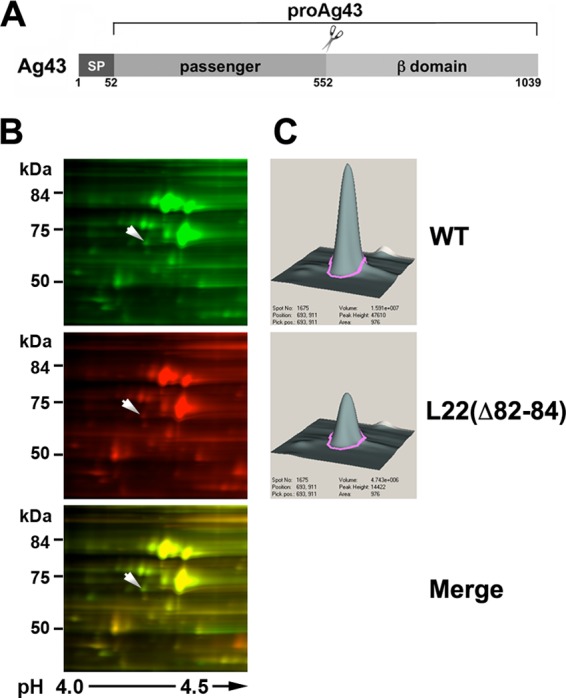
Level of Ag43 is reduced in an L22(Δ82-84) mutant strain. (A) Illustration of Ag43 showing the signal peptide (SP; residues 1 to 52), the passenger domain (residues 53 to 552), and the β domain (residues 553 to 1039). The pro form of the protein contains covalently linked passenger and β domains and is observed prior to the secretion and proteolytic processing of the passenger domain. (B) Soluble proteins from MG1655 and MNY15 [MG1655 rplV(Δ82-84)] were labeled with the fluorescent dyes Cy3 and Cy5, respectively, and analyzed by 2D-DIGE. Images of a section of each gel run in a representative experiment and an overlay image are shown. The spot that corresponds to Ag43 is denoted by an arrow. (C) Quantitative DeCyder analysis of the spot indicated in panel B reveals a 2.7-fold reduction of Ag43.
Fig 2.

Reduced synthesis of SecA in L22 mutant strains leads to a dramatic loss of Ag43 transcripts. (A) MG1655 (WT) and MG1655 derivatives containing the indicated mutation were grown at 37°C, and the steady-state level of Ag43 was measured by Western blotting using an antiserum raised against the Ag43 β domain. The cells shown in lane 6 were transformed with plasmid pMF8 (encoding secM-secA). pro, proAg43; β, Ag43 β domain. (B) Autoaggregation of the strains shown in panel A in static cultures. (C) Quantitative RT-PCR analysis of the level of the Ag43 gene, secA, and tnaA transcripts in MNY15 relative to MG1655. (D) The steady-state level of SecA in MG1655 (WT) and MG1655 derivatives containing the indicated mutation was measured by Western blotting. The cells shown in the last lane were transformed with plasmid pMF8.
We next used quantitative RT-PCR to determine whether the steady-state level of Ag43 gene mRNA was altered in the MG1655 derivative that harbored the L22(Δ82-84) mutation. Interestingly, we found that the level of the Ag43 gene transcript was 34-fold lower in MNY15 than in MG1655 (Fig. 2C). In contrast, we did not detect a significant difference in the level of mRNAs that encode proteins such as Ndk, whose abundance changed ∼2- to 3-fold in the mutant strain (see Table S2 in the supplemental material). Consistent with previous results showing that L22 mutations reduce secA expression at the translational level but strongly reduce tnaA transcription (16, 25), MNY15 contained essentially the same amount of secA mRNA as the wild-type strain but 67-fold less tnaA mRNA (Fig. 2C). These data imply that the strong decrease in the level of Ag43 in MNY15 is due to a corresponding decrease in the level of its mRNA.
The L22 mutation specifically inhibits Ag43 gene expression by reducing the level of SecA.
Because Ag43 export is dependent on the Sec machinery (30), we hypothesized that L22 mutations indirectly affect the level of Ag43 by impairing SecM-mediated ribosome stalling and thereby slightly reducing the level of SecA. Consistent with previous results, we found that the L22(Δ82-84) mutation [but not the L23(loopΔ10) deletion] reduced the level of SecA by ∼2-fold (Fig. 2D). To determine whether this effect of the L22 mutation accounts for the sharp decline in the level of Ag43, we transformed MNY15 with pMF8, a multicopy plasmid that contains the secM-secA locus (33). Overexpression of secA completely restored Ag43 production and suppressed the flu phenotype (Fig. 2A, lane 6, and B). A derivative of pMF8 containing a point mutation (D133N) that abolishes SecA ATPase and protein translocation activities (45), however, failed to rescue Ag43 synthesis (see Fig. S2 in the supplemental material). Taken together with the results described above, the data suggest that modest changes in SecA concentration lead to a defect in the secretion of Ag43 (or possibly other proteins) that in turn reduces Ag43 gene expression.
To gain further insight into the consequences of reducing the SecA concentration modestly, we next examined the fate of a variety of model presecretory proteins and IM proteins whose insertion is SecA dependent in MNY15. Interestingly, Western blot analysis revealed that the decrease in secA expression did not affect the production of any of the proteins we tested and did not cause a detectable accumulation of the precursor form in the cytoplasm (Fig. 3A). These results strongly suggest that wild-type cells produce more SecA than they need to drive the efficient secretion or membrane insertion of most proteins that travel through the Sec pathway. Consistent with this conclusion, the overproduction of SecA did not offset a growth defect exhibited by MNY15 (Fig. 3B). The relatively slow growth of the L22 mutant strain is therefore presumably caused by changes in the level of cytoplasmic proteins and not by a general secretion defect caused by a reduced concentration of SecA. In any case, the results suggested that the decreased expression of secA in the L22 mutant strain generates a specific signal that affects the expression of the Ag43 gene.
Fig 3.

L22(Δ82-84) mutation does not affect the steady-state level of model envelope proteins. (A) MG1655 (WT) and MG1655 derivatives containing the indicated mutation were grown at 37°C, and the steady-state levels of various proteins were measured by Western blotting. The cells shown in the last lane were transformed with plasmid pMF8 (encoding secM-secA). (B) The growth of MG1655 (WT) and MG1655 derivatives containing the indicated mutation is shown.
The expression of the Ag43 gene is highly sensitive to the secretion status of the cell.
We reasoned that if the sharp decline in Ag43 gene expression in MNY15 results from a decrease in the secretion activity of SecA, then other defects in the Sec pathway should generate the same effect. To test this prediction, we examined the level of Ag43 in a strain that harbors the cold-sensitive secY39 allele (MNY37; see reference 46) at both permissive (41°C) and nonpermissive (22°C) temperatures. The precursor form of OmpA could not be detected in MNY37 at the permissive temperature, and only a modest amount of precursor accumulated at the nonpermissive temperature (Fig. 4, lanes 3 and 4). In contrast, the synthesis of Ag43 was severely reduced at 41°C and almost completely eliminated at 22°C (Fig. 4, compare lanes 1 and 2 to lanes 3 and 4). These results support the hypothesis that even minor defects in the Sec pathway can strongly affect Ag43 gene expression.
Fig 4.
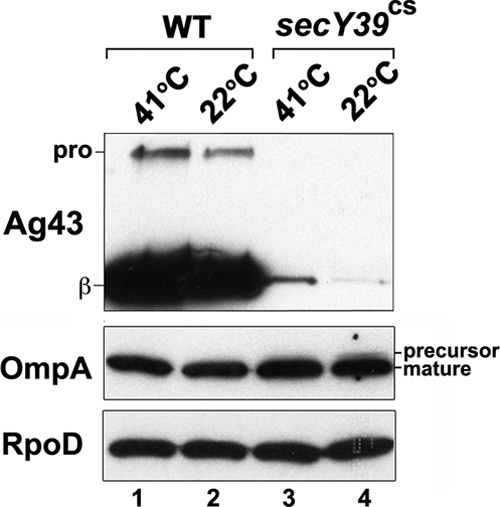
secY mutation dramatically reduces the steady-state level of Ag43. MG1655 (WT) and MNY37 (MG1655 secY39cs) were grown at 41°C and diluted into fresh medium. At an OD600 of 0.2, cultures were divided in half, and one half was shifted to 22°C. Cells were harvested 2 h later. The steady-state levels of Ag43, OmpA, and RpoD were determined by Western blotting. pro, proAg43; β, Ag43 β domain.
To gain further insight into the phenomenon of Ag43 gene downregulation in strains that harbor L22 mutations, we used a random transposon mutagenesis strategy to identify suppressor mutations that restored autoaggregation in MNY15. We isolated 10 mutants from a library of 3,578 clones that had the desired phenotype and that produced a wild-type level of Ag43 (Fig. 5). We expected that at least some of the insertions would inactivate genes that encode components of a signaling pathway that regulates Ag43 gene expression. To our surprise, however, nine of the insertions resided in genes that encode either a membrane protein (ompL, yaiT, fepG, araE, glvC, rhtA, mrcA, or gspC) or a presecretory protein (prc). The tenth insertion resided in a cryptic locus (intQ). Although more complicated scenarios can be envisioned, it seems likely that the inactivation of these genes increases the availability of SecA and alleviates the minor secretion defect caused by the reduced SecA concentration. This interpretation of the results is consistent with the conclusion that Ag43 gene expression is exquisitely sensitive to conditions that influence secretion efficiency.
Fig 5.
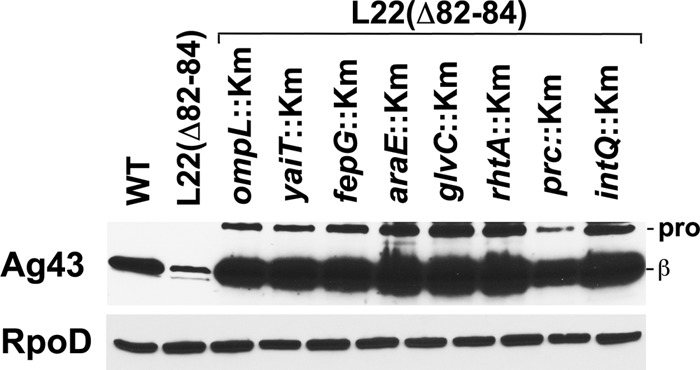
Second-site mutations that disrupt genes encoding envelope proteins suppress the effect of L22 mutation on Ag43 production. The steady-state levels of Ag43 and RpoD were determined by Western blotting in MG1655 (WT) and MG1655 containing the indicated mutation(s). pro, proAg43; β, Ag43 β domain.
Secretion stress reduces the stability of Ag43 gene mRNA.
In principle, the decrease in the steady-state level of Ag43 gene mRNA that we observed in MNY15 might be due to a reduction in the initiation of transcription. Indeed, it is conceivable that the L22(Δ82-84) mutation (or the resulting reduction in secA expression) leads to an increase in the level of the transcriptional repressor OxyR, which has been shown to inhibit Ag43 gene transcription. Western blot analysis revealed, however, that the steady-state level of OxyR in MG1655, MNY15, and a strain that overexpressed secA was indistinguishable (see Fig. S3A in the supplemental material). Although none of the mutant strains that we isolated in the suppressor screen described above contained an insertion in oxyR, we found that the deletion of oxyR could partially compensate for the sharp reduction of Ag43 in MNY15 (see Fig. S3B in the supplemental material). This finding raises the formal possibility that enhanced binding of OxyR to target sequences upstream of the Ag43 gene accounts for part of the decrease in Ag43 mRNA observed in L22 mutant strains.
Alternatively, mutations in L22 might lead to a destabilization of Ag43 mRNA. To explore this possibility, we devised an experimental strategy to rapidly induce a secretion defect in wild-type cells. We first constructed a strain (MNY40) in which the chromosomal copy of the Ag43 gene was replaced with a C-terminally His-tagged version of the gene (Ag43-His6). The steady-state level of the His-tagged version of Ag43 was lower than that of the wild-type protein, but the introduction of the L22(Δ82-84) mutation into MNY40 caused the same relative decline in Ag43 that we observed when we introduced the mutation into MG1655 (see Fig. S4 in the supplemental material). Subsequently, MNY40 was transformed with a plasmid that encodes an HA-tagged version of the Ag43 passenger domain (Ag43PD-HA) under the control of the IPTG-inducible trc promoter. The addition of IPTG led to the accumulation of a significant amount of the precursor form of Ag43PD-HA, a marked decrease in the level of Ag43-His6, and a slight increase in the level of SecA (Fig. 6A, lane 4). These results strongly suggest that Ag43PD-HA overloads the Sec machinery when it is overproduced and thereby creates a secretion defect that triggers both an increase in secA expression and a decrease in Ag43 gene expression. In contrast, a version of the passenger domain that lacks the signal peptide (ΔSP-Ag43PD-HA) did not affect the level of either Ag43-His6 or SecA, presumably because it failed to engage the Sec machinery and cause a secretion defect (Fig. 6A, lane 6).
Fig 6.
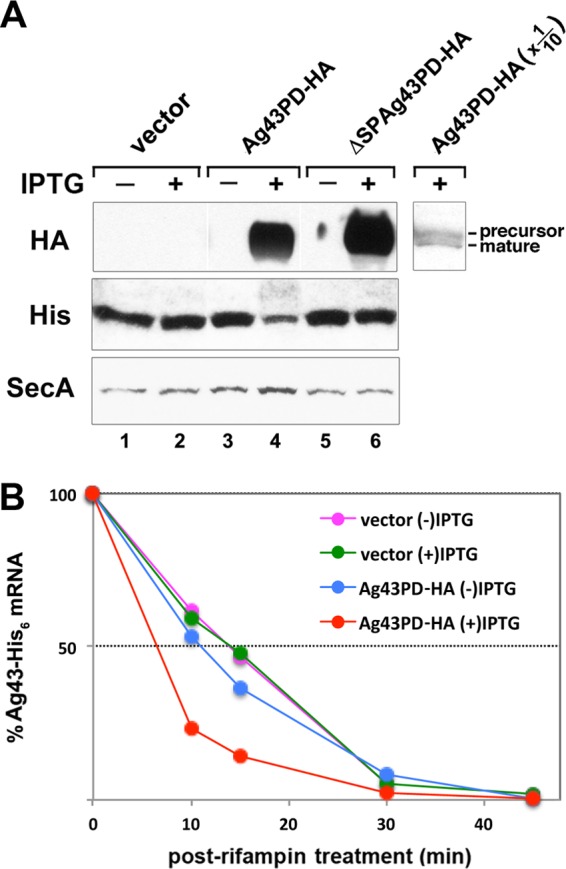
Secretion stress reduces the stability of Ag43 mRNA. (A) MNY40 (MG1655 Ag43::His6) transformed with plasmid pMY28 (encoding Ag43PD-HA) or pMY29 (encoding ΔSP-Ag43PD-HA) or the cloning vector (pTrc99a) were grown to an OD600 of 0.4. Cultures were divided in half, and IPTG was added to one half to induce the expression of the plasmid-borne gene. After 30 min, steady-state levels of HA- and His-tagged proteins and SecA were determined by Western blotting. Analysis of one-tenth as much protein on a separate Western blot revealed the presence of precursor and mature forms of Ag43PD-HA. (B) Rifampin was added to cultures following the 30-min IPTG induction, and quantitative RT-PCR was performed to determine the percentage of the Ag43 gene transcripts present at t0 that remained at the indicated time points. gapA mRNA served as an internal control. The averages from two independent experiments are shown.
We next performed a rifampin chase to examine the stability of the Ag43-His6 transcript in cells transformed with the plasmid that encodes Ag43PD-HA both in the absence and presence of IPTG. After dividing cultures in two and inducing the synthesis of Ag43PD-HA in half of the cells for 30 min, rifampin was added to block further transcription. The decay of Ag43-His6 mRNA was then monitored by quantitative RT-PCR using β domain-specific primers. Interestingly, we found that while the half-life of Ag43 gene-His6 mRNA in control cells was ∼15 min, the half-life was significantly shorter in cells that overproduced Ag43PD-HA (Fig. 6B). These results provide evidence that the loss of Ag43 that we observed in cells that have a reduced basal level of SecA or functional defects in the Sec pathway is due at least in part to the transmission of a signal that leads to the degradation of Ag43 mRNA.
DISCUSSION
In this study, we describe an unanticipated link between the regulation of expression of a gene encoding a secreted bacterial virulence factor and the secretion status of the cell. In light of the finding that the L22(Δ82-84) mutation perturbs the recognition of several translation arrest peptides and the folding of at least one polypeptide in E. coli, we sought to determine whether this mutation exerted a broader effect on the proteome. We found that the mutation causes a >10-fold reduction in the steady-state level of Ag43. Our results strongly suggested that the change in the level of Ag43 was an indirect effect of the reduction in secA expression that has been attributed to the L22 mutation. Consistent with this conclusion, another defect in the Sec pathway also strongly suppressed Ag43 production. Further analysis revealed that the decline in the level of Ag43 correlated with a sharp drop in the stability and steady-state level of its mRNA. Presumably, the reduced flux of one or more proteins through the Sec machinery generated a signal that led to the destabilization of the Ag43 transcript.
Besides drastically affecting the synthesis of Ag43, the L22 mutation produced surprisingly few effects on the E. coli proteome. The reduction in SecA did not alter the levels of a variety of secreted and membrane proteins and did not cause a detectable accumulation of unprocessed secretory precursors. Furthermore, we observed only modest changes in the levels of a few soluble proteins in the L22 mutant strain. While our analysis was by no means exhaustive, the results suggest that the regulation of gene expression by peptide-mediated translation arrest is relatively uncommon, and that the folding of most polypeptides is not significantly affected by their passage through the ribosome tunnel. It is possible, however, that the L22(Δ82-84) mutation only slightly perturbs that tunnel environment, and that widespread effects on the proteome can only be produced by more severe mutations that likely reduce cell viability. In any case, unlike the level of Ag43 mRNA, the level of the mRNAs encoding several proteins whose abundance rose or fell ∼2- to 3-fold did not change significantly. This observation suggests that the efficiency of translation of specific mRNAs or the efficiency of folding of specific proteins may be affected by the L22 mutation. Although the mutation does not impair decoding (47) or overall translation rate (42), it might affect local translation rates in a few mRNAs or slightly alter the conformation of a few nascent polypeptides.
Our results provide at least a partial explanation for the tight regulation of secA expression that has previously been reported. The observation that the secretion of most proteins is unaffected by L22 mutations implies that SecA is normally present in excess and suggests that the basal level of Sec pathway components is fundamentally capable of handling an increased burden of presecretory proteins that would hypothetically be produced under conditions of secretion stress. An increase in the level of SecA might still be required, however, because a subset of proteins is inherently more difficult to secrete or to maintain in a secretion-competent state and therefore is more sensitive to the availability of critical components of the Sec machinery. Under conditions in which an increase in the level of SecA is insufficient to promote efficient secretion of these proteins (which might include Ag43), it makes perfect biological sense to shut down their production. Indeed, such proteins might jam the Sec machinery and create a toxic secretion block. It is also possible that secretion stress typically occurs under conditions when biofilm disassembly is advantageous and is used to generate a signal to degrade Ag43 mRNA. Interestingly, the coupling of protein export efficiency to gene expression has previously been observed in both bacteria and eukaryotes. The type III secretion systems of Pseudomonas and Yersinia and the type VII secretion system of Mycobacteria are transcriptionally repressed or activated by a feedback mechanism in which the expression of genes that encode components of the secretion channel is dictated by the secretion of a specific regulatory protein (48–50). In eukaryotes, secretion stress induces the unfolded protein response and the activation of a nuclease that rapidly degrades a subset of mRNAs that encode presecretory proteins (51).
The mechanism by which secretion stress leads to the destabilization of Ag43 mRNA is unclear. One possibility is that an impairment of secretion activates the σE envelope stress pathway, which has been shown to promote small RNA-mediated decay of mRNAs encoding many outer membrane proteins (52). Although a small RNA (isrC or IS201) is embedded in the promoter region of the Ag43 gene (53), its role in the turnover of Ag43 gene mRNA is unknown. Alternatively, failure to translocate Ag43 or other proteins across the IM may lead to the recruitment or synthesis of an RNase that degrades Ag43 mRNA. The observation that the overproduction of an Ag43 derivative that lacks a signal peptide had no effect on Ag43 gene mRNA stability suggests that any signal generated by Ag43 itself would arise from the slow transit of the protein through the Sec apparatus and not from the accumulation of the precursor form in the cytoplasm. Regardless of the mechanism by which the mRNA is destabilized, chromosomal context appears to be important, because we did not observe a change in the level of Ag43 when we cloned the Ag43 locus into a plasmid and introduced it into strains that have L22 mutations or defects in the Sec pathway (data not shown). Finally, it should be noted that destabilization of the mRNA probably evolved to halt Ag43 synthesis rapidly. Like mRNAs that encode other abundant outer membrane proteins, Ag43 mRNA is unusually long lived (∼15 min; Fig. 6B) (54). Especially in light of the finding that the transcriptional repressor OxyR only binds to unmethylated DNA (which is produced only during DNA replication), mRNA degradation would almost certainly provide a more effective means of turning off Ag43 expression than the inhibition of transcription.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and by grant R00GM094212 (National Institute of General Medical Sciences) to M.-N.F.Y.
Footnotes
Published ahead of print 26 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00211-13.
REFERENCES
- 1. Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. 2000. The structural basis of ribosome activity in peptide bond synthesis. Science 289:920–930 [DOI] [PubMed] [Google Scholar]
- 2. Lu J, Deutsch C. 2008. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J. Mol. Biol. 384:73–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malkin LI, Rich A. 1967. Partial resistance of nascent polypeptide chains to proteolytic digestion due to ribosomal shielding. J. Mol. Biol. 26:329–346 [DOI] [PubMed] [Google Scholar]
- 4. Blobel G, Sabatini DD. 1970. Controlled proteolysis of nascent polypeptides in rat liver cell fractions. I. Location of the polypeptides within ribosomes. J. Cell Biol. 45:130–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Woolhead CA, McCormick PJ, Johnson AE. 2004. Nascent membrane and secretory proteins differ in FRET-detected folding far inside the ribosome and in their exposure to ribosomal proteins. Cell 116:725–736 [DOI] [PubMed] [Google Scholar]
- 6. Lu J, Deutsch C. 2005. Secondary structure formation of a transmembrane segment in Kv channels. Biochemistry 44:823–8243 [DOI] [PubMed] [Google Scholar]
- 7. Woolhead CA, Johnson AE, Bernstein HD. 2006. Translation arrest requires two-way communication between a nascent polypeptide and the ribosome. Mol. Cell 22:587–598 [DOI] [PubMed] [Google Scholar]
- 8. Bhushan S, Meyer H, Starosta AL, Becker T, Mielke T, Berninghausen O, Sattler M, Wilson DN, Beckmann R. 2010. Structural basis for translational stalling by human cytomegalovirus and fungal arginine attenuator peptide. Mol. Cell 40:138–146 [DOI] [PubMed] [Google Scholar]
- 9. Peterson JH, Woolhead CA, Bernstein HD. 2010. The conformation of a nascent polypeptide inside the ribosome tunnel affects protein targeting and protein folding. Mol. Microbiol. 78:203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berndt U, Oellerer S, Zhang Y, Johnson AE, Rospert S. 2009. A signal-anchor sequence stimulates signal recognition particle binding to ribosomes from inside the exit tunnel. Proc. Natl. Acad. Sci. U. S. A. 106:1398–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pool MR. 2009. A trans-membrane segment inside the ribosome exit tunnel triggers RAMP4 recruitment to the Sec61p translocase. J. Cell Biol. 185:889–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tenson T, Ehrenberg M. 2002. Regulatory nascent peptides in the ribosomal tunnel. Cell 108:591–594 [DOI] [PubMed] [Google Scholar]
- 13. Gong F, Yanofsky C. 2002. Instruction of translating ribosome by nascent peptide. Science 297:1864–1867 [DOI] [PubMed] [Google Scholar]
- 14. Lovett PS, Rogers EJ. 1996. Ribosome regulation by the nascent peptide. Microbiol. Rev. 60:366–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oliver DB, Beckwith J. 1982. Regulation of a membrane component required for protein secretion in Escherichia coli. Cell 30:311–319 [DOI] [PubMed] [Google Scholar]
- 16. Nakatogawa H, Ito K. 2002. The ribosomal exit tunnel functions as a discriminating gate. Cell 108:629–636 [DOI] [PubMed] [Google Scholar]
- 17. Butkus ME, Prundeanu LB, Oliver DB. 2003. Translocon “pulling” of nascent SecM controls the duration of its translational pause and secretion-responsive secA regulation. J. Bacteriol. 185:6719–6722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yap MN, Bernstein HD. 2011. The translational regulatory function of SecM requires the precise timing of membrane targeting. Mol. Microbiol. 81:540–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murakami A, Nakatogawa H, Ito K. 2004. Translation arrest of SecM is essential for the basal and regulated expression of SecA. Proc. Natl. Acad. Sci. U. S. A. 101:12330–12335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oliver D, Norman J, Sarker S. 1998. Regulation of Escherichia coli secA by cellular protein secretion proficiency requires an intact gene X signal sequence and an active translocon. J. Bacteriol. 180:5240–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakatogawa H, Ito K. 2001. Secretion monitor, SecM, undergoes self-translation arrest in the cytosol. Mol. Cell 7:185–192 [DOI] [PubMed] [Google Scholar]
- 22. Sarker S, Oliver D. 2002. Critical regions of secM that control its translation and secretion and promote secretion-specific secA regulation. J. Bacteriol. 184:2360–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gabashvili IS, Gregory ST, Valle M, Grassucci R, Worbs M, Wahl MC, Dahlberg AE, Frank J. 2001. The polypeptide tunnel system in the ribosome and its gating in erythromycin resistance mutants of L4 and L22. Mol. Cell 8:181–188 [DOI] [PubMed] [Google Scholar]
- 24. Chittum HS, Champney WS. 1994. Ribosomal protein gene sequence changes in erythromycin-resistant mutants of Escherichia coli. J. Bacteriol. 176:6192–6198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cruz-Vera LR, Rajagopal S, Squires C, Yanofsky C. 2005. Features of ribosome-peptidyl-tRNA interactions essential for tryptophan induction of tna operon expression. Mol. Cell 19:333–343 [DOI] [PubMed] [Google Scholar]
- 26. Vazquez-Laslop N, Thum C, Mankin AS. 2008. Molecular mechanism of drug-dependent ribosome stalling. Mol. Cell 30:190–202 [DOI] [PubMed] [Google Scholar]
- 27. Lawrence M, Lindahl L, Zengel JM. 2008. Effects on translation pausing of alterations in protein and RNA components of the ribosome exit tunnel. J. Bacteriol. 190:5862–5869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yap MN, Bernstein HD. 2009. The plasticity of a translation arrest motif yields insights into nascent polypeptide recognition inside the ribosome tunnel. Mol. Cell 34:201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chiba S, Lamsa A, Pogliano K. 2009. A ribosome-nascent chain sensor of membrane protein biogenesis in Bacillus subtilis. EMBO J. 18:3461–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leyton DL, Rossiter AE, Henderson IR. 2012. From self sufficiency to dependence: mechanisms and factors important for autotransporter biogenesis. Nat. Rev. Microbiol. 10:213–225 [DOI] [PubMed] [Google Scholar]
- 31. van der Woude MW, Henderson IR. 2008. Regulation and function of Ag43 (flu). Annu. Rev. Microbiol. 62:153–169 [DOI] [PubMed] [Google Scholar]
- 32. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt MG, Oliver DB. 1989. SecA protein autogenously represses its own translation during normal protein secretion in Escherichia coli. J. Bacteriol. 171:643–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amann E, Ochs B, Abel KJ. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315 [DOI] [PubMed] [Google Scholar]
- 35. Tannu NS, Hemby SE. 2006. Two-dimensional fluorescence difference gel electrophoresis for comparative proteomics profiling. Nat. Protoc. 1:1732–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chuang SE, Daniels DL, Blattner FR. 1993. Global regulation of gene expression in Escherichia coli. J. Bacteriol. 175:2026–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vandesompele J, de Preter K, Pattyn F, Poppe B, van Roy N, de Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034. 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta CT) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 39. Blewett N, Coller J, Goldstrohm A. 2011. A quantitative assay for measuring mRNA decapping by splinted ligation reverse transcription polymerase chain reaction: qSL-RT-PCR. RNA 17:535–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bernstein HD, Zopf D, Freymann DM, Walter P. 1993. Functional substitution of the signal recognition particle 54-kDa subunit by its Escherichia coli homolog. Proc. Natl. Acad. Sci. U. S. A. 90:5229–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ieva R, Bernstein HD. 2009. Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc. Natl. Acad. Sci. U. S. A. 106:19120–19125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choi PS, Bernstein HD. 2010. Sequential translocation of an Escherichia coli two-partner secretion pathway exoprotein across the inner and outer membranes. Mol. Microbiol. 75:440–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zaman S, Fitzpatrick M, Lindahl L, Zengel J. 2007. Novel mutations in ribosomal proteins L4 and L22 that confer erythromycin resistance in Escherichia coli. Mol. Microbiol. 66:1039–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Owen P, Meehan M, DeLoughry-Doherty H, Henderson I. 1996. Phase-variable outer membrane proteins in Escherichia coli. FEMS Immunol. Med. Microbiol. 16:63–76 [DOI] [PubMed] [Google Scholar]
- 45. Zito CR, Antony E, Hunt JF, Oliver DB, Hingorani MM. 2005. Role of a conserved glutamate residue in the Escherichia coli SecA ATPase mechanism. J. Biol. Chem. 280:14611–14619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baba T, Jacq A, Brickman E, Beckwith J, Taura T, Ueguchi C, Akiyama Y, Ito K. 1990. Characterization of cold-sensitive secY mutants of Escherichia coli. J. Bacteriol. 172:7005–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. O'Connor M, Gregory ST, Dahlberg AE. 2004. Multiple defects in translation associated with altered ribosomal protein L4. Nucleic Acids Res. 32:5750–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Urbanowski ML, Lykken GL, Yahr TL. 2005. A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc. Natl. Acad. Sci. U. S. A. 102:9930–9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen Y, Anderson DM. 2011. Expression hierarchy in the Yersinia type III secretion system established through YopD recognition of RNA. Mol. Microbiol. 80:966–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Raghavan S, Manzanillo P, Chan K, Dovey C, Cox JS. 2008. Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature 454:717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hollien J, Weissman JS. 2006. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313:104–107 [DOI] [PubMed] [Google Scholar]
- 52. Gogol EB, Rhodius VA, Papenfort K, Vogel J, Gross CA. 2011. Small RNAs endow a transcriptional activator with essential repressor functions for single-tier control of a global stress regulon. Proc. Natl. Acad. Sci. U. S. A. 108:12875–12880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen S, Lesnik EA, Hall TA, Sampath R, Griffey RH, Ecker DJ, Blyn LB. 2002. A bioinformatics based approach to discover small RNA genes in the Escherichia coli genome. Biosystems 65:157–177 [DOI] [PubMed] [Google Scholar]
- 54. von Gabain A, Belasco JG, Schottel JL, Chang AC, Cohen SN. 1983. Decay of mRNA in Escherichia coli: investigation of the fate of specific segments of transcripts. Proc. Natl. Acad. Sci. U. S. A. 80:653–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.