

Abstract
The Enterococcus faecalis leucine-rich protein ElrA promotes virulence by stimulating bacterial persistence in macrophages and production of the interleukin-6 (IL-6) cytokine. The ElrA protein is encoded within an operon that is poorly expressed under laboratory conditions but induced in vivo. In this study, we identify ef2687 (renamed elrR), which encodes a member of the Rgg (regulator gene for glucosyltransferase) family of putative regulatory proteins. Using quantitative reverse transcription-PCR, translational lacZ fusions, and electrophoretic mobility shift assays, we demonstrate that ElrR positively regulates expression of elrA. These results correlate with the attenuated virulence of the ΔelrR strain in a mouse peritonitis model. Virulence of simple and double elrR and elrA deletion mutants also suggests a remaining ElrR-independent expression of elrA in vivo and additional virulence-related genes controlled by ElrR.
INTRODUCTION
Enterococcus faecalis is a commensal bacterium of the intestinal tract and an opportunistic pathogen responsible for a variety of community-acquired and health care-associated infections (1, 2). The dual lifestyle of E. faecalis relies on multiple factors, including adhesins, secreted proteases, surface polysaccharides, and transcriptional regulators (3–5). One of the E. faecalis genes reported to be involved in virulence, elrA, encodes a protein with an N-terminal domain containing leucine-rich repeats (LRRs) and a C-terminal WxL domain, which promotes noncovalent association with the bacterial cell surface (6, 7). LRR-containing proteins are ubiquitous and are frequently involved in protein-protein interactions in a variety of functions such as ligand-receptor interactions, signal transduction, and adhesion processes (8). ElrA shares structural features with the members of the internalin multigenic family of Listeria monocytogenes (9). We previously demonstrated that inactivation of the elrA gene significantly reduces E. faecalis virulence in a mouse peritonitis model and that ElrA contributes to bacterial persistence in macrophages and increases the host inflammatory response by stimulating the production of the cytokine interleukin-6 (IL-6) in vivo (6). We also reported that elrA is the first gene of an operon that is poorly transcribed under laboratory growth conditions and is induced in vivo in mice (6), suggesting tight environmental control of elrA expression.
Quorum sensing in low-G+C Gram-positive bacteria seems to rely on short peptides, which either interact with two-component systems or are internalized and interact with peptide-responsive transcription factors (10). The peptide-associated transcriptional regulators described to date belong to the RNPP superfamily, which includes the Rap (aspartyl phosphate phosphatase), NprR (neutral protease regulator), and PlcR (phospholipase C regulator) regulators of Bacillus species and PrgX of E. faecalis. All of these proteins contain tandem tetratricopeptide repeats (TPRs), which interact with cytoplasmic peptide signals (11, 12). Recently, based on predicted structural similarities, Rgg family transcription factors have been proposed as candidates for members of the RNPP superfamily (13, 14). Rgg stands for regulator gene for glucosyltransferase of Streptococcus gordonii (15), the first member of a large family of regulators exclusively found in the order Lactobacillales and the family Listeriaceae (14). Rgg regulators are characterized by an N-terminal XRE-type helix-turn-helix (HTH) DNA-binding domain and a C-terminal region rich in predicted alpha-helices, and they share structural similarity with PlcR and PrgX of the RNPP family (13, 16). In addition to participating in various physiological functions, such as the biosynthesis of extracellular polysaccharides, the stress response, and natural competence (13, 17–23), Rgg-type regulators control the expression of genes involved in virulence in Streptococcus pyogenes, Streptococcus agalactiae, and Streptococcus suis (21, 24–28). Most Rgg-type regulators activate the transcription of adjacent genes (29–31), and the direct interaction between the activator and the corresponding promoter region has been demonstrated in several cases (14, 20, 24, 25, 30). However, some Rgg-type proteins in S. pyogenes, S. agalactiae, and S. suis are global regulators acting both as activators and as repressors (25, 26, 28, 32).
The present study reports the characterization of the first E. faecalis chromosomally encoded Rgg-like protein, named ElrR for enterococcal leucine-rich protein regulator, which is identified as a positive regulator of elrA expression and most likely controls additional virulence-related genes in E. faecalis.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The E. faecalis strain used in this study was OG1RF (33). Escherichia coli strains TG1 (34) and TG1 repA (35) were used for cloning and plasmid propagation. Plasmids used in this study are listed in Table 1. Enterococci were grown in M17 broth or agar supplemented with 0.5% glucose (M17G) at 37°C without aeration. E. coli strains were grown aerobically in Luria-Bertani broth or agar at 37°C. Antibiotics used were ampicillin (80 μg/ml), tetracycline (12.5 μg/ml for E. coli and 5 μg/ml for E. faecalis), and erythromycin (150 μg/ml for E. coli and 30 μg/ml for E. faecalis).
Table 1.
Plasmids used in this study
| Plasmid | Relevant characteristicsa,b | Reference or source |
|---|---|---|
| p3TET | Tetr (tetM), ori ColE1 | 36 |
| pET2818 | Ampr, ori ColE1 | 37 |
| pGhost9 | Ermr, ori pWV01, repA(Ts) | 38 |
| pJIM2242 | Ermr, ori pWV01 | 35 |
| pIL253 | Ermr, ori pAMβ1 | 39 |
| pMutin2 | rrnBt1t2-λt0 (Term) | 40 |
| pMutin4 | lacZ | 40 |
| pUC18 | Ampr, ori ColE1 | 41 |
| pVE3916 | Cmr, ori pWV01, repA | 42 |
| pVE14040 | Cmr, ori pWV01, repA with PaphA3::elrR | This study |
| pVE14041 | Ampr, pUC18 with PaphA3 | This study |
| pVE14042 | Ampr, pUC18 with PaphA3::elrR | This study |
| pVE14043 | Ampr Ermr, pUC18/pIL253 with PaphA3::elrR | This study |
| pVE14044 | Ermr, ori pWV01, repA(Ts) with ΔelrR | This study |
| pVE14045 | Ampr, ori ColE1with elrR::6×His | This study |
| pVE14147 | Ermr, ori pWV01 with elrR::6×His | This study |
| pVE14161 | Tetr, ori ColE1, ori pAMβ1 | This study |
| pVE14162 | Ampr Ermr, pUC18/pIL253 with PaphA3::elrR::6×His | This study |
| pVE14165 | Tetr, ori pWV01, Term with lacZ | This study |
| pVE14167 | Tetr, ori pWV01, Term, PelrA::lacZ | This study |
| pVE14173 | Tetr, ori pWV01, Term 0, UperlR-elrR-PelrA::lacZ | This study |
| pVE14174 | Tetr, ori ColE1, ori pAMβ1, Term, lacZ | This study |
| pVE14181 | Tetr, ori ColE1, ori pWV01, Term, PaphA3-elrR-PelrA::lacZ | This study |
| pVE14182 | Tetr, ori ColE1, ori pAMβ1, Term, PelrA::lacZ | This study |
| pVE14183 | Tetr, ori ColE1, ori pAMβ1, Term, PaphA3-elrR-PelrA::lacZ | This study |
| pVE14186 | Tetr, ori ColE1, ori pAMβ1, Term, UperlR-elrR-PelrA::lacZ | This study |
| pVE14188 | Tetr, ori ColE1, ori pAMβ1, Term, PaphA3-elrRstop-PelrA::lacZ | This study |
| pVE14189 | Tetr, ori ColE1, ori pAMβ1, Term, lacZ | This study |
| pVE14190 | Tetr, ori ColE1, ori pAMβ1, Term, PelrA[−82 to +3]::lacZ | This study |
| pVE14191 | Tetr, ori ColE1, ori pAMβ1, Term, PelrA[−190 to +3]::lacZ | This study |
| pVE14194 | Tetr, ori ColE1, ori pAMβ1, Term, PelrA[−583 to +3]::lacZ | This study |
| pVE14195 | Tetr, ori ColE1, ori pAMβ1, lacZ | This study |
| pVE14196 | Tetr, ori ColE1, ori pAMβ1, PelrA[−82 to +3]::lacZ | This study |
| pVE14197 | Tetr, ori ColE1, ori pAMβ1, PelrA[−190 to +3]::lacZ | This study |
| pVE14198 | Tetr, ori ColE1, ori pAMβ1, PelrA[−583 to +3]::lacZ | This study |
Term corresponds to t1, t2, and t0 transcriptional terminators from pMutin2.
Brackets indicate positions relative to the elrA start codon.
General DNA techniques.
General molecular biology techniques were performed by standard methods (43). Restriction enzymes, T4 DNA ligase, and the Klenow DNA polymerase fragment were used according to the manufacturer's instructions. PCR amplification was performed using a Mastercycler apparatus (Eppendorf). Oligonucleotides used in this work are listed in Table 2. When necessary, PCR products and DNA restriction fragments were purified with QIAquick kits (Qiagen). Plasmids were purified using QIAprep Miniprep or Midiprep kits (Qiagen). Electrotransformation of E. coli and E. faecalis was carried out as described previously (7), using a Gene Pulser apparatus (Bio-Rad Laboratories). Plasmid inserts were sequenced by GATC Biotech.
Table 2.
Primers used in this study
| Primer | Sequence (5′–3′)a |
|---|---|
| OEF212 | CTCTTCTGCCGATGAAGTTTCTGG |
| OEF262 | TTTCCAGAGGATCATTCGCCGTTCAATTTCTTCACC |
| OEF264 | TCTCGAGGTATTGAACTCCTTCCAC |
| OEF273 | AAGGATCCTGACGGAAGAAGATATTCGC |
| OEF274 | TGGCATGCGACAAATTCTCAATTATGCC |
| OEF279 | TCCCATGGAACAAAAAGAATATATGGAATATCGG |
| OEF280 | TTAGGATCCGTTTTCTTTTATTTCTTTTAAAC |
| OEF281 | CCTGCAGACAAGAAAACGGTGCTC |
| OEF324 | CGAATGATCCTCTGGAAA |
| OEF333 | TTCACCTAGCGGCCGATATTC |
| OEF339 | GCAACGAAATGGTGGAACAG |
| OEF340 | AAGGCATCGGCAATCTCTAAG |
| OEF353 | FAM-AGGCATAATTGAGAATTTGTC |
| OEF354 | FAM-GGAGGATGCGATTGTTTCG |
| OEF355 | FAM-ATTAATTTGGACAGTTTGTCG |
| OEF501 | TTGTCGACCATAAACTTCACCTCCTG |
| OEF502 | AAGGATCCGGGAGGTTCTGGCAAAG |
| OEF504 | AAGCATGCCGTTTCCAACTTCGTTAC |
| OEF594 | FAM-CGAATGATCCTCTGGAAA |
| OEF651 | FAM-GCCGCTAGGTGAAGAAATTG |
| OEF652 | FAM-CGATAATCTGCAACGATTCG |
| OEF658 | GATGCCACAAAAGGACTGTTTGA |
| OEF659 | CAGGTTGGACGACTGTTCCA |
| OEF689 | AAGCATGCGGCATAATTGAGAATTTGTC |
| OEF690 | AAGCATGCTTAATTTGGACAGTTTGTCG |
| OEF694 | AAGCATGCGAGTGTGTTGATAGTGC |
| OEF695 | TTTTCTTAAGCTCCTTTCGCTTTCTTC |
| FR.lac5 | ATGGGATAGGTCACGTTGGTGTAGAT |
Underlined nucleotides specify the restriction enzyme site used for cloning. FAM indicates coupling with 6-carboxyfluorescein.
Labeling elrR with a 6×His coding sequence tag and Western blotting.
The chromosomal elrR gene was tagged by integrating the conditionally replicating plasmid pVE14147. A 1.28-kb EaeI-EcoRI fragment from pVE14045 (see below) was cloned in pJIM2242, generating pVE14147, which was introduced into strain OG1RFΔgelE (44) by electroporation. An integrant of pVE14147 was obtained as previously described (45), generating a strain expressing chromosomal ElrR-6×His under the control of its own promoter. Another OG1RF strain expressing ElrR-6×His under the control of the constitutively expressed E. faecalis PaphA3 promoter (46) on a replicating plasmid was used as a positive control. The ElrR-6×His-encoding sequence was fused to the PaphA3 promoter. For this purpose, plasmids pVE14045 and pVE14043 (see below) were ligated at their BglI and EagI sites to generate pVE14162. Protein extracts and Western blotting were performed as previously described (7). ElrR-6×His was detected with a mouse monoclonal antipolyhistidine antibody (Zymed Laboratories) diluted 1:5,000. Bound primary antibodies were detected with horseradish peroxidase-coupled anti-mouse antibodies diluted 1:5,000 (GE Healthcare).
Deletion of elrR in OG1RF and in ΔelrA strains.
Strain ΔelrR was constructed by deleting the elrR coding sequence in strain OG1RF by double-crossover recombination, using pVE14044, a pG+host9-derivative plasmid (38). Two DNA fragments encompassing the 5′ and 3′ ends of the elrR gene were amplified by PCR from OG1RF chromosomal DNA with primer pairs OEF281-OEF262 and OEF324-OEF264, respectively. The two PCR products were fused by PCR using the external primers OEF281 and OEF264. The resulting product was digested with PstI and XhoI and cloned in pG+host9, yielding pVE14044, which was then introduced into OG1RF. A markerless in-frame elrR deletion mutant was selected as previously described (7), and the deletion of elrR was confirmed by sequencing of the chromosomal locus.
The ΔelrR ΔelrA strain, with a markerless in-frame ΔelrR ΔelrA deletion, was constructed by deleting the elrR coding sequence in the ΔelrA strain (6). For this purpose, an 853-bp AhdI-KpnI fragment from the pG+host9 derivative plasmid that carried the ΔelrA construct (7) was ligated with a 4.88-kb AhdI-KpnI fragment of pVE14044, generating pVE14143, which carries the double deletion of elrR and elrA. Plasmid pVE14143 was introduced into the ΔelrA strain, and the ΔelrR ΔelrA deletion mutant was obtained as described above.
Construction of strain OG1RF overproducing ElrR.
To overproduce ElrR, the corresponding gene was cloned on a plasmid under the control of the E. faecalis PaphA3 promoter. The entire elrR gene was amplified by PCR using primers OEF273 and OEF274, and the resulting product was digested with BamHI and SphI and cloned in pVE14041 (Table 1). The resulting plasmid, pVE14042, was fused to plasmid pIL253 at the EcoRI site, to obtain pVE14043, which was introduced into the OG1RF strain, yielding the elrR+ strain.
Construction of lacZ reporter plasmids.
Translational lacZ fusions were constructed using pVE14165, which carries a promoterless lacZ gene devoid of the first 15 codons and preceded by rho-independent transcription terminators (Term) (63) and a pWV01 replication origin and a tetM tetracycline resistance marker (36). The region encompassing the elrA promoter region from positions −583 to +3 with respect to the elrA start codon was recovered by PCR using oligonucleotides OEF501 and OEF504 as primers. The region from positions −1688 to +3 with respect to the elrA start codon, which includes the entire elrR gene plus the elrA promoter region and start codon, was amplified by PCR using primers OEF501 and OEF502. The resulting fragments were digested with SphI and SalI and ligated to the SphI and SalI sites of pVE14165 to obtain plasmids pVE14167 and pVE14173, which carried the PelrA::lacZ and UpelrR-elrR-PelrA::lacZ translational fusions, respectively. To ensure the expression of the elrR gene in the latter plasmid construct, the EagI-EcoRI upstream region of the elrR gene in pVE14173 was replaced by the 0.5-kb EagI-EcoRI fragment from pVE14042, which contained the constitutive PaphA3 promoter (46). The resulting plasmid pVE14181 carried the translational fusion PaphA3-elrR-PelrA::lacZ.
To convert these plasmids into E. faecalis replicative plasmids, the pWV01 origin was replaced by a pAMβ1 replicon from pVE14161 (Table 1). The 4.67-kb SnaBI-BstEII fragment from pVE14161 was ligated to BstEII-SwaI fragments from pVE14167 (6.26 kb), pVE14173 (7.267 kb), and pVE14181 (7.56 kb) to yield pVE14182, pVE14186, and pVE14183, respectively. The plasmid pVE14186, which carries the fusion PaphA3-elrRstop-PelrA::lacZ, in which the elrR open reading frame (ORF) was disrupted, was obtained by digesting pVE14183 with EagI and self-ligating after filling in the ends with the Klenow fragment.
The plasmids pVE14174, pVE14189, and pVE14195 were used as negative controls for the lacZ fusions. pVE14174 was constructed by ligating a 4.67-kb BstEII and SnaBI fragment of pVE14161 to a 5.7-kb BstEII and SwaI fragment of pVE14165. pVE14189 resulted from the replacement of the 875-bp SapI-Nhe fragment of pVE14174 with the 551-bp EcoRI-NheI fragment of pVE14161 after filling in the SapI and EcoRI ends with the Klenow fragment. The 7.25-kb SphI-AflII fragment from pVE14189 was ligated to the tetM gene amplified by PCR using primers OEF694 and OEF695. The resulting plasmid pVE14195 lacks the transcriptional terminators and has the tetM and lacZ genes in opposite orientation.
Plasmids pVE14190 and pVE14196, pVE14191 and pVE14197, and pVE14194 and pVE14198 contained fragments of the elrA promoter region from positions −80, −190, and −583 bp relative to the elrA start codon fused to lacZ, respectively. They were generated by cloning PCR-amplified fragments using primer OEF690, OEF689, or OEF504 paired with OEF501 in pVE14189 and pVE14195, respectively.
Ectopic expression of ElrR was performed by cloning elrR on the pWV01-derivative plasmid pVE3916 (42). For this purpose, the 1.5-kb PvuII-SphI fragment from pVE14042 was ligated to pVE3916 to generate pVE14040.
RNA isolation, Northern blotting, quantitative reverse transcription-PCR (qRT-PCR), and transcription start site (TSS) mapping.
Total RNA was extracted as described previously (47). Northern blotting assays were performed on 40 μg of total RNA separated on a 0.9% denaturing agarose gel as previously described (48). Specific oligonucleotides OEF212 and OEF333 were used to detect elrA and elrR RNAs, respectively. Oligonucleotides were labeled with [γ-32P]ATP and T4 polynucleotide kinase (NEB Biolabs) according to the recommendations of the manufacturer (NEB Biolabs). An RNA molecular weight ladder was from Ambion (Millenium RNA; Ambion).
Single-stranded cDNA libraries to analyze RNA samples via quantitative reverse transcription-PCR (qRT-PCR) amplification were prepared as described previously (47). The RNA and cDNA quality and concentration were assessed using a ND-100 spectrophotometer (Nanodrop) and the 2100 Bioanalyzer (Agilent). Primers for qRT-PCR were designed using Primer Express software (Applied Biosystems) and used to amplify PCR fragments of about 150 bp. elrA primers were OEF658 and OEF659, and recA primers were OEF339 and OEF340 (Table 2). PCRs were performed on an ABI Prism 7900 HT (Applied Biosystems) in triplicate with two amounts (1 ng or 0.2 ng) of cDNA in a 20-μl volume containing forward and reverse primers at 300 nM and Power SYBR Green PCR Master Mix (Applied Biosystems), according to the manufacturer's recommendations. Results were normalized by using the recA gene. The relative mRNA expression level of the elrA gene in each sample was calculated using the comparative cycle threshold (CT) as described previously (49). Analysis was performed from cDNA synthesized from RNA extracted from three independent cultures.
Expression of the elrA gene was assessed in vitro using RNA extracted from liquid cultures of E. faecalis strains with an RNeasy minikit (Qiagen) including an RNase-free DNase treatment step and in vivo using RNA extracted from peritoneal cavity fluids of E. faecalis-infected mice (see below) with the RNeasy minikit, after the cells were suspended in RNAlater solution (Qiagen) as described previously (6). Real-time qRT-PCR was performed in an iCycler iQ system (Bio-Rad Laboratories), using rpoB as the normalization gene. The elrA primers and probe (OEF246, OEF247, and OEF248) and the rpoB primers and probe (OEF249, OEF250, and OEF251) were used in RT-PCRs as previously reported (6). The relative mRNA expression level of the elrA gene in each sample was tested in triplicate and calculated using the abovementioned comparative cycle threshold method. For all qRT-PCR analyses, statistical significance between the mean ratios of genes was evaluated by a two-sided paired t test by using GraphPad Prism version 4.03 for Windows (GraphPad Software). A P value of <0.05 was considered significant.
The elrA transcription start site was mapped in strains that carried lacZ fusions using the 5′-tag RACE (rapid amplification of cDNA ends) method (47). Specific amplification of lacZ transcripts from the tagged cDNA library was performed using the FR.lac5 and FR.DNA5 oligonucleotides (47). The authenticity of amplicons was confirmed by sequencing after cloning the PCR products in the pCRII-Topo vector (Invitrogen), and the transcription start site was deduced as the first nucleotide after the FR.DNA5 sequence.
β-Galactosidase assays.
E. faecalis strains were grown as described for RNA isolation. Frozen pellets of cells harvested in mid-exponential phase (optical density at 600 nm [OD600], ∼1) were thawed and permeabilized by vigorous mixing in Z buffer (50) containing 1% (vol/vol) toluene. β-Galactosidase activity was assayed at 37°C and expressed in Miller units (MU), which were calculated as follows: OD420 × 1,000/(t × v × OD600), where OD420 is optical density at 420 nm of o-nitrophenol released, t is time of assay in minutes, v is volume of the cell culture in milliliters, and OD600 is optical density at 600 nm of the cell culture used (50).
Overexpression and purification of recombinant ElrR-6×His in E. coli.
A DNA fragment containing the full-length elrR open reading frame was amplified by PCR from E. faecalis OG1RF genomic DNA using primers OEF279 and OEF280. The resulting PCR fragment was digested with NcoI and BamHI and inserted into the pET2818 vector to generate pVE14045, which encodes the ElrR-6×His tag. After verification by sequencing, pVE14045 was introduced into E. coli BL21(DE3). Protein expression was induced at an OD600 of 0.6 by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h at 37°C. Cells were harvested by centrifugation at 5,700 × g for 8 min at 4°C and resuspended in binding buffer (50 mM NaHPO4, pH 8, 300 mM NaCl, 10 mM imidazole) containing 1 mM AEBSF [4-(2-aminoethyl)-benzenesulfonyl fluoride], 10 μg ml−1 RNase A, and 5 μg ml−1 DNase I. The cells were broken using a cell disrupter system (Constant Systems Ltd.). Cell debris was removed by centrifugation at 3,000 × g for 15 min at 4°C, and the supernatant fluid was loaded on a Select affinity column (Sigma) equilibrated with binding buffer according to the supplier's instructions. The elution was carried out with binding buffer containing an imidazole gradient (20 to 250 mM). Eluted fractions were analyzed individually by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). ElrR-containing fractions were dialyzed overnight in a Slide-A-Lyzer (10-kDa cutoff; Pierce Perbio) against Tris-HCl, pH 7.5, and 1 mM dithiothreitol (DTT). Protein concentrations were determined by the Bradford assay (Bio-Rad), and purified ElrR was stored in 15% glycerol at −20°C.
Electrophoretic mobility shift assay (EMSA).
5′ fluorescent oligonucleotides labeled with 6-carboxyfluorescein (FAM) (Eurogentec) were used in PCRs to create labeled DNA fragments. The 188-bp, 296-bp, and 492-bp fragments contained the elrA promoter region from positions −83, −191, and −387 to +105 bp relative to the elrA start codon, respectively. They were generated from OG1RF DNA using the primer OEF355, OEF353, or OEF594 paired with primer OEF354, respectively, and purified using the PureLink PCR purification kit (Invitrogen). Nonspecific labeled DNA, which corresponds to a 288-bp internal fragment of the elrR coding sequence, was amplified with primers OEF651 and OEF652. Binding reactions were performed with purified protein and 5 to 3 ng of probe for 30 min at room temperature in binding buffer [5% glycerol, 40 mM Tris-HCl, pH 7.0, 1 mM EDTA, 1 mM DTT, 0.3 mM bovine serum albumin, and 10 μg/ml poly(dI-dC)] in a final volume of 20 μl. Reaction mixtures were resolved at room temperature on a 12% polyacrylamide gel in Tris-buffered EDTA (TBE) buffer (Invitrogen) and visualized using a Typhoon fluorescence imager (GE Healthcare). Where specified, reactions were carried out in the presence of a more-than-100-fold excess of nonlabeled competitor DNA. Gel shift experiments were performed with two independent batches of purified ElrR.
Mouse peritonitis model of virulence.
The mouse experiments were performed under a protocol approved by the Institutional Animal Use and Care Committee at the Università Cattolica del Sacro Cuore, Rome, Italy (permit number Z21, 1 November 2010), and authorized by the Italian Ministry of Health, according to the Legislative Decree 116/92, which implemented the European Directive 86/609/EEC on laboratory animal protection in Italy. Animal welfare was routinely checked by veterinarians of the Service for Animal Welfare.
Testing of the OG1RF, ΔelrA, ΔelrR, ΔelrR ΔelrA, and ΔelrR-complemented strains was performed as described previously (6). The inoculum size was confirmed by determining the number of CFU on brain heart infusion (BHI) agar. Each inoculum was 10-fold diluted in 25% sterile rat fecal extract prepared from a single batch as previously described (51). Groups of 4- to 6-week-old (22 to 25 g) outbred (ICR) female mice (Harlan Italy SRL) were challenged intraperitoneally with 1 ml of each bacterial inoculum, housed five per cage, and fed ad libitum. A control group of mice was injected with 25% sterile rat fecal extract only. Survival was monitored every 3 to 6 h. Survival estimates were constructed by the Kaplan-Meier method and compared by log rank analysis. In another set of experiments, groups of mice were killed 20 h postinfection, and fluids recovered from the animal peritoneal cavities were serially diluted in saline solution for RT-PCR analyses as described above. All statistical analyses were performed using the aforementioned GraphPad Prism software, and comparisons with P values of <0.05 were considered to be significant.
RESULTS AND DISCUSSION
elrR, an Rgg-like regulator-encoding gene upstream of the elrA operon.
PCR screening of our laboratory collection of enterococcal isolates using an array of primers nested and outside the elrA operon had revealed several strains that lacked the entire operon (from ef2686 to ef2682) (6). In those strains, only primers located upstream of the ef2687 gene and downstream of the ef2682 gene yielded a PCR product corresponding to the junction between ef2688 and ef2681 (Fig. 1A). Despite the predicted presence of a rho-independent transcription terminator (according to the programs TransTerm [52] and ARNold finding terminators [53]) between them, the systematic association of the ef2687 gene and the elrA operon (88% of the strains tested) suggested that these genes are functionally related. The EF2687 product, here referred to as ElrR for enterococcal leucine-rich protein regulator, is annotated as a conserved domain protein in the V583 genome at the J. Craig Venter Institute website (http://cmr.jcvi.org/tigr-scripts/CMR/shared/GenePage.cgi?locus=EF2687) and is predicted to be a 307-residue polypeptide, of which the first 230 amino acids share 36% identity with those of S. gordonii Rgg. In silico analysis using NCBI PSI-BLAST revealed a significant similarity of the first 174 amino acids with the N-terminal putative DNA-binding helix-turn-helix motif of the Rgg-like regulators. Moreover, ElrR contains the invariant glycine (G13), the arginine (R20), and the aromatic residue tyrosine (Y160) corresponding to G4, R11, and W149 of S. pyogenes RopB (16) (Fig. 1B). The putative presence of a DNA-binding domain in ElrR was supported by other predictions using Protein Fold Recognition servers PHYRE2 (54) and COMPASS (55) on the GeneSilico structure prediction Metaserver, which identified PrgX of E. faecalis as the best match to ElrR over the first 220 amino acid residues. Moreover, during the course of this work ElrR was reported to be a member of the Rgg-like proteins (14). Interestingly, the E. faecalis V583 genome has 5 chromosomally encoded (EF0073, EF1224, EF1316, EF1599, and ElrR) and two plasmid-encoded (EFA0004 and EFB0005) Rgg-like proteins. Among them, ElrR is phylogenetically related to EF0073, EFA0004, and EFB0005 (14), supporting the hypothesis that ElrR is an Rgg family member. Structure prediction algorithms have led to the proposal that Rgg proteins are members of the RNPP family (11, 14, 18). The carboxy-terminal regions of RNPP family proteins have predicted folds consistent with TPRs which mediate protein-protein or protein-peptide interactions (12, 56). As observed for PrgX, PlcR, and Rgg proteins, no tetratricopeptide repeats (TPRs) could be predicted from the primary sequence of ElrR; however, the secondary structure of ElrR predicted by PSIPRED (57) reveals alpha-helices as reported for other Rgg-like proteins that align with the TPR helices of PlcR and PrgX (13). Both PrgX and PlcR regulate the transcription of their target genes in response to a signal peptide. Peptide binding to PlcR enhances DNA binding through drastic conformational and oligomerization changes promoting transcription activation of cognate target genes (58, 59). In contrast, PrgX responds to two competing signaling peptides and acts as a transcriptional repressor. The change of PrgX from the tetrameric to the dimeric state allows access to the promoter region of the PrgX-regulated gene (58, 60). With the exception of RopB, which binds an N-terminal secretion signal sequence of a distantly encoded lipoprotein (61), the Rgg signaling molecules characterized so far are short peptides encoded by small adjacent genes of the peptide-associated Rgg subfamilies (14, 18, 62). In the case of elrR, no short coding sequence was detected upstream or downstream, as for the majority of predicted Rggs, suggesting that if any signaling molecule is involved, it is encoded at another locus or imported from the extracellular medium. All these characteristics suggest that ElrR belongs to the RNPP family and could act as a transcriptional regulator.
Fig 1.
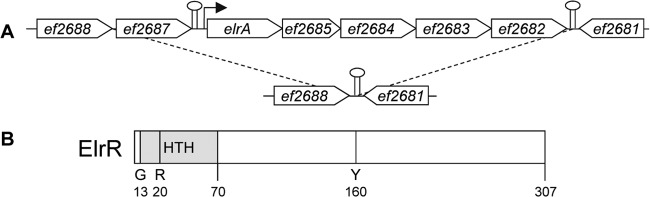
Gene organization and natural deletion of the elrA operon in E. faecalis isolates. (A) Schematic representation of the chromosomal region of the elrA operon. Putative rho-independent transcriptional terminators are indicated as lollipops. The promoter region of the elrA operon is represented by a black-headed arrow. (B) Schematic representation of the predicted ElrR product. HTH indicates the helix-turn-helix domain. G, R, and Y indicate glycine, arginine, and tyrosine residues, respectively. Numbers refer to the amino acid positions in ElrR.
ElrR expression under laboratory conditions.
To get some insight into elrR expression in E. faecalis, we attempted to characterize the elrR mRNA by Northern blotting of RNA extracted from bacteria grown under standard laboratory conditions. elrR mRNA could not be detected when expressed from its chromosomal locus (data not shown). To rule out the possibility that the elrR transcript might be particularly unstable, we inserted a 6×His tag coding sequence at the 5′ end of the elrR gene and searched for ElrR-6×His by Western blotting using anti-His tag antibodies (Fig. 2). We also failed to detect the ElrR-6×His protein expressed from the chromosomal locus. In contrast, ElrR-6×His was detected under the same growth conditions in a strain that carries pVE14162, a plasmid which contains ElrR-6×His under the control of the constitutively expressed E. faecalis PaphA3 promoter (46). These observations indicated that ElrR expression is extremely low under in vitro conditions. They also validated the use of PaphA3 to overcome the lack of ElrR expression under these conditions.
Fig 2.
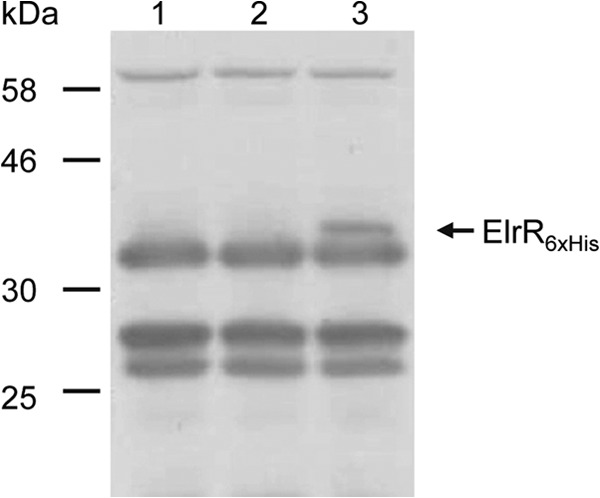
Western immunoblot analysis of ElrR-6×His under laboratory growth conditions from OG1RF derivative strains. Cytoplasmic and cell wall proteins from strains expressing chromosomal ElrR (lane 1), chromosomal ElrR-6×His (lane 2), and ElrR-6×His under the control of PaphA3 on a plasmid (lane 3) were extracted. Equivalent amounts of proteins were analyzed using an antipolyhistidine polyclonal rabbit antibody. The band corresponding to ElrR-6×His is indicated by an arrow. Other bands are nonspecific and correspond to cross-reacting signal.
ElrR activates elrA expression.
To determine whether ElrR has any impact on elrA expression, an in-frame deletion mutant of elrR (ΔelrR) and a strain that expresses elrR in trans under the control of PaphA3 (elrR+) were constructed in E. faecalis strain OG1RF. Levels of elrA transcripts were first analyzed by Northern blotting hybridization on total RNA prepared from the wild-type (wt), ΔelrR, and elrR+ strains. No elrA transcript was detected in any of the strains analyzed (data not shown). Since elrA was previously shown to be induced in vivo, it is likely that the expression level of elrA transcript is too low to be detected by Northern blotting under laboratory growth conditions. We then compared the levels of elrA transcript in the wt, ΔelrR, and elrR+ strains using quantitative RT-PCR. The recA transcript tested as an internal control gave CT values from 21.2 to 24.2; high CT values from 29.4 to 34.4 were systematically obtained with elrA-specific primers, confirming a very low level of elrA transcript. The specific amplification of elrA cDNA was ascertained by the lack of amplification using cDNA extracted from the ΔelrA strain. The transcript level of elrA was increased 2.31- ± 0.39-fold (P < 0.000001) in the elrR+ strain, indicating that the constitutive expression of elrR increases the level of elrA transcripts. In contrast, no significant difference in the levels of elrA transcript was noticed between the ΔelrR and wt strains, further supporting the idea that elrR is not expressed at significant levels during growth under the tested conditions.
To get further insights into the role of ElrR in elrA expression, we created an array of translational elrA::lacZ fusions (Fig. 3). We used a multicopy plasmid to compare the levels of expression of the PelrA::lacZ and the promoterless lacZ fusions, carried by pVE14182 and pVE14174, respectively. Although the β-galactosidase activity of the PelrA::lacZ fusion was low (1.29 ± 0.24 Miller units), it was still more than 8-fold higher than the basal level detected for the control strain carrying the promoterless lacZ fusion, i.e., the background level of the lacZ reporter system used, 0.15 ± 0.04 Miller units. This result shows the functionality of the PelrA, enabling us to further investigate the role of elrR in PelrA::lacZ activity. β-Galactosidase activities of the other constructs were compared and normalized with that of PelrA::lacZ (Fig. 3). A comparable activity ratio was detected in the strain that harbored the elrR gene and 483 bp of upstream DNA, suggesting that elrR may not be expressed (UperlR-elrR-PelrA::lacZ). In contrast, when elrR was expressed under the control of the constitutive promoter PaphA3, the β-galactosidase ratio was increased more than 10-fold, indicating that the PaphA3-driven expression of elrR stimulated PelrA::lacZ expression. Due to the potential read-through of the elrR transcription terminator from PaphA3-driven expression and to demonstrate the activator effect of ElrR on the expression of PelrA::lacZ, a translational stop codon was introduced into the ElrR-encoding sequence, leading to the plasmid construct pVE14188. The β-galactosidase ratio of this new construct (PaphA3::elrRstop-PelrA::lacZ) was only 2-fold higher than that with PelrA::lacZ, demonstrating that most of the activity of the PaphA3::elrRstop-PelrA::lacZ fusion is due to the activator effect of ElrR on PelrA::lacZ (∼10-fold) and that the possible transcriptional read-through at the elrR terminator has a minor effect on the PelrA::lacZ expression (∼2-fold). To confirm elrA transactivation by ElrR, we expressed the elrR gene in trans under the control of the PaphA3 promoter on plasmid pVE14040, which is compatible with the PelrA::lacZ plasmid. The β-galactosidase activity of the strain that expresses both elrR and PelrA::lacZ on compatible plasmids (8.8 ± 1.2 MU) was ∼14-fold higher than that of the control strain, which carries pVE3916 and PelrA::lacZ (0.6 ± 0.1 MU). These data further demonstrate that ElrR activates elrA expression in trans.
Fig 3.

Schematic representation of lacZ translational fusions and their relative β-galactosidase activities. At top is shown the genomic organization of the elrR-elrA region with the PelrA promoter, and the potential rho-independent transcription terminator is indicated by a lollipop. DNA fragments encompassing the elrA upstream region were fused to the lacZ coding sequence. Transcription from tetM of the vector pVE14174 was arrested by rho-independent transcription terminators, Term (black lollipop). Ratios of β-galactosidase activities were normalized to the activity measured for PelrA::lacZ (1.29 ± 0.24 Miller units). Data provided are the averages of five independent experiments.
Rgg family regulators positively regulate their own expression, and their activity is modulated by short hydrophobic peptides in S. mutans, S. thermophilus, and S. pyogenes (13, 14, 62–65). Recently, Shelburne et al. identified a lipoprotein-derived inhibitory peptide that interacts with RopB of S. pyogenes and therefore could modulate RopB-dependent expression of speB (61). Our data indicate that the 483-bp DNA region upstream of elrR is not sufficient either to express elrR or to reach a detectable effect on elrA expression under laboratory conditions. However, we show that the expression of elrR from a strong surrogate promoter activates elrA expression regardless of the need for a signaling molecule. Based on the structural predictions and despite the absence of reliable prediction of a short coding sequence adjacent to elrR, we cannot exclude the possibility that ElrR regulatory activity relies on a signaling molecule that may not be expressed or available under laboratory conditions.
ElrR specifically binds to the elrA promoter region.
The suggestion that ElrR is an Rgg family member, i.e., the presence of the HTH DNA-binding motif and the positive effect of the ElrR protein on the PelrA::lacZ fusion (see above), prompted us to assay the direct binding of ElrR to the elrA promoter region by electrophoretic mobility shift assays (EMSAs). The assays were performed using purified ElrR and a 492-bp DNA fragment (probe S) encompassing the elrA promoter region from positions −387 to +105 bp relative to the start codon of elrA (6) or a 288-bp DNA fragment internal to the ElrR coding sequence as a negative control (Fig. 4). ElrR provoked a mobility shift of the DNA fragment containing the elrA promoter region in a protein concentration-dependent manner (Fig. 4, left panel, lanes 2 to 4). No band shift was observed with the control DNA fragment (NS in Fig. 4). The ElrR binding specificity was further confirmed by showing that addition of unlabeled competitor DNA reduced the amount of shifting of the fluorescently labeled S probe DNA (Fig. 4, left panel, lane 6), whereas an excess of the nonspecific DNA fragment had no effect on ElrR binding to the elrA promoter region (Fig. 4, left panel, lane 8). This result shows that ElrR binds specifically to the elrA promoter and suggests that the positive effect of ElrR on elrA expression is due to direct interaction with the elrA promoter region.
Fig 4.

ElrR specifically binds the elrA promoter region. (Top) Map of the intergenic region upstream of the E. faecalis elrA operon, with the ORFs elrR and elrA. The transcription orientation of elrA and elrR is indicated by thick arrows, and the thin arrow indicates the promoter of the elrA operon (PelrA). The lollipop indicates a 50-bp inverted repeat at the DNA level and the predicted transcription terminator. Lines below represent DNA probes used for EMSAs; numbers correspond to the location of the extremities relative to the start codon of the elrA gene. (Bottom) EMSA of elrA promoter region with ElrR. In the left panel, 3 ng of a 0.5-kb fluorescently labeled DNA fragment encompassing the upstream region of PelrA (S for “specific”) and 5 ng of a fluorescently labeled DNA control fragment (NS for “nonspecific”), corresponding to a 0.3-kb internal fragment of the elrR gene, were incubated with 0, 1, 1.5, and 2 μM purified ElrR (lanes 1 to 4, respectively). An excess of cold S (lanes 5 and 6) and NS (lanes 7 and 8) DNA probes was added to the binding reaction mixtures in the absence (lanes 5 and 7) or presence (lanes 6 and 8) of 1.5 μM ElrR. In the right panel, EMSA of DNA probes encompassing nested fragments of the PelrA region that were incubated without (−) or with (+) 1.5 μM ElrR. S and 1 to 4 correspond to the DNA probes shown on the map above.
To narrow down the elrA promoter region recognized by ElrR, an EMSA was performed with four additional DNA probes, which covered the elrA promoter region from positions −387 to +105 bp relative to the start codon of elrA (Fig. 4, right panel). A shift was observed with probes that encompassed the region from positions −387 to −56 bp (probe 1) and from −191 to +105 bp (probe 3). It is noteworthy, however, that the retardation efficacy was decreased compared to that for probe S. Given the lack of mobility shift for probes corresponding to positions −387 to −144 bp (probe 2) and −83 to +105 bp (probe 4), the shift of probes 1 and 3 points toward the 136-bp region from positions −191 to −56 bp, which includes an inverted repeat DNA sequence predicted to be the transcription terminator of elrR, as the region required for binding. However, attempts to observe a mobility shift for the postulated minimal ElrR binding region failed (data not shown), indicating that ElrR binding in vitro requires sequences flanking both sides of the elrA promoter region from positions −191 to −56 bp or a specific DNA conformation. This observation agrees with the apparent decreased binding efficiency of ElrR on probes 1 and 3 compared to that on the extended probe S (Fig. 4), suggesting that the tightness of ElrR binding is influenced by the position of the binding site within the probe. As is the case for other Rgg-like regulators (14, 20, 24–26, 30), our data demonstrate that ElrR does not require the presence of a signaling molecule to bind DNA, and yet further experiments are needed to determine if ElrR binding in the absence of any signal is sufficient to promote transcription.
To correlate ElrR binding and transcriptional activity, we constructed additional lacZ translational fusions, which contained 583, 190, or 82 bp upstream of the elrA start codon (Fig. 5). The resulting plasmids were introduced into a strain expressing elrR in trans from pVE14040 or harboring the empty plasmid pVE3916, used as a negative control. The strains carrying fusions from pVE14194, pVE14191, and pVE14190 that include different elements of the elrA promoter region displayed similar levels of β-galactosidase activity in response to ElrR expression compared to the control strains (Fig. 5, top). Since we hypothesized that a terminator-like structure (i.e., inverted repeat sequence on DNA) could be recognized by ElrR, we cannot rule out the possibility that the terminator (“Term” in Fig. 5) used to silence transcription from the tetM promoter upstream of the lacZ fusions may be targeted by ElrR. We therefore constructed a second vector carrying lacZ fusions where “Term” was removed and the tetM cassette was inverted (Fig. 5, bottom). In the two sets of vectors, the elrA::lacZ fusions responded specifically to the presence of ElrR and with the same magnitude, demonstrating that the 82 bp upstream of the elrA ORF are specifically targeted by ElrR in vivo and indicating that ElrR-dependent activation of elrA occurs at the previously mapped proximal promoter of elrA (6). Transcription start site (TSS) mapping was performed using the 5′-tag RACE method (47) on PelrA::lacZ transcripts expressed from fusions containing the elrA promoter region and ElrR provided in cis (pVE14183) or in trans (pVE14194). Regardless of how ElrR was provided, all TSSs mapped for elrA::lacZ transcripts were located at 21 nucleotides upstream of the translation initiation codon of ElrA corresponding to t1, the elrA proximal promoter previously mapped (6). An identical TSS was mapped in strains expressing the PelrA::lacZ fusion where ElrR was either inactivated by a stop codon (pVE14188, cis construct) or absent from the vector (pVE3916) when provided in trans. We previously mapped a second elrA transcription start site (t2) within the 50-bp inverted repeat at position −104 relative to the start codon of elrA (6). The absence of detectable t2 in these experiments further supports the idea that the long inverted repeat is not required for ElrR-dependent activation of elrA (Fig. 5). Thus, our data show that ElrR acts on the elrA proximal promoter (Fig. 6). The two predicted boxes for this promoter, −35 (TTGAAC) and −10 (TATAAT), are separated by 21 bp, a fairly long element (66). Interestingly, regulators of the MerR family bind between the −35 and −10 boxes separated by long spacers and induce DNA distortion, activating transcription (67, 68). It is conceivable that ElrR may act in a similar manner. Although we cannot rule out the possibility that the N-terminal histidine tag modifies ElrR binding capacities, the apparent discrepancy between the in vivo ElrR-dependent activation of elrA expression and the in vitro ElrR binding region may reflect changes in conformation or oligomerization of ElrR, which would act differently on the elrA promoter region. Noticeably, in vitro binding of Rgg1358 of S. thermophilus to the promoter region of its target genes does not require the small hydrophobic activating peptide that is necessary for transcription activation in vivo (14), suggesting that binding does not necessarily imply transcription activation. In the case of PrgX, which acts as a repressor by binding as a tetramer to two operator sites upstream of the prgQ operon, its binding capacity is modified by the activating pheromone cCF10, which switches the oligomeric status of the regulator from a tetramer to a dimer and relieves repression (60, 69, 70). More recently, it was also proposed that Rgg3-mediated repression in S. pyogenes was relieved upon interaction with the signaling peptide (71). At this stage of our knowledge, further investigation is required to analyze the elrR-dependent activation of elrA at the molecular level.
Fig 5.

Schematic representation of the lacZ translational fusions and their corresponding β-galactosidase activities with elrR provided in trans. At the top, the elrA promoter region is schematized with the two transcription start sites t1 and t2 and the 50-bp inverted repeat indicated by the lollipop as defined previously (6). DNA fragments encompassing elrA promoter regions were fused to the lacZ coding sequence in two vectors. Transcription from tetM of vector pVE14189 was stopped by rho-independent transcription terminators, Term (black lollipop). tetM of vector pVE14195 was reversed and Term terminators were removed compared to pVE14189. β-Galactosidase activities of strains expressing elrR (ElrR++) and plasmid control (Vector) are the averages of at least three independent experiments. nd, not done.
Fig 6.

Nucleotide sequence of elrA promoter region. Lowercase letters indicate elrR and elrA coding sequences; the stop and start codons are in bold. The 50-bp inverted repeat is indicated by convergent arrows. Numbering refers to the translation start site of elrA (+1). The two transcriptional start sites t1 and t2 of elrA previously mapped are shown (6); t2 was not found under the conditions used in this work. The putative −35 and −10 boxes are underlined. The ribosome binding site for elrA is shown by italics. Shaded letters correspond to extremities of DNA probes used for EMSA (Fig. 4).
ElrR inactivation attenuates E. faecalis virulence in a mouse peritonitis model.
In several bacterial species, Rgg family members participate in virulence by regulating expression of virulence factors (72–75). We previously showed that elrA expression is enhanced in vivo, stimulates the host inflammatory response, and contributes to virulence (6). Added to the positive effect of ElrR on elrA expression (see above), these data led us to investigate the effects of ElrR on E. faecalis virulence. Using a mouse peritonitis model (76), the virulence of the isogenic single-mutant ΔelrR and ΔelrA strains and the double-deletion ΔelrR ΔelrA mutant was compared to that of the parental strain OG1RF and the ΔelrR-complemented strain. For each infection, mice were injected intraperitoneally with ∼109 CFU and mortality rates were compared. Mortality was delayed and significantly reduced for mice infected with the ΔelrR strain; 20% of the mice were still alive after 120 h, whereas 100% of mice died after 75 h when infected with the OG1RF strain (Fig. 7). Survival analysis after infection with the ΔelrR ΔelrA and ΔelrA strains showed that only 40% of the mice infected with the ΔelrR ΔelrA mutant died within 5 days, while 65% of the mice died after infection with the ΔelrA strain. Attenuation of a strain deleted for elrR could be expected from its positive regulatory effect on ElrA, previously implicated in E. faecalis virulence in the same virulence model. Although not significant, the slight delay in virulence of the ΔelrA strain compared with the ΔelrR strain suggests that inactivation of elrR does not totally abrogate elrA expression in vivo. Unexpectedly, concomitant inactivation of the elrA and elrR genes had a cumulative effect on virulence attenuation, strongly suggesting that elrR regulates genes other than elrA. As elrA belongs to an operon, the other genes of the operon (ef2685 to ef2682) are obvious candidates that may participate in increased virulence attenuation of the ΔelrR ΔelrA strain. Since we had previously reported that elrA was upregulated in vivo in mice (6), we compared the elrA transcript levels of OG1RF and ΔelrR strains in mice at 20 h postinfection. Under these experimental conditions, the level of elrA transcript found in the enterococci recovered from peritoneal cavity fluids of mice infected with ∼1 × 109 CFU was about 4-fold higher in the wild-type strain than in the ΔelrR strain, indicating that ElrR activates elrA expression also in vivo.
Fig 7.

Effect of elrR inactivation on E. faecalis virulence. Kaplan-Meier survival analysis in a mouse peritonitis model with the OG1RF wild-type strain (open circles), the ΔelrA mutant strain (squares), the ΔelrR mutant strain (inverted triangles), the ΔelrR ΔelrA double mutant strain (diamonds), and the elrR-complemented strain (closed circles). Data were pooled from two independent experiments, except for the ΔelrR-complemented strain. A total of 20 mice were infected intraperitoneally with 1.4 × 109 or 0.85 × 109, 1.3 × 109 or 0.9 × 109, 1.5 × 109 or 1.1 × 109, and 1.3 × 109 or 0.98 × 109 CFU of OG1RF, ΔelrA, ΔelrR, and ΔelrR ΔelrA strains, respectively. A total of 10 mice were infected with 1.6 × 109 CFU of the ΔelrR-complemented strain. For each pairwise comparison, OG1RF/ΔelrR, ΔelrR/ΔelrR ΔelrA, and ΔelrA/ΔelrR ΔelrA, P values were <0.0001, <0.005, and <0.15, respectively.
WxL proteins are present in a subset of low-GC Gram-positive species, including E. faecalis, Enterococcus faecium, Lactobacillus plantarum, Lactobacillus sakei, Listeria monocytogenes, and Bacillus cereus (7, 77, 78). We previously showed that the C-terminal WxL domain, which contains two conserved motifs, Trp-x-Leu, of secreted proteins including ElrA, promotes noncovalent association to the bacterial cell surface (7). The elrA operon is representative of the organization of other WxL protein-encoding clusters, which encode at least two WxL domain proteins, a small LPXTG protein, and a predicted membrane-anchored protein with a conserved DUF916 domain. These encoded surface proteins were hypothesized to form a cell surface complex at the bacterial surface (7, 77). Proteins EF2685 to EF2682 encoded by the elrA operon could contribute to virulence independently of ElrA. Functional investigation of the complete elrA operon should help to answer this question. In addition, as reported for ropB of S. pyogenes (26) and rovS of S. agalactiae (25), elrR may regulate loci other than solely its adjacent elrA target locus, as suggested by in vivo data (Fig. 7).
In conclusion, we identified ElrR, an E. faecalis member of the Rgg regulator family, which activates elrA expression. We showed that the elrR gene is functionally linked to the contiguous elrA operon and that ElrR is not expressed at detectable levels under laboratory conditions in strain OG1RF. Constitutive expression of elrR increases the RNA levels of elrA, and the ElrR protein binds specifically to the elrA promoter region, suggesting that ElrR acts directly as an activator of elrA transcription. Moreover, these data provide the first evidence for the involvement of ElrR in E. faecalis virulence. Identification of a signal that could trigger natural expression of elrR and/or elrA remains a challenging question in order to provide means to undertake mechanistic studies of ElrR effects and to delineate the ElrR regulon.
ACKNOWLEDGMENTS
We are very grateful to L. Rigottier-Gois for helpful discussions, to V. Juillard for in silico prediction of the short coding sequences, and to the reviewers for their constructive comments. We warmly thank A. L. Sonenshein for careful reading of the manuscript. We thank A. Salomon for technical assistance and M. Perego and S. Mesnage for providing p3TET and pET2818, respectively. We also thank C. Bevilacqua and C. Beauvallet from the ICE platform (INRA-GABI, Jouy-en-Josas) for their help in qRT-PCR experiments and the use of the Typhoon imager.
R.D. was supported by a fellowship from the Région Ile-de-France in the framework of the Dim Malfin-2008.
Footnotes
Published ahead of print 3 May 2013
REFERENCES
- 1. Bar K, Wisplinghoff H, Wenzel RP, Bearman GM, Edmond MB. 2006. Systemic inflammatory response syndrome in adult patients with nosocomial bloodstream infections due to enterococci. BMC Infect. Dis. 6:145. 10.1186/1471-2334-6-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mundy LM, Sahm DF, Gilmore M. 2000. Relationships between enterococcal virulence and antimicrobial resistance. Clin. Microbiol. Rev. 13:513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gilmore MS, Ferretti JJ. 2003. Microbiology. The thin line between gut commensal and pathogen. Science 299:1999–2002 [DOI] [PubMed] [Google Scholar]
- 4. Ogier JC, Serror P. 2008. Safety assessment of dairy microorganisms: the Enterococcus genus. Int. J. Food Microbiol. 126:291–301 [DOI] [PubMed] [Google Scholar]
- 5. Kayaoglu G, Orstavik D. 2004. Virulence factors of Enterococcus faecalis: relationship to endodontic disease. Crit. Rev. Oral Biol. Med. 15:308–320 [DOI] [PubMed] [Google Scholar]
- 6. Brinster S, Posteraro B, Bierne H, Alberti A, Makhzami S, Sanguinetti M, Serror P. 2007. Enterococcal leucine-rich repeat-containing protein involved in virulence and host inflammatory response. Infect. Immun. 75:4463–4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brinster S, Furlan S, Serror P. 2007. C-terminal WxL domain mediates cell wall binding in Enterococcus faecalis and other gram-positive bacteria. J. Bacteriol. 189:1244–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pancer Z, Cooper MD. 2006. The evolution of adaptive immunity. Annu. Rev. Immunol. 24:497–518 [DOI] [PubMed] [Google Scholar]
- 9. Bierne H, Sabet C, Personnic N, Cossart P. 2007. Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect. 9:1156–1166 [DOI] [PubMed] [Google Scholar]
- 10. Bassler BL, Losick R. 2006. Bacterially speaking. Cell 125:237–246 [DOI] [PubMed] [Google Scholar]
- 11. Declerck N, Bouillaut L, Chaix D, Rugani N, Slamti L, Hoh F, Lereclus D, Arold ST. 2007. Structure of PlcR: insights into virulence regulation and evolution of quorum sensing in Gram-positive bacteria. Proc. Natl. Acad. Sci. U. S. A. 104:18490–18495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rocha-Estrada J, Aceves-Diez AE, Guarneros G, de la Torre M. 2010. The RNPP family of quorum-sensing proteins in Gram-positive bacteria. Appl. Microbiol. Biotechnol. 87:913–923 [DOI] [PubMed] [Google Scholar]
- 13. Mashburn-Warren L, Morrison DA, Federle MJ. 2010. A novel double-tryptophan peptide pheromone controls competence in Streptococcus spp. via an Rgg regulator. Mol. Microbiol. 78:589–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fleuchot B, Gitton C, Guillot A, Vidic J, Nicolas P, Besset C, Fontaine L, Hols P, Leblond-Bourget N, Monnet V, Gardan R. 2011. Rgg proteins associated with internalized small hydrophobic peptides: a new quorum-sensing mechanism in streptococci. Mol. Microbiol. 80:1102–1119 [DOI] [PubMed] [Google Scholar]
- 15. Sulavik MC, Tardif G, Clewell DB. 1992. Identification of a gene, rgg, which regulates expression of glucosyltransferase and influences the Spp phenotype of Streptococcus gordonii Challis. J. Bacteriol. 174:3577–3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loughman JA, Caparon MG. 2007. Contribution of invariant residues to the function of Rgg family transcription regulators. J. Bacteriol. 189:650–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gardan R, Besset C, Guillot A, Gitton C, Monnet V. 2009. The oligopeptide transport system is essential for the development of natural competence in Streptococcus thermophilus strain LMD-9. J. Bacteriol. 191:4647–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fontaine L, Boutry C, de Frahan MH, Delplace B, Fremaux C, Horvath P, Boyaval P, Hols P. 2010. A novel pheromone quorum-sensing system controls the development of natural competence in Streptococcus thermophilus and Streptococcus salivarius. J. Bacteriol. 192:1444–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sulavik MC, Clewell DB. 1996. Rgg is a positive transcriptional regulator of the Streptococcus gordonii gtfG gene. J. Bacteriol. 178:5826–5830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vickerman MM, Minick PE. 2002. Genetic analysis of the rgg-gtfG junctional region and its role in Streptococcus gordonii glucosyltransferase activity. Infect. Immun. 70:1703–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dmitriev AV, McDowell EJ, Kappeler KV, Chaussee MA, Rieck LD, Chaussee MS. 2006. The Rgg regulator of Streptococcus pyogenes influences utilization of nonglucose carbohydrates, prophage induction, and expression of the NAD-glycohydrolase virulence operon. J. Bacteriol. 188:7230–7241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanders JW, Leenhouts K, Burghoorn J, Brands JR, Venema G, Kok J. 1998. A chloride-inducible acid resistance mechanism in Lactococcus lactis and its regulation. Mol. Microbiol. 27:299–310 [DOI] [PubMed] [Google Scholar]
- 23. Fernandez A, Borges F, Gintz B, Decaris B, Leblond-Bourget N. 2006. The rggC locus, with a frameshift mutation, is involved in oxidative stress response by Streptococcus thermophilus. Arch. Microbiol. 186:161–169 [DOI] [PubMed] [Google Scholar]
- 24. Neely MN, Lyon WR, Runft DL, Caparon M. 2003. Role of RopB in growth phase expression of the SpeB cysteine protease of Streptococcus pyogenes. J. Bacteriol. 185:5166–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Samen UM, Eikmanns BJ, Reinscheid DJ. 2006. The transcriptional regulator RovS controls the attachment of Streptococcus agalactiae to human epithelial cells and the expression of virulence genes. Infect. Immun. 74:5625–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anbalagan S, McShan WM, Dunman PM, Chaussee MS. 2011. Identification of Rgg binding sites in the Streptococcus pyogenes chromosome. J. Bacteriol. 193:4933–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chaussee MS, Watson RO, Smoot JC, Musser JM. 2001. Identification of Rgg-regulated exoproteins of Streptococcus pyogenes. Infect. Immun. 69:822–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zheng F, Ji H, Cao M, Wang C, Feng Y, Li M, Pan X, Wang J, Qin Y, Hu F, Tang J. 2011. Contribution of the Rgg transcription regulator to metabolism and virulence of Streptococcus suis serotype 2. Infect. Immun. 79:1319–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rawlinson EL, Nes IF, Skaugen M. 2002. LasX, a transcriptional regulator of the lactocin S biosynthetic genes in Lactobacillus sakei L45, acts both as an activator and a repressor. Biochimie 84:559–567 [DOI] [PubMed] [Google Scholar]
- 30. Rawlinson EL, Nes IF, Skaugen M. 2005. Identification of the DNA-binding site of the Rgg-like regulator LasX within the lactocin S promoter region. Microbiology 151:813–823 [DOI] [PubMed] [Google Scholar]
- 31. Lyon WR, Gibson CM, Caparon MG. 1998. A role for trigger factor and an rgg-like regulator in the transcription, secretion and processing of the cysteine proteinase of Streptococcus pyogenes. EMBO J. 17:6263–6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaussee MS, Sylva GL, Sturdevant DE, Smoot LM, Graham MR, Watson RO, Musser JM. 2002. Rgg influences the expression of multiple regulatory loci to coregulate virulence factor expression in Streptococcus pyogenes. Infect. Immun. 70:762–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dunny GM, Brown BL, Clewell DB. 1978. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc. Natl. Acad. Sci. U. S. A. 75:3479–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gibson TJ. 1984. Studies on the Epstein-Barr virus genome. Ph.D. thesisUniversity of Cambridge, Cambridge, United Kingdom [Google Scholar]
- 35. Guedon E, Serror P, Ehrlich SD, Renault P, Delorme C. 2001. Pleiotropic transcriptional repressor CodY senses the intracellular pool of branched-chain amino acids in Lactococcus lactis. Mol. Microbiol. 40:1227–1239 [DOI] [PubMed] [Google Scholar]
- 36. Hancock LE, Perego M. 2004. Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J. Bacteriol. 186:7951–7958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chastanet A, Fert J, Msadek T. 2003. Comparative genomics reveal novel heat shock regulatory mechanisms in Staphylococcus aureus and other Gram-positive bacteria. Mol. Microbiol. 47:1061–1073 [DOI] [PubMed] [Google Scholar]
- 38. Maguin E, Duwat P, Hege T, Ehrlich D, Gruss A. 1992. New thermosensitive plasmid for gram-positive bacteria. J. Bacteriol. 174:5633–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Simon D, Chopin A. 1988. Construction of a vector plasmid family and its use for molecular cloning in Streptococcus lactis. Biochimie 70:559–566 [DOI] [PubMed] [Google Scholar]
- 40. Vagner V, Dervyn E, Ehrlich SD. 1998. A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144:3097–3104 [DOI] [PubMed] [Google Scholar]
- 41. Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
- 42. Veiga P, Bulbarela-Sampieri C, Furlan S, Maisons A, Chapot-Chartier MP, Erkelenz M, Mervelet P, Noirot P, Frees D, Kuipers OP, Kok J, Gruss A, Buist G, Kulakauskas S. 2007. SpxB regulates O-acetylation-dependent resistance of Lactococcus lactis peptidoglycan to hydrolysis. J. Biol. Chem. 282:19342–19354 [DOI] [PubMed] [Google Scholar]
- 43. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 44. Sifri CD, Mylonakis E, Singh KV, Qin X, Garsin DA, Murray BE, Ausubel FM, Calderwood SB. 2002. Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and mice. Infect. Immun. 70:5647–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rigottier-Gois L, Alberti A, Houel A, Taly JF, Palcy P, Manson J, Pinto D, Matos RC, Carrilero L, Montero N, Tariq M, Karsens H, Repp C, Kropec A, Budin-Verneuil A, Benachour A, Sauvageot N, Bizzini A, Gilmore MS, Bessieres P, Kok J, Huebner J, Lopes F, Gonzalez-Zorn B, Hartke A, Serror P. 2011. Large-scale screening of a targeted Enterococcus faecalis mutant library identifies envelope fitness factors. PLoS One 6:e29023. 10.1371/journal.pone.0029023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poyart C, Trieu-Cuot P. 1997. A broad-host-range mobilizable shuttle vector for the construction of transcriptional fusions to β-galactosidase in Gram-positive bacteria. FEMS Microbiol. Lett. 156:193–198 [DOI] [PubMed] [Google Scholar]
- 47. Fouquier d'Herouel A, Wessner F, Halpern D, Ly-Vu J, Kennedy SP, Serror P, Aurell E, Repoila F. 2011. A simple and efficient method to search for selected primary transcripts: noncoding and antisense RNAs in the human pathogen Enterococcus faecalis. Nucleic Acids Res. 39:e46. 10.1093/nar/gkr012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mandin P, Repoila F, Vergassola M, Geissmann T, Cossart P. 2007. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res. 35:962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meijerink J, Mandigers C, van de Locht L, Tonnissen E, Goodsaid F, Raemaekers J. 2001. A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J. Mol. Diagn. 3:55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 51. Pai SR, Singh KV, Murray BE. 2003. In vivo efficacy of the ketolide ABT-773 (cethromycin) against enterococci in a mouse peritonitis model. Antimicrob. Agents Chemother. 47:2706–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ermolaeva MD, Khalak HG, White O, Smith HO, Salzberg SL. 2000. Prediction of transcription terminators in bacterial genomes. J. Mol. Biol. 301:27–33 [DOI] [PubMed] [Google Scholar]
- 53. Naville M, Ghuillot-Gaudeffroy A, Marchais A, Gautheret D. 2011. ARNold: a web tool for the prediction of Rho-independent transcription terminators. RNA Biol. 8:11–13 [DOI] [PubMed] [Google Scholar]
- 54. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 55. Sadreyev RI, Grishin NV. 2004. Quality of alignment comparison by COMPASS improves with inclusion of diverse confident homologs. Bioinformatics 20:818–828 [DOI] [PubMed] [Google Scholar]
- 56. Blatch GL, Lassle M. 1999. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21:932–939 [DOI] [PubMed] [Google Scholar]
- 57. McGuffin LJ, Bryson K, Jones DT. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16:404–405 [DOI] [PubMed] [Google Scholar]
- 58. Bouillaut L, Perchat S, Arold S, Zorrilla S, Slamti L, Henry C, Gohar M, Declerck N, Lereclus D. 2008. Molecular basis for group-specific activation of the virulence regulator PlcR by PapR heptapeptides. Nucleic Acids Res. 36:3791–3801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grenha R, Slamti L, Nicaise M, Refes Y, Lereclus D, Nessler S. 2013. Structural basis for the activation mechanism of the PlcR virulence regulator by the quorum-sensing signal peptide PapR. Proc. Natl. Acad. Sci. U. S. A. 110:1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shi K, Brown CK, Gu ZY, Kozlowicz BK, Dunny GM, Ohlendorf DH, Earhart CA. 2005. Structure of peptide sex pheromone receptor PrgX and PrgX/pheromone complexes and regulation of conjugation in Enterococcus faecalis. Proc. Natl. Acad. Sci. U. S. A. 102:18596–18601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shelburne SA, Olsen RJ, Makthal N, Brown NG, Sahasrabhojane P, Watkins EM, Palzkill T, Musser JM, Kumaraswami M. 2011. An amino-terminal signal peptide of Vfr protein negatively influences RopB-dependent SpeB expression and attenuates virulence in Streptococcus pyogenes. Mol. Microbiol. 82:1481–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chang JC, LaSarre B, Jimenez JC, Aggarwal C, Federle MJ. 2011. Two group A streptococcal peptide pheromones act through opposing Rgg regulators to control biofilm development. PLoS Pathog. 7:e1002190. 10.1371/journal.ppat.1002190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bae T, Clerc-Bardin S, Dunny GM. 2000. Analysis of expression of prgX, a key negative regulator of the transfer of the Enterococcus faecalis pheromone-inducible plasmid pCF10. J. Mol. Biol. 297:861–875 [DOI] [PubMed] [Google Scholar]
- 64. Ibrahim M, Guillot A, Wessner F, Algaron F, Besset C, Courtin P, Gardan R, Monnet V. 2007. Control of the transcription of a short gene encoding a cyclic peptide in Streptococcus thermophilus: a new quorum-sensing system? J. Bacteriol. 189:8844–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lereclus D, Agaisse H, Gominet M, Salamitou S, Sanchis V. 1996. Identification of a Bacillus thuringiensis gene that positively regulates transcription of the phosphatidylinositol-specific phospholipase C gene at the onset of the stationary phase. J. Bacteriol. 178:2749–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Haugen SP, Ross W, Gourse RL. 2008. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 6:507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brown NL, Stoyanov JV, Kidd SP, Hobman JL. 2003. The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 27:145–163 [DOI] [PubMed] [Google Scholar]
- 68. Joshi CP, Panda D, Martell DJ, Andoy NM, Chen TY, Gaballa A, Helmann JD, Chen P. 2012. Direct substitution and assisted dissociation pathways for turning off transcription by a MerR-family metalloregulator. Proc. Natl. Acad. Sci. U. S. A. 109:15121–15126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kozlowicz BK, Shi K, Gu ZY, Ohlendorf DH, Earhart CA, Dunny GM. 2006. Molecular basis for control of conjugation by bacterial pheromone and inhibitor peptides. Mol. Microbiol. 62:958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Caserta E, Haemig HA, Manias DA, Tomsic J, Grundy FJ, Henkin TM, Dunny GM. 2012. In vivo and in vitro analyses of regulation of the pheromone-responsive prgQ promoter by the PrgX pheromone receptor protein. J. Bacteriol. 194:3386–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lasarre B, Aggarwal C, Federle MJ. 2013. Antagonistic Rgg regulators mediate quorum sensing via competitive DNA binding in Streptococcus pyogenes. mBio 3(6):e00333–12. 10.1128/mBio.00333_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hollands A, Aziz RK, Kansal R, Kotb M, Nizet V, Walker MJ. 2008. A naturally occurring mutation in ropB suppresses SpeB expression and reduces M1T1 group A streptococcal systemic virulence. PLoS One 3:e4102. 10.1371/journal.pone.0004102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Carroll RK, Shelburne SA, III, Olsen RJ, Suber B, Sahasrabhojane P, Kumaraswami M, Beres SB, Shea PR, Flores AR, Musser JM. 2011. Naturally occurring single amino acid replacements in a regulatory protein alter streptococcal gene expression and virulence in mice. J. Clin. Invest. 121:1956–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bortoni ME, Terra VS, Hinds J, Andrew PW, Yesilkaya H. 2009. The pneumococcal response to oxidative stress includes a role for Rgg. Microbiology 155:4123–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Neely MN, Pfeifer JD, Caparon M. 2002. Streptococcus-zebrafish model of bacterial pathogenesis. Infect. Immun. 70:3904–3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Singh KV, Coque TM, Weinstock GM, Murray BE. 1998. In vivo testing of an Enterococcus faecalis efaA mutant and use of efaA homologs for species identification. FEMS Immunol. Med. Microbiol. 21:323–331 [DOI] [PubMed] [Google Scholar]
- 77. Siezen R, Boekhorst J, Muscariello L, Molenaar D, Renckens B, Kleerebezem M. 2006. Lactobacillus plantarum gene clusters encoding putative cell-surface protein complexes for carbohydrate utilization are conserved in specific gram-positive bacteria. BMC Genomics 7:126. 10.1186/1471-2164-7-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chaillou S, Champomier-Verges MC, Cornet M, Crutz-Le Coq AM, Dudez AM, Martin V, Beaufils S, Darbon-Rongere E, Bossy R, Loux V, Zagorec M. 2005. The complete genome sequence of the meat-borne lactic acid bacterium Lactobacillus sakei 23K. Nat. Biotechnol. 23:1527–1533 [DOI] [PubMed] [Google Scholar]