

Abstract
A crucial element in the horizontal transfer of mobilizable and conjugative plasmids is the relaxase, a single-stranded endonuclease that nicks the origin of transfer (oriT) of the plasmid DNA. The relaxase of the pMV158 mobilizable plasmid is MobM (494 residues). In solution, MobM forms a dimer through its C-terminal domain, which is proposed to anchor the protein to the cell membrane and to participate in type 4 secretion system (T4SS) protein-protein interactions. In order to gain a deeper insight into the structural MobM requirements for efficient DNA catalysis, we studied two endonuclease domain variants that include the first 199 or 243 amino acid residues (MobMN199 and MobMN243, respectively). Our results confirmed that the two proteins behaved as monomers in solution. Interestingly, MobMN243 relaxed supercoiled DNA and cleaved single-stranded oligonucleotides harboring oriTpMV158, whereas MobMN199 was active only on supercoiled DNA. Protein stability studies using gel electrophoresis and mass spectrometry showed increased susceptibility to degradation at the domain boundary between the N- and C-terminal domains, suggesting that the domains change their relative orientation upon DNA binding. Overall, these results demonstrate that MobMN243 is capable of nicking the DNA substrate independently of its topology and that the amino acids 200 to 243 modulate substrate specificity but not the nicking activity per se. These findings suggest that these amino acids are involved in positioning the DNA for the nuclease reaction rather than in the nicking mechanism itself.
INTRODUCTION
Conjugation is one of three fundamentally different ways, in addition to transformation and transduction, by which the genetic content of a cell can be altered (1, 2). The conjugative process involves active DNA transfer from one living organism to another through the type 4 secretion system (T4SS), which is encoded on the donor cell side (3, 4). The shuttling of plasmids and integrative and conjugative elements (ICEs) between organisms has proven to be a major contributor to evolutionary diversity, as many genes have been exchanged rather than evolved by mutation during evolution (5). Although conjugation is generally associated with the exchange of genetic material, it most probably originated as a protein transport mechanism, since DNA transfer is always preceded by protein transfer and T4SS transfer is frequently used for translocation of proteinaceous virulence factors (6). The protein that plays a central role in the conjugational transfer of DNA is termed relaxase (7, 8). It associates with other factors to form a complex called the relaxosome (8), which provides the context for the controlled cleavage and processing of transferable DNA. The relaxase recognizes a specific DNA sequence, the origin of transfer (oriT), which generally contains one or more inverted repeats (9). The relaxase then nicks the DNA at a specific position and forms a stable complex with the oriT-harboring strand in the 5′ terminus, leaving a free 3′-OH end. The DNA-protein adduct is then transferred to the recipient cell, while the opposite strand is replicated in the donor cell using the available 3′ oxygen, generally through a rolling-circle replication mechanism (10). Finally, the single-stranded DNA (ssDNA) in the recipient cell is replicated, possibly through lagging-strand synthesis (11).
The nuclease functionality of relaxases is provided by a domain that is conserved among relaxases and replication initiators of rolling-circle replication from various genetic entities (7, 12, 13). The nuclease domains representing the MOBF (TraI_pUC1, TrwC_R388, and TraI_F) and MOBQ (MobA_R1162 and NES_pLW1043) families have been structurally characterized (14–19). A comparison of these structures shows a similar fold of the nuclease domain and suggests a like mechanism of action. All relaxases studied up to now were found to use a metal-dependent nucleophilic attack of a catalytic residue to the phosphodiester bond. The metal ion is believed to polarize the phosphodiester group, facilitating the attack by the nucleophile. The nucleophilic amino acid of the relaxases that have been characterized to date was found to be a tyrosine. Despite the general similarity between the nuclease domains of the different relaxases, they can be classified into families on the basis of their sequences and properties (7). The classification is likely to be related to details in the DNA processing reactions, e.g., to the type of metal used for bond polarization. Structural information on the Mob families other than MOBF or MOBQ is scarce, especially for relaxases of plasmids isolated from Gram-positive (G+) bacteria (20). Recently, the structure of NES, the MOBQ relaxase from the staphylococcal plasmid pLW1043, has been determined (19). This represents the only structure of a relaxase isolated from G+ bacteria to date. The MobM relaxase from the mobilizable streptococcal plasmid pMV158 (20, 21) is the prototype of the MOBV family of relaxases, which holds more than 100 members (7). The full-length MobM protein (Ser2-Phe494; Met1 is removed during production) has a predicted molecular mass of 57,692 Da and behaves as a dimer in solution (21). Its nuclease activity resides in the N-terminal domain, in which three conserved motifs can be identified: (i) motif I (HxxR), of unknown function; (ii) motif II (NYD/EL), which contains the proposed catalytic tyrosine (22); and (iii) motif III (HxDExxPHuH), also known as the 3H motif and probably involved in the coordination of a divalent metal (7, 22). The conjugative transfer is initiated by nicking of oriTpMV158 at the phosphodiester linkage between guanidine 3595 and thymine 3596 (Fig. 1 and Table 1) (23). oriT of pMV158 has an intricate pattern of inverted repeats (20), allowing the formation of at least three mutually exclusive hairpin-loop structures (Fig. 1). Using ssDNAs and a truncated MobM protein containing the first 199 residues, we have recently defined a minimal oriT binding sequence of 26 nucleotides (nt) (20).
Fig 1.

Genetic map and origin of transfer (oriT) of plasmid pMV158. (A) Scheme of pMV158. Arrowheads indicate the direction of transcription of the four protein-coding genes (mobM, copG, repB, and tetL). The positions of the oriT and the origins of the leading-strand (dso) and lagging-strand (ssoU and ssoA) syntheses are shown. (B) DNA sequence of the oriT of pMV158 (coordinates 3564 to 3605). The three identified inverted repeats (IR1, IR2, and IR3) are indicated by arrows, and the nick site is indicated by a vertical arrowhead. The oligonucleotides used in this study are also depicted (Table 1).
Table 1.
Oligonucleotides used in this study
| Primer namea | Sequenceb (5′to 3′) | Coordinatesc (nt) |
|---|---|---|
| Nde-F | AAGGAGGGAAACATATGAGTTACA | 3718–3741 |
| P244-Stop-Xho | CTTGGTTAGTTGTCTCGAGTATCTAAGATTGAAC | 4485–4452 |
| 19nic4d | GAATATAAAGTATAGTGTG/TTAT | 3577–3599 |
| IR1+12pAd | ACTTTATGAATATAAAGTATAGTGTG/TTATGG(A15) | 3570–3599 |
| IR1+8 | ACTTTATGAATATAAAGTATAGTGTG | 3570–3595 |
| IR1+12 | ACTTTATGAATATAAAGTATAGTGTG/TTAT | 3570–3599 |
The oligonucleotide IR1+12pA contains a 15-nt-long poly(A) tail.
Restriction sites are in boldface, and base changes that generate restriction sites are underlined. The double-base change introduced in the P244-Stop-Xho oligonucleotide to obtain the stop codon (P244Stop) is shown in italics. The oriT cleavage site is indicated by a slash.
Coordinates are given with respect to the pMV158 sequence (accession number NC_010096).
These oligonucleotides were labeled with the Cy5 fluorophore at its 5′ end.
Here, we report the DNA nicking properties, oligomeric state, and degradation behavior of the full-length MobM protein and two N-terminal constructs that included the first 199 (MobMN199) or 243 (MobMN243) amino acid residues. The substrate specificities, physiochemical properties, and stabilities differed depending on the construct length. A comparative functional assay demonstrated that the extra 44 amino acid residues of MobMN243 protein (compared to MobMN199) are essential for efficient ssDNA processing. To the best of our knowledge, this study provides the first in-depth characterization of the functional properties and structural requirements of a DNA relaxase domain belonging to the MOBV family.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Streptococcus pneumoniae 708 (end-1 exo-2 trt-1 hex-4 malM594) harboring pMV158 was used to purify plasmid DNA. Escherichia coli BL21(DE3) (rB− mB− gal ompT int::PlacUV5-T7 gen1 imm21 nin5; a gift of F. W. Studier) was used for the purification of MobM, MobMN199, and MobMN243. For overexpression of the mobM gene, plasmid pLGM2 was used; it contains two copies of the mobM gene under the control of the φ10 promoter of phage T7 (23). In the case of MobMN199 and MobMN243 proteins, plasmids pMobMN199 and pMobMN243 (pET24b-based vectors; see below) were used; they contain one copy of the mobMN199 or mobMN243 gene under the control of the φ10 promoter of phage T7 (23). E. coli was grown on tryptone-yeast extract (TY) medium (Pronadisa) at 37°C, whereas S. pneumoniae was grown in AGCH medium supplemented with 0.2% yeast extract and 0.3% sucrose (24, 25). In the case of plasmid-harboring cells, the medium was supplemented with tetracycline (Tet; 1 μg/ml) for S. pneumoniae/pMV158, with ampicillin (Amp; 100 μg/ml) for E. coli/pLGM2, and with kanamycin (Kan; 30 μg/ml) for E. coli/pMobMN199 and E. coli/pMobMN243.
DNA purification, amplification, and cloning.
Plasmid DNA from pMV158 (26) was purified from S. pneumoniae by two consecutive CsCl gradients as described previously (27). Plasmids pET24b, pMobMN199, pMobMN243, and pLGM2 were purified by minipreparations using the High Pure plasmid isolation kit (Roche Applied Science). The oligonucleotides used for nicking and stability studies were synthesized and purified by high-performance liquid chromatography (HPLC) by Integrated DNA Technologies (Coralville, IA) and are listed in Table 1.
Plasmids pLGM2 and pMobMN199 were constructed as described previously (20, 23). To obtain plasmid pMobMN243, a 768-bp fragment that encodes the first 243 residues of MobM protein was amplified by PCR on pMV158 DNA (GenBank accession number NC_010096) with the primers Nde-F and P244-Stop-Xho (Table 1). To facilitate cloning, forward and reverse primers included changes to generate recognition sites for NdeI and XhoI restriction enzymes, respectively. Furthermore, a double-base change was also introduced in the reverse primer to introduce an amber stop codon instead of Pro244 (P244Stop). PCR amplifications were done in 50-μl reaction mixtures containing 1× reaction buffer [16 mM (NH4)2SO4, 6 mM Tris-HCl, pH 8.8, 1.5 mM MgCl2], 0.2 mM each deoxynucleoside triphosphate (dNTP) (Roche), 0.4 mM each primer, 0.65 U of Phusion DNA polymerase (Finnzymes), and 1 ng of DNA template. The conditions were the following: an initial denaturing step at 98°C (30 s); 30 cycles of denaturation at 98°C (10 s), annealing at 55°C (20 s), and extension at 72°C (30 s); followed by a final extension at 72°C (10 min). PCR products were purified with the QIAquick gel extraction kit (Qiagen). After digestion with NdeI and XhoI enzymes, the fragments were ligated into pET24b previously digested with the same enzymes. All of the constructs were confirmed by Sanger DNA sequencing (Secugen S.L., Madrid, Spain).
Protein purification and secondary structure prediction.
MobM, MobMN199, and MobMN243 proteins were purified as described previously (20, 21, 23). After cell disruption of the cell pellet of the induced culture (4 liters), the sample was centrifuged to remove cell debris. The nucleic acids were precipitated by 0.2% (vol/vol) polyethylenimine (Sigma), and proteins of the supernatant were precipitated at 70% (wt/vol) ammonium sulfate saturation. Proteins in the precipitate were dissolved in buffer A (25 mM Tris-HCl, pH 7.6, 1 mM dithiothreitol, 1% [vol/vol] glycerol, 1 mM EDTA) with 300 mM NaCl, and after equilibration in the same buffer by dialysis, the sample was loaded in a 100-ml heparin-agarose (Bio-Rad) column. The protein was eluted with 400 ml of a 0.3 to 0.8 M NaCl gradient, and fractions were analyzed by 12% sodium dodecyl sulfate-glycine polyacrylamide gel electrophoresis (SDS-PAGE) using Bio-safe Coomassie (Bio-Rad Laboratories) as the staining agent. Fractions containing the peaks of MobM, MobMN243, or MobMN199 were pooled and concentrated by filtering them through 3-kDa cutoff membranes (Pall). The sample was subjected to a gel filtration column (HiLoad Superdex 200; GE-Healthcare Life Science) connected to a fast-pressure liquid chromatography system (FPLC; Biologic DuoFlow; Bio-Rad). Fractions were analyzed and concentrated as described above until reaching a concentration of 2 mg/ml and then were stored at −80°C. The concentration of MobM, MobMN243, and MobMN199 was estimated by spectrophotometric measurements considering theoretical molar extinction coefficients of 37,485, 24,410, and 17,420 M−1 cm−1, respectively. After running a purified MobMN243 sample in a 12% SDS-PAGE gel, the protein bands were transferred to a polyvinylidene difluoride (PVDF) blotting membrane. Edman's sequential degradation was performed directly from the PVDF membrane in order to analyze N-terminal sequences in a Procise 494 protein sequencer (PerkinElmer) as reported previously (23).
Secondary structure predictions of MobMN243 were carried out with the programs PSIPRED (28), Jpred (29), NPS@ (30), and SABLE (31) using the website http://www.expasy.ch/. The structure prediction was manually aligned to the secondary structure elements of the published structure of NES_pLW1043 (19).
Analytical ultracentrifugation.
Sedimentation equilibrium experiments were performed at 20°C in an Optima XL-A (Beckman-Coulter) analytical ultracentrifuge (CIB, Madrid, Spain). MobMN243 (10 and 20 μM) was examined in buffer UA (20 mM Tris-HCl, pH 7.6, 500 mM NaCl, 1 mM EDTA, 1% [vol/vol] glycerol). Samples were centrifuged at two successive speeds (20,000 and 35,000 rpm), and absorbance was measured at 3-h intervals to determine that samples had reached equilibrium. Apparent average molecular masses of MobMN243 were obtained using the program Heteroanalysis v1.1.44 (University of Connecticut, Storrs, CT). Sedimentation velocity experiments were performed at 48,000 rpm and 20°C with samples of MobMN243 (10 and 20 μM, in buffer UA). The sedimentation coefficient distribution for this protein was calculated by least-squares boundary modeling of sedimentation velocity data using the sedimentation coefficient distribution method, as implemented in the SEDFIT 12.1b program (32). The coefficient was corrected to standard conditions to obtain the corresponding standardized sedimentation coefficient (S20,w) value using the SEDNTERP program (33). The translational frictional coefficient of MobMN243 (f) was determined from the molecular mass and sedimentation coefficient of the protein (34), whereas the frictional coefficient of the equivalent hydrated sphere (f0) was estimated using a hydration of 0.4506 g H2O per g of protein (35).
scDNA and ssDNA nicking.
Nicking of supercoiled DNA (scDNA) by purified MobMN199 and MobMN243 proteins was performed as reported previously (20, 23).
Cleavage activity of ssDNA by purified MobMN199 or MobMN243 proteins was tested in standard reaction mixtures (20 μl) containing fluorophore-labeled oligonucleotide (60 nM) in buffer (25 mM Tris-HCl [pH 7.6], 0.1 mM EDTA, 10% [vol/vol] glycerol, 1 mM dithiothreitol) to which different concentrations of purified MobMN199 or MobMN243 was added. Standard reactions were performed at 30°C for 20 min in the presence of 8 mM MnCl2 and were stopped by addition of SDS (0.5%) and proteinase K (1 mg/ml), followed by incubation for 30 min at 37°C. DNA was precipitated by adding 0.3 M Na acetate and 100% ethanol and incubation for 30 min at −20°C. After centrifugation at 13,000 rpm for 10 min at 4°C, the pellet was dried (10 min at room temperature) and dissolved in distilled water with formamide. Samples were heated to 90°C for 3 min, after which they were flash-cooled to 4°C. Nicking of oligonucleotides was monitored by electrophoresis on 20% polyacrylamide (PAA) plus 7 M urea and visualized by a phosphorimager (Fuji). Band intensities were measured using Bio-Rad Laboratories software.
To test the capacity of MobMN243 to form stable complexes with ssDNA, 10 μM protein was incubated in a mix (20 μl) containing buffer (25 mM Tris-HCl [pH 7.6], 0.1 mM EDTA, 10% [vol/vol] glycerol, 1 mM dithiothreitol) and the oligonucleotide (10 or 20 μM). After 20 min at 30°C, samples were loaded in a 12% SDS-PAGE gel and stained with SYPRO Ruby (Bio-Rad) by following the manufacturer's recommendations.
Protein stability assays.
The time-dependent stability of MobMN243 was assayed by SDS-PAGE as follows. Purified protein (180 μM) from a single batch in buffer A plus 100 mM NaCl was supplemented with either (i) MnCl2 (15 mM final concentration), (ii) oligonucleotide IR1+8 (in 10% excess to MobMN243), or (iii) MnCl2 (15 mM final concentration) plus oligonucleotide IR1+8 (in 10% excess to MobMN243). The three samples were incubated at 4°C for 2 months and then analyzed by SDS-PAGE (12%). The IR1+8 oligonucleotide (Table 1) represents the minimal oriT recognized with a high binding affinity by MobMN199 (20). For comparison purposes, full-length MobM, MobMN243, and MobMN199 were also included in the gels.
DNA oligonucleotide IR1+12, encompassing the minimal oriT binding site (IR1+8) and 4 nucleotides downstream of the cleavage site (Table 1), was used for mass spectrometry (MS) stability experiments. MobMN243 (40 μM) in buffer A plus 100 mM NaCl was supplemented with either phosphoramidate or with a mixture of 15 mM MnCl2 or MgCl2 and the IR1+12 oligonucleotide (50 μM). Phosphoramidate was synthesized as described previously (36). The samples were then incubated at 30°C for 20 min and subsequently supplemented with 40 mM EDTA (final concentration) and placed on ice. The resulting samples then were loaded on a Superdex 200 column or a HiTrap desalting 5-ml column (Sephadex G-25 Superfine), both preequilibrated with MS buffer (100 mM ammonium acetate, pH 6.8). Size-exclusion chromatography was performed at 4°C. Fractions containing protein-DNA complex were concentrated using centrifuge concentrators with a 10-kDa cutoff and subjected to MS analysis.
Electrospray mass spectrometry (ESI-MS) was performed on an LTQ-FT Ultra instrument (Thermo Scientific), which was equipped with a nanoflow ESI source. The NanoMate (Advion BioSciences, Ithaca, NY) aspirated the samples from a 384-well plate (protein Lobind) with disposable, conductive pipette tips and infused the samples through the ESI chip, which consists of 400 nozzles in a 20 by 20 array. The spray voltage was 1.7 to 1.85 kV, and the delivery pressure was 0.3 to 0.35 lb/in2. Samples, provided in 100 mM ammonium acetate, pH 6.8, were diluted either in 0.1% acetonitrile-formic acid or 1% acetonitrile-water-formic acid to a final concentration of 1 to 5 μM in order to perform MS analysis (final formic acid concentration of 0.1 to 0.5%).
RESULTS AND DISCUSSION
Purification of MobMN243.
To gain a deeper insight into the structure-function properties of the MobM endonuclease domain and its binding to the pMV158 oriT (Fig. 1), we designed a strategy to obtain a protein containing the first 243 N-terminal residues. A truncated MobMN243 protein was obtained by replacing the P244 amino acid residue with a stop codon. To purify MobMN243, the protocol used to obtain the MobMN199 protein was followed (20). The fractions recovered after the various purification stages are shown in Fig. 2A. After the ammonium sulfate precipitation (lanes 6 and 7), the sample was subjected to heparin-affinity chromatography, and the pure fractions were collected and subjected to a final gel filtration step. The purest fractions were concentrated to 2 mg/ml (lane 8). The MobMN243 gel filtration profile then was compared to the chromatograms of MobMN199 and full-length MobM (Fig. 2B). MobM eluted in a single peak at 61.5 ml, while MobMN243 and MobMN199 eluted at 80.5 and 87.1 ml, respectively. Since MobM and MobMN199 behave as dimer and monomer, respectively (20, 21), the elution behavior suggests that MobMN243 is a monomer in solution. Under denaturing conditions, MobMN243 migrated with a relative molecular mass between 30 and 45 kDa, which differs slightly from the theoretical mass of 28,393.6 Da (Table 2). An increase in the amount of SDS in the gels abolished this effect (not shown), resulting in band migration corresponding to the molecular mass (between 20 and 30 kDa).
Fig 2.
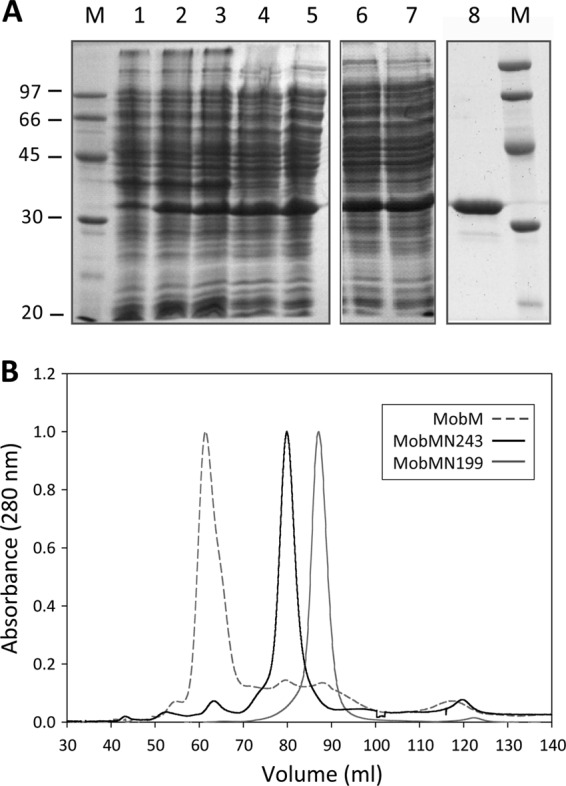
Overexpression and purification of the MobMN243 protein. (A) Fractions from the different purification steps of MobMN243 were analyzed by electrophoresis on 12% SDS-PAGE gels. Samples were loaded in the following order: noninduced cultures (lane 1), cultures after induction with IPTG (lane 2) and rifampin (lane 3), supernatant of a total cell lysate (lane 4), supernatant after polyethyleneimine precipitation (lane 5), supernatant of the ammonium sulfate precipitation step dialysis against buffer A plus 300 mM NaCl (lane 6), and protein soluble fraction after dialysis (lane 7). The sample was loaded onto a heparin-agarose column and then subjected to a gel filtration column (lane 8). M indicates the molecular mass standards (in kDa). (B) Comparative analysis of the gel filtration elution profiles of MobM (gray dashed line), MobMN243 (continuous black line), and MobMN199 (continuous gray line) subjected to 1 ml/min isocratic flow (buffer A plus 300 mM NaCl) in a HiLoad Superdex 200 column.
Table 2.
Substrate processivity and masses of MobM protein and its truncated versions
| Protein | No. of residuesa | Calculated massb (Da) | Measured massc (Da) | Activity on DNA |
|
|---|---|---|---|---|---|
| Supercoiled | Single stranded | ||||
| MobM | 493 | 57,692.3 | ND | Yes | Yes |
| MobMN199 | 198 | 23,115.2 | 23,195.3d | Yes | No |
| MobMN243 | 242 | 28,393.6 | 28,409.7e | Yes | Yes |
Number of total amino acid residues, excluding Met1, which is processed in the expression systems used in this study.
Monoisotopic, excluding Met1 (see footnote a).
Masses as determined by ESI-MS (see the text).
Mass corresponds to MobMN199 with one phosphorylated residue.
Mass corresponds to MobMN243 oxidized at one position.
MobMN243 oligomerization state.
Analytical ultracentrifugation analyses showed that full-length MobM behaved as an ellipsoidal dimer in solution (21), whereas MobMN199 was observed to be a monomer (20). Since the shorter version, MobMN243, lacks the last 251 C-terminal amino acids, we examined its oligomeric state and hydrodynamic properties. The elution volume of the truncated protein in the size-exclusion chromatography used for purification (Fig. 2B) suggested that MobMN243 is a monomer. To corroborate this, analytical ultracentrifugation assays were performed at two protein concentrations (10 and 20 μM). The results of sedimentation equilibrium and velocity assays are shown in Fig. 3. Sedimentation velocity profiles fit well with a model of single sedimenting species, with an S20,w of 2.5 S and an average apparent molecular mass of 27,503 ± 1,000 Da. These findings are consistent with the theoretical monomer mass. At 10 and 20 μM, the experimental sedimentation equilibrium data fitted best with an average apparent molecular mass of 30,500 ± 1,000 Da, which is near the value obtained for the sedimentation velocity and is also consistent with a monomeric species. The frictional ratio (f/f0) calculated was 1.29, indicating that MobMN243 deviates from the behavior expected for globular particles (f/f0 ≈ 1). Clearly, the dimerization interface resides in the C-terminal domain, perhaps through self-interaction of two protomers through a putative leucine zipper motif (from Leu317 to Leu338) present in this part of the protein (37).
Fig 3.

MobMN243 oligomerization state studied by analytical ultracentrifugation profile. The sedimentation equilibrium profile (35,000 rpm and 20°C) of 20 μM MobMN243 in buffer UA is shown. The lower part shows the experimental data (circles) and the best fit (solid line) to a single species with a molecular size of 31,000 ± 1,000 Da. The upper part shows residuals of the theoretical fit. The inset shows the distribution of sedimentation coefficients of the same MobMN243 protein sample in sedimentation velocity experiments (48,000 rpm at 20°C).
Substrates of MobMN243.
The oriT of pMV158 contains the MobM-mediated nick site (located between coordinates 3595 and 3596) and three inverted repeats (IR1, IR2, and IR3). The capacity of MobMN199 and MobMN243 to convert scDNA (form FI) into open circular species (form FII) was tested at several protein concentrations (from 8 to 256 nM) in the presence of 8 mM MnCl2. The samples were incubated at 30°C for 20 min, as reported for the full-length protein (23). No differences in scDNA nicking efficiency between MobMN199 and MobMN243 were detected. Indeed, the results showed that both truncated proteins relaxed pMV158 DNA with the same efficiency as full-length MobM, converting up to 60% of supercoiled plasmid into the FII form (Fig. 4A). The failure to reach 100% cleavage could be explained by the equilibrium between the nicking and closing activities of MobMN243, which has been observed for several relaxases (38–40).
Fig 4.
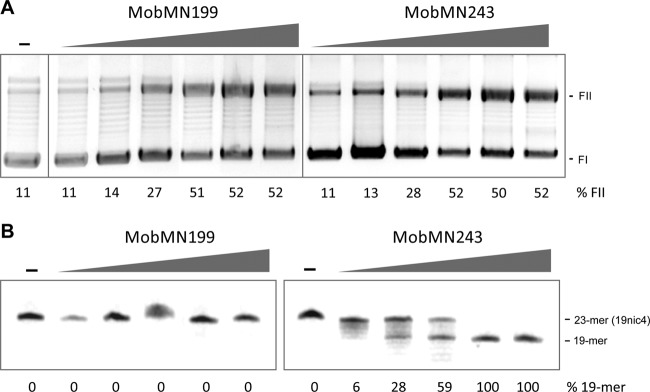
Nicking activity of MobMN199 and MobMN243. (A) Relaxation assays of pMV158 by MobMN199 and MobMN243. Supercoiled pMV158 DNA samples (8 nM) were incubated with (8, 16, 32, 64, 128, or 256 nM) or without (−) MobMN199 or MobMN243 and supplemented with 8 mM MnCl2 at 30°C for 20 min. Generation of relaxed forms (FII) from scDNA forms (FI) was analyzed by electrophoresis on 1% agarose gels stained with ethidium bromide (1 μg/ml). The weak band above relaxed forms (FII) has been observed before (20) and might correspond to relaxed DNA dimers. (B) Nicking activity of MobMN199 and MobMN243 on single-strand DNA harboring regions of the pMV158 oriT (19nic4). The nicking reaction yields 19- and 4-mer (not shown) oligonucleotide products. 19nic4 (60 nM) was incubated with a range of protein concentrations (60, 120, 240, 480, and 960 nM) at 30°C for 20 min in the presence of 8 mM MnCl2. The reaction was stopped with 0.1% SDS and proteinase K (1 mg/ml) for 30 min at 37°C, and samples were electrophoretically analyzed in a denaturing 20% PAA–7 M urea gel and visualized by a phosphorimager. Band quantification was done using the QuantityOne software (Bio-Rad) for both panels.
Nicking assays using oligonucleotide 19nic4 (Table 1) were performed in order to study the capacity of MobMN199 and MobMN243 to cleave ssDNA. This oligonucleotide encompasses the right arm of IR3 (19 nt) and 4 nt after the nick site (Fig. 1). A control with no proteins and different concentrations of the two proteins (60 to 480 nM range) were incubated with 60 nM the 19nic4 ssDNA (Table 1) in the presence of 8 mM MnCl2 as described in Materials and Methods and the legend to Fig. 4B. The expected products after a specific nick of the 19nic4 substrate are a 19-mer and a 4-mer. Contrary to the results obtained in the scDNA relaxation assays, no ssDNA cleaving activity was observed when the reaction was carried out with MobMN199 (Fig. 4B) or with an N-terminal His-tagged version of MobM243 (results not shown). However, a 19-mer band appeared in the presence of MobMN243 even at the lowest concentration tested (Fig. 4B).
Interestingly, all of the N-terminal truncations studied retained nuclease activity, indicating that all of the residues required for nicking activity reside in the first 199 amino acids, which was the shortest construct tested here. As the MobM C terminus is truncated beyond S243, the substrate range that can be cleaved becomes more restricted (Table 2). Thus, MobMN243 behaves like full-length MobM as far as the substrate nicking is concerned, and both are able to cleave ssDNA as well as scDNA. Removal of the 200- to 243-amino-acid region abolishes activity on ssDNA, although MobMN199 still cleaved the scDNA. The observation that the presence of the C-terminal 44 amino acid residues of MobMN243 are essential for efficient ssDNA cleavage suggests that the minimal endonuclease requirements for an efficient nicking depend on the DNA topology.
The minimal requirements for efficient DNA nicking have been analyzed for other conjugative relaxases belonging to the MOBF and MOBQ families. The first 275 amino acid residues of the TrwC relaxase (R388 plasmid) catalyzed ssDNA cleavage, although a longer fragment (348 amino acid residues) was required to produce the nick on a supercoiled double-stranded DNA substrate (41). In the case of MobA (R1162 plasmid), it has been demonstrated that a 184-residue amino-terminal fragment cleaves ssDNA as well as scDNA (17). Without high-resolution structural information, we can only speculate about whether the substrate specificity of the different constructs has a structural origin or whether other mechanisms are involved. However, a plausible explanation reconciles the experimental observations and assumes that MobM residues beyond amino acid 200 participate in DNA processing, inducing a conformation amenable to ssDNA nicking. The finding that MobMN199 cuts scDNA can be attributed to the topology of the supercoiled plasmid, allowing nicking to occur in the absence of the 200- to 243-amino-acid region. However, further processing of the DNA is expected to depend on these residues. Overall, the data provided here show that the mechanistic nuclease activity depends solely on the N-terminal domain, whereas DNA processivity depends on a region closer to the C-terminal domain.
MobMN243-DNA stable complexes.
The conjugative relaxases carry out cleavage reactions of a specific phosphodiester bond (nick site) in order to initiate DNA transfer. Such a reaction is based on a transesterification mechanism that produces a stable protein-DNA intermediate and a free 3′ OH terminus. To visualize the protein-ssDNA adducts expected after the nicking reaction, we incubated MobMN243 with the oligonucleotide IR1+12pA, which starts with the IR1+12 sequence and ends with a 15-mer-long poly(A) tail (Table 1 and Fig. 1B) to increase the molecular size of the protein-DNA complexes and facilitate the separation of the complex from the free MobM protein by electrophoretic analysis. The predicted products after the IR1+12pA oligonucleotide cleavage are two shorter DNA fragments; one of them (a 21-mer) attaches to the MobMN243 protein (Fig. 5A). Highly efficient ssDNA nicking was observed for MobMN243 when the IR1+12pA oligonucleotide was used as the substrate (Fig. 5B). Under the same experimental conditions, the protein-DNA adducts were solved by SDS-PAGE followed by SYPRO Ruby staining. Two bands were detected, one corresponding to the free protein (MobMN243) and the other to the MobMN243-DNA21 complex (Fig. 5C). However, the ratio for covalently bound complex was quite small, which can be explained in part by the equilibrium between the nicking and closing activities of MobMN243 (42, 43). Furthermore, similar results were previously reported for the relaxase domain of TrwC, where the molecules involved in the formation of stable complexes represented less than 10% of the total protein (44).
Fig 5.
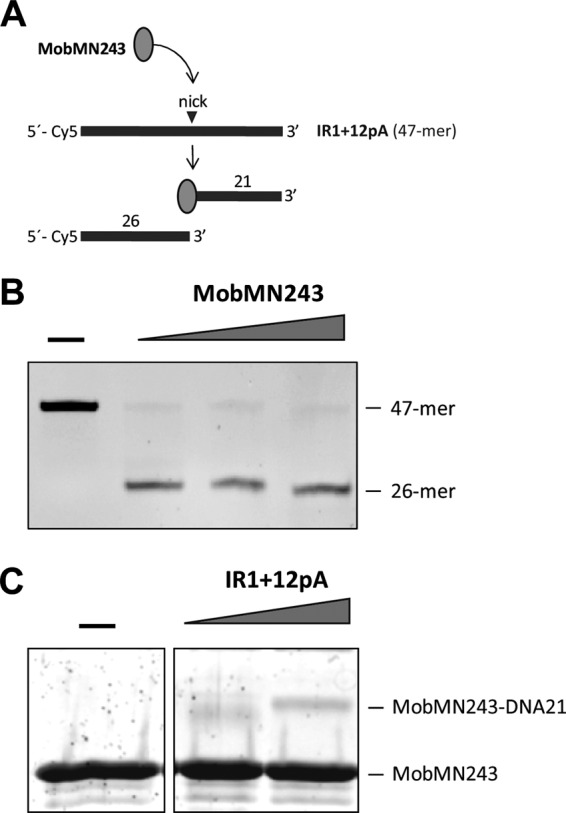
Isolation of the ssDNA-MobMN243 stable complexes after IR1+12pA oligonucleotide cleavage. (A) Scheme of the substrate and reaction products expected after IR1+12pA nicking by MobMN243. (B) DNA analysis. IR1+12pA 5′-labeled (Cy5) oligonucleotide (60 nM) was incubated with different concentrations of MobMN243 (128, 256, and 512 μM) for 20 min at 30°C in the presence of 8 mM MnCl2. The reaction was stopped by incubation with 0.1% SDS and proteinase K (100 μg/ml) for 30 min at 37°C. Samples were loaded in a denaturing 20% PAA gel (7 M urea), and DNA was visualized in a phosphorimager platform. (C) Protein analysis. MobMN243 (10 μM) was incubated with DNA (10 or 20 μM) in the same conditions used for the DNA analysis. Samples were visualized by SDS-PAGE (12%) and SYPRO Ruby staining. A stable adduct corresponding to the MobMN243 protein attached to a 21-mer ssDNA fragment was identified (MobMN243-DNA21).
MobM stability.
The recently determined structures of NES, a relaxase found in Gram-positive bacteria (19), indicate that MobM-like nucleases are divided into two functional domains, which are separated by a boundary region that encompasses part of the C terminus of MobMN243. The predicted secondary structure of the MobMN243 protein obtained by computational methods (not shown) shows that the C-terminal domain of MobM contains a sequence of secondary structure elements similar to NES. Although the structure of the C-terminal domain may differ from that of NES (21), it is likely that MobM has a domain architecture similar to that of this relaxase. The long-term storage stability of MobM under a range of conditions was found to reflect the domain boundary. The electrophoretic profiles of MobMN243 showed an extra band with a smaller apparent molecular size (not shown). In order to determine whether this band corresponds to a partner protein that copurifies with MobM derivatives, an Edman sequencing approach was followed after protein transfer and band isolation from nylon membranes. Interestingly, the sequence obtained after 8 cycles was SYMVARMQ, which corresponds to the N-terminal residues of the MobM protein (excluding the initial Met residue). This result indicates that MobMN243 presents intrinsic instability, as does the entire MobM protein. For this reason, the stability of various MobM constructs after 2 months of storage in the absence and presence of ligands was studied by gel electrophoresis under denaturing conditions (Fig. 6). Incubation of full-length MobM at 4°C for 2 months showed that the nondegraded species was predominant for the full-length protein, although the occurrence of bands of lower molecular mass indicated some degradation (Fig. 6, lane 1). Incubation of MobMN243 without ligands at 4°C in buffer A did not show appreciable degradation (Fig. 6, lane 3) compared to fresh sample (lane 2). Likewise, the presence of Mn2+ (lane 4) or IR1+8 DNA (lane 5) slightly enhanced MobMN243 proteolysis; however, the main species was still the nondegraded sample. A remarkable increase in degradation was observed in the presence of all three components, i.e., protein, metal cation, and DNA, for MobMN243 (lane 6) but not for MobMN199 (not shown). The main product of degradation showed a migration similar to that of MobMN199 (lane 7; fresh sample). Thus, the DNA had a remarkable effect on the stability of the amino acid region beyond residue 199.
Fig 6.
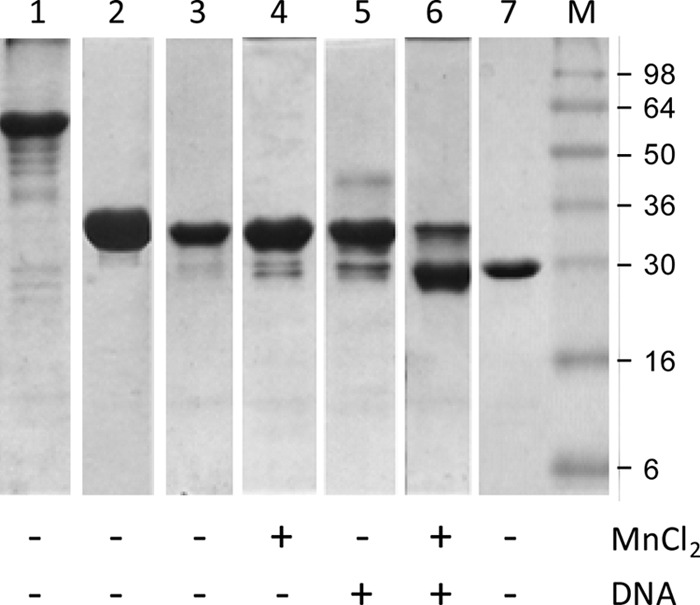
MobMN243 ligand-dependent proteolysis as analyzed by 12% SDS-PAGE. Lane 1, full-length MobM (57.9 kDa), incubated for 2 months at 4°C. Lane 2, MobMN243 (28.4 kDa), fresh sample. Lane 3, MobMN243 incubated for 2 months at 4°C. Lane 4, MobMN243, incubated for 2 months at 4°C in the presence of MnCl2 (15 mM final concentration). Lane 5, MobMN243 incubated for 2 months at 4°C in the presence of IR1+8 (180 μM). Lane 6, MobMN243 incubated for 2 months at 4°C in the presence of MnCl2 plus IR1+8. Lane 7, MobMN199 (23.1 kDa), nondegraded fresh sample, included for comparison to the degraded samples in lanes 1 to 6. The marker lane (M) was loaded with SeeBlue prestained standards (Invitrogen).
To determine where degradation occurs, the masses of the degradation products were checked with ESI-MS experiments on fresh and pure samples of MobMN199 and MobMN243. The monoisotopic masses of both intact proteins corresponded well to the theoretical values (Table 2) when taking into account that Met1 was cleaved off and that oxidation and phosphorylation of the sample occurred during production and purification (not shown). Degradation was observed for MobMN243 and occurred exclusively at the C terminus, at amino acids Y209 and K202, and to a lesser extent at K193, A190, and E176. Degradation in the presence of Mg2+ was diminished under the given condition with respect to the presence of Mn2+, but similar results were obtained for Mg2+ when we extended the incubation time 2-fold. The results confirm that degradation occurs predominantly around residue 199, in the region of 190 to 210.
Susceptibility to degradation has been linked to flexibility (45); therefore, the increase in degradation susceptibility of the boundary region upon substrate binding points to an increase in solvent exposure as a result of a substrate-induced change in conformation. The degradation behavior suggests that the N-terminal domains are flexible appendices attached to the dimerized C-terminal domains of MobM. How this localized degradation affects or modulates the conjugative process is not known. It has been proposed that the C-terminal domain of MobM is involved in the interactions with other elements, such as the coupling protein (20), which mediates the interactions between the relaxosome with the secretion system (3, 4). Once this interaction takes place, it is reasonable to assume that the C-terminal part of MobM is not essential. In this regard, it would be interesting to determine whether the protein-DNA complex that enters the recipient cell (as demonstrated for TrwC_R388 [46]) contains the full-length MobM protein or a natural truncated version similar to MobMN199. Further in vivo experiments are needed to address this question and will allow us a deeper understanding of the pMV158 transfer mechanism.
In summary, we propose that the E200-S243 region where degradation occurs is important for DNA processivity and functions as a signal transducer of substrate binding to the C-terminal domain (21). Our data imply that the N- and C-terminal domains are loosely connected and that the intervening region is crucial for substrate processivity. Thus, we have dissected the MobM N-terminal sequence into at least two functionally distinct regions, where the first 190 to 200 amino acids are relevant for DNA nicking activity while the region of amino acids 200 to 243 acts as a nicking modulator and a hinge that allows for a change in relative orientations of the N- and C-terminal domains of MobM during substrate processing.
ACKNOWLEDGMENTS
This work was supported by the Spanish Ministry of Science and Innovation (grants BFU2008-02372/BMC, CONSOLIDER CSD 2006-23, and BFU2011-22588 to M.C. and grants CSD2008-00013, INTERMODS, and BFU2010-19597 to M.E.), the Generalitat de Catalunya (grant SGR2009-1309 to M.C.), the “La Caixa”/IRB Barcelona International PhD Programme Fellowship (IRB Barcelona call 01/09/FLC to R.P.), and the European Commission (FP7 Cooperation Project SILVER-GA no. 260644 to M.C.).
We thank Marina Gay and Marta Vilaseca of the Mass Spectrometry Core Facility at IRB Barcelona.
Footnotes
Published ahead of print 26 April 2013
REFERENCES
- 1. de la Cruz F, Frost LS, Meyer RJ, Zechner EL. 2010. Conjugative DNA metabolism in Gram-negative bacteria. FEMS Microbiol. Rev. 34:18–40 [DOI] [PubMed] [Google Scholar]
- 2. Lanka E, Wilkins BM. 1995. DNA processing reactions in bacterial conjugation. Annu. Rev. Biochem. 64:141–169 [DOI] [PubMed] [Google Scholar]
- 3. Alvarez-Martinez CE, Christie PJ. 2009. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 73:775–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wallden K, Rivera-Calzada A, Waksman G. 2010. Type IV secretion systems: versatility and diversity in function. Cell. Microbiol. 12:1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wozniak RAF, Waldor MK. 2010. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8:552–563 [DOI] [PubMed] [Google Scholar]
- 6. Shrivastava R, Miller JF. 2009. Virulence factor secretion and translocation by Bordetella species. Curr. Opin. Microbiol. 12:88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcillán-Barcia MP, Francia MV, de la Cruz F. 2009. The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 33:657–687 [DOI] [PubMed] [Google Scholar]
- 8. Gomis-Rüth FX, Coll M. 2006. Cut and move: protein machinery for DNA processing in bacterial conjugation. Curr. Opin. Struct. Biol. 16:744–752 [DOI] [PubMed] [Google Scholar]
- 9. Parker C, Becker E, Zhang X, Jandle S, Meyer R. 2005. Elements in the co-evolution of relaxases and their origins of transfer. Plasmid 53:113–118 [DOI] [PubMed] [Google Scholar]
- 10. Llosa M, Gomis-Ruth FX, Coll M, de la Cruz Fd F. 2002. Bacterial conjugation: a two-step mechanism for DNA transport. Mol. Microbiol. 45:1–8 [DOI] [PubMed] [Google Scholar]
- 11. Lorenzo-Díaz F, Espinosa M. 2009. Lagging strand DNA replication origins are required for conjugal transfer of the promiscuous plasmid pMV158. J. Bacteriol. 191:720–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibbs MJ, Smeianov VV, Steele JL, Upcroft P, Efimov BA. 2006. Two families of rep-like genes that probably originated by interspecies recombination are represented in viral, plasmid, bacterial, and parasitic protozoan genomes. Mol. Biol. Evol. 23:1097–1100 [DOI] [PubMed] [Google Scholar]
- 13. Grohmann E, Muth G, Espinosa M. 2003. Conjugative plasmid transfer in Gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67:277–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boer R, Russi S, Guasch A, Lucas M, Blanco AG, Perez-Luque R, Coll M, de la Cruz F. 2006. Unveiling the molecular mechanism of a conjugative relaxase: the structure of TrwC complexed with a 27-mer DNA comprising the recognition hairpin and the cleavage site. J. Mol. Biol. 358:857–869 [DOI] [PubMed] [Google Scholar]
- 15. Guasch A, Lucas M, Moncalian G, Cabezas M, Perez-Luque R, Gomis-Ruth FX, de la Cruz F, Coll M. 2003. Recognition and processing of the origin of transfer DNA by conjugative relaxase TrwC. Nat. Struct. Biol. 10:1002–1010 [DOI] [PubMed] [Google Scholar]
- 16. Larkin C, Datta S, Harley MJ, Anderson BJ, Ebie A, Hargreaves V, Schildbach JF. 2005. Inter- and intramolecular determinants of the specificity of single-stranded DNA binding and cleavage by the F factor relaxase. Structure 13:1533–1544 [DOI] [PubMed] [Google Scholar]
- 17. Monzingo AF, Ozburn A, Xia S, Meyer RJ, Robertus JD. 2007. The structure of the minimal relaxase domain of MobA at 2.1 Å resolution. J. Mol. Biol. 366:165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nash RP, Habibi S, Cheng Y, Lujan SA, Redinbo MR. 2010. The mechanism and control of DNA transfer by the conjugative relaxase of resistance plasmid pCU1. Nucleic Acids Res. 38:5929–5943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Edwards JS, Betts L, Frazier ML, Pollet RM, Kwong SM, Walton WG, Ballentine WK, III, Huang JJ, Habibi S, Del Campo M, Meier JL, Dervan PB, Firth N, Redinbo MR. 2013. Molecular basis of antibiotic multiresistance transfer in Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 110:2804–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lorenzo-Diaz F, Dostal L, Coll M, Schildbach JF, Menendez M, Espinosa M. 2011. The MobM relaxase domain of plasmid pMV158: thermal stability and activity upon Mn2+ and specific DNA binding. Nucleic Acids Res. 39:4315–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Antonio C, Farias ME, de Lacoba MG, Espinosa M. 2004. Features of the plasmid pMV158-encoded MobM, a protein involved in its mobilization. J. Mol. Biol. 335:733–743 [DOI] [PubMed] [Google Scholar]
- 22. Francia MV, Varsaki A, Garcillan-Barcia MP, Latorre A, Drainas C, de la Cruz F. 2004. A classification scheme for mobilization regions of bacterial plasmids. FEMS Microbiol. Rev. 28:79–100 [DOI] [PubMed] [Google Scholar]
- 23. Guzmán LM, Espinosa M. 1997. The mobilization protein MobM, of the streptoccocal plasmid pMV158, specifically cleaves supercoiled DNA at the plasmid oriT. J. Mol. Biol. 266:688–702 [DOI] [PubMed] [Google Scholar]
- 24. Lacks S. 1966. Integration efficiency and genetic recombination in pneumococcal transformation. Genetics 53:207–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruiz-Cruz S, Solano-Collado V, Espinosa M, Bravo A. 2010. Novel plasmid-based genetic tools for the study of promoters and terminators in Streptococcus pneumoniae and Enterococcus faecalis. J. Microb. Methods 83:156–163 [DOI] [PubMed] [Google Scholar]
- 26. Lacks SA, López P, Greenberg B, Espinosa M. 1986. Identification and analysis of genes for tetracycline resistance and replication functions in the broad-host-range plasmid pLS1. J. Mol. Biol. 192:753–765 [DOI] [PubMed] [Google Scholar]
- 27. del Solar G, Díaz R, Espinosa M. 1987. Replication of the streptococcal plasmid pMV158 and derivatives in cell-free extracts of Escherichia coli. Mol. Gen. Genet. 206:428–435 [DOI] [PubMed] [Google Scholar]
- 28. Buchan DW, Ward SM, Lobley AE, Nugent TC, Bryson K, Jones DT. 2010. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 38:W563–W568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cole C, Barber JD, Barton GJ. 2008. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36:W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Combet C, Blanchet C, Geourjon C, Deléage G. 2000. NPS@: network protein sequence analysis. Trends Biochem. Sci. 25:147–150 [DOI] [PubMed] [Google Scholar]
- 31. Adamczak R, Porollo A, Meller J. 2005. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins 59:467–475 [DOI] [PubMed] [Google Scholar]
- 32. Schuck P, Rossmanith P. 2000. Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers 54:328–341 [DOI] [PubMed] [Google Scholar]
- 33. Laue TM, Shah BD, Ridgeway TM, Pelletier SL. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. In Harding SE, Rowe A, Horton JC. (ed), Analytical ultracentrifugation in biochemistry and polymer sciences. Royal Society of Chemistry, Cambridge, United Kingdom [Google Scholar]
- 34. van Holde KE. 1985. Physical Biochemistry, 2nd ed Prentice Hall, Upper Saddle River, NJ [Google Scholar]
- 35. Pessen H, Kumosinsky TF. 1985. Measurement of protein hydration by various techniques. Methods Enzymol. 117:219–255 [DOI] [PubMed] [Google Scholar]
- 36. Wei YF, Matthews HR. 1991. Identification of phosphohistidine in proteins and purification of protein-histidine kinases. Methods Enzymol. 200:388–414 [DOI] [PubMed] [Google Scholar]
- 37. Fernández-López C, Lorenzo-Díaz F, Pérez-Luque R, Rodríguez-González L, Boer R, Lurz R, Bravo A, Coll M, Espinosa M. Nicking activity of the pMV158 MobM relaxase on cognate and heterologous origins of transfer. Plasmid. 2013 Apr 3; doi: 10.1016/j.plasmid.2013.03.004. article-title>. [Epub ahead of print.] [DOI] [PubMed] [Google Scholar]
- 38. Llosa M, Grandoso G, de la Cruz F. 1995. Nicking activity of TrwC directed against the origin of transfer of the IncW plasmid R388. J. Mol. Biol. 246:54–62 [DOI] [PubMed] [Google Scholar]
- 39. Matson SW, Morton BS. 1991. Escherichia coli DNA helicase I catalyzes a site- and strand-specific nicking reaction at the F plasmid oriT. J. Biol. Chem. 266:16232–16237 [PubMed] [Google Scholar]
- 40. Pansegrau W, Balzer D, Kruft V, Lurz R, Lanka E. 1990. In vitro assembly of relaxosomes at the transfer origin of plasmid RP4. Proc. Natl. Acad. Sci. U. S. A. 87:6555–6559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Llosa M, Grandoso G, Hernando MA, de la Cruz F. 1996. Functional domains in protein TrwC of plasmid R388: dissected DNA strand transferase and DNA helicase activities reconstitute protein function. J. Mol. Biol. 264:56–67 [DOI] [PubMed] [Google Scholar]
- 42. Pansegrau W, Schroder W, Lanka E. 1993. Relaxase (TraI) of IncP alpha plasmid RP4 catalyzes a site-specific cleaving-joining reaction of single-stranded DNA. Proc. Natl. Acad. Sci. U. S. A. 90:2925–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grandoso G, Avila P, Cayon A, Hernando MA, Llosa M, de la Cruz F. 2000. Two active-site tyrosyl residues of protein TrwC act sequentially at the origin of transfer during plasmid R388 conjugation. J. Mol. Biol. 295:1163–1172 [DOI] [PubMed] [Google Scholar]
- 44. González-Pérez B, Lucas M, Cooke LA, Vyle JS, de la Cruz F, Moncalián G. 2007. Analysis of DNA processing reactions in bacterial conjugation by using suicide oligonucleotides. EMBO J. 26:3847–3857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, Shaul Y. 2008. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins 70:1357–1366 [DOI] [PubMed] [Google Scholar]
- 46. Garcillán-Barcia MP, Jurado P, González-Pérez B, Moncalián G, Fernández LA, de la Cruz F. 2007. Conjugative transfer can be inhibited by blocking relaxase activity within recipient cells with intrabodies. Mol. Microbiol. 63:404–416 [DOI] [PubMed] [Google Scholar]