

Abstract
The oral bacterium Streptococcus mutans, strain JH1140, produces the antibiotic mutacin 1140. Mutacin 1140 belongs to a group of antibiotics called lanthipeptides. More specifically, mutacin 1140 is related to the epidermin type A(I) lanthipeptides. Mutagenesis experiments of this group of lanthipeptides have been primarily restricted to the posttranslationally modified meso-lanthionine and 3-methyllanthionine residues. Site-directed mutagenesis of the core peptide of mutacin 1140 was performed using the suicide vector pVA891. Substitutions of the N-terminal residue, the charged residue in the hinge region, and residues in ring A and intertwined rings C and D were investigated. A truncation and insertion of residues in ring A and intertwined rings C and D were also performed to determine whether or not they would alter the antimicrobial activity of the producing strain. Bioassays revealed that five of 14 mutants studied had improved antimicrobial activity against the indicator strain Micrococcus luteus ATCC 10240. MICs against Streptococcus mutans UA159, Streptococcus pneumoniae ATCC 27336, Staphylococcus aureus ATCC 25923, Clostridium difficile UK1, and Micrococcus luteus ATCC 10240 were determined for three mutacin 1140 variants that had the most significant increases in bioactivity in the M. luteus bioassay. This mutagenesis study of the epidermin group of lanthipeptides shows that antimicrobial activity can be significantly improved.
INTRODUCTION
Lanthipeptides, an important class of antibiotics with potential clinical relevance (1–3), are so called because of the characteristic lanthionine rings that are present. The term lanthipeptides is a relatively new name for the group of peptide antibiotics formerly called lantibiotics (4). The name needed updating given that some of these peptides were found to not have antibacterial activity (5). Lanthipeptides are ribosomally synthesized, containing a leader peptide that is cleaved off by a protease and a core peptide that is responsible for the bioactivity (4). Particular features of lanthipeptides, such as their novel and diverse mechanisms of action and the difficulty for sensitive bacteria to acquire resistance, have aroused considerable interest in these molecules as potential therapeutic agents (3, 6). Lanthipeptides are also known to have various unusual amino acids, such as 2,3-didehydroalanine (Dha), 2,3-didehydrobutyrine (Dhb), S-aminovinyl-d-cysteine (AviCys), aminobutyrate (Abu), 2-oxopropionyl, 2-oxobutyryl, and hydroxypropionyl (3, 7, 8). The molecular structure of mutacin 1140 contains four macrocyclic rings (Fig. 1A), each of which contains a meso-lanthionine or 3-methyllanthionine residue (9). The alanine moieties of the lanthionine rings are distinguished as AlaS and SAla for their N- and C-terminal positions in the core peptide, respectively. Mutacin 1140's rings A and B are similar to nisin's rings A and B (Fig. 1B), as well as those of other type A(I) lanthipeptides. Nisin, one of the most studied lanthipeptides, is produced by Lactococcus lactis and has been used in the food industry for over 50 years. It was discovered that both nisin and mutacin 1140 abduct lipid II from the site of new cell wall synthesis, ultimately causing cell death (10–13). Rings A and B provide the structural motif for binding to lipid II and are referred to as the lipid II binding domain (14).
Fig 1.
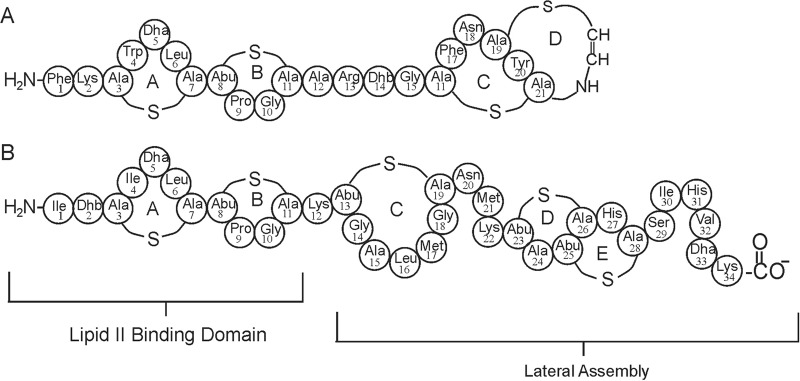
Covalent structures of related type A(I) lanthipeptides. (A) Mutacin 1140. (B) Nisin A. The brackets signify functional regions of the two lanthipeptides.
Mutacin 1140 is a key feature in the development of an approach called “replacement therapy” for the prevention of dental caries (15–17). In this approach, decay-causing strains of Streptococcus mutans are displaced from dental plaque by introduction of a nonpathogenic, superior colonizing “effector” strain of S. mutans. In one application of this approach, the cariogenic potential of strain JH1140 was essentially eliminated by inactivating the lactate dehydrogenase gene. A spontaneous mutation that increased mutacin 1140 production 2-fold gave JH1140 a selective advantage in colonization, enabling it to persistently colonize dental plaque following a single infection regimen and to aggressively displace indigenous, disease-causing S. mutans strains from the teeth. The combination of mutacin 1140 production with a decrease in lactic acid production may provide lifelong protection against tooth decay (16, 17). Mutacin 1140 has also been shown to be effective at inhibiting multidrug-resistant Staphylococcus aureus and at treating S. aureus infection in rats (18, 19). Preliminary tests indicate that it may be well suited to serve as an antibiotic for the treatment of this and other important infectious diseases. Thus, for these several reasons, it is important to obtain a comprehensive understanding of mutacin 1140.
Work performed by Smith et al. using the membrane mimetic solvent acetonitrile/water (80:20) showed that mutacin 1140 has an overall horseshoe-like shape that is kinked at the “hinge region” between rings B and C (20). This shape is the result of a turn-like motif in the hinge region that folds the N-terminal rings A and B (the lipid II binding domain) toward the C-terminal overlapped rings C and D (20). Interestingly, the flexibility of the hinge region is believed to be important for promoting lateral assembly of mutacin 1140 monomers. The Ψ angle of Trp4 and Φ angle of Dha5 in ring A help contribute to ring A's flexibility (20). Also, it was determined that the Ψ bond of SAla7 (a residue that is not confined by the thioether ring) freely rotates 360°, allowing ring A to spin freely with respect to ring B (20). This flexibility and the flexibility of ring A are thought to be important in orientating rings A and B during lipid II binding. Resistance to hydrolysis may also be, in part, a reflection of the unusual, horseshoe-shaped three-dimensional structure of mutacin 1140 (20, 21). The hinge region contains a potentially protease-susceptible arginine at residue 13. This residue appears to be protected, as evidenced by the relative difficulty of inactivating mutacin 1140 by trypsin or pronase treatment (20–22).
There have been over 80 different directed mutations reported on the nisin core peptide (23, 24). In addition, a large bank of randomly mutated nisin derivatives was screened, which resulted in the identification of three mutants that significantly improved the bioactivity of nisin (23). There are a total of nine mutations in nisin that appear to improve its bioactivity. Four of these mutations occur in rings A and B, which are structurally similar to mutacin 1140 (Fig. 1). Currently, there are a total of 15 site-directed mutations reported in the literature on the epidermin group of lanthipeptides (25, 26), and none contribute to a statistically significant improvement in bioactivity (Table 1). The identification of mutacin 1140 variants with increased bioactivity would be important for possible improvements of the “effector” strain used in replacement therapy. Moreover, some variants may prove to be superior to the native compound for the control of infectious microorganisms. Other variants of mutacin 1140 may also further our understanding of the importance of structural regions within the core peptide. In this study, residues within the N-terminal region comprising rings A and B, charged residues within the hinge region, and residues within the C-terminal region comprising intertwined rings C and D were exchanged to determine their importance for bioactivity.
Table 1.
Site-directed mutagenesis studies on the lanthipeptides epidermin and gallidermin
| Epidermin/gallidermin mutation | Effect of mutationa | Reference(s) |
|---|---|---|
| Ser3Asn | Elimination of activity | 26 |
| Leu6Val | 5-fold reduction in activity against S. aureus Cowan I | 26 |
| Leu6Gly | >7-fold reduction in activity against S. aureus Cowan I | 26 |
| Gly10Glu | Elimination of activity | 26 |
| Ala12Leu | Loss of pore formation; 3-fold reduction in activity against S. aureus Cowan I | 25, 26 |
| Dhb14Dha | No change in activity against S. aureus Cowan I | 26 |
| Dhb14S | 6-fold reduction in activity against S. aureus Cowan I | 26 |
| Dhb14A | >7-fold reduction in activity against S. aureus Cowan I | 26 |
| Dhb14P | >7-fold reduction in activity against S. aureus Cowan I | 26 |
| Ser16Thr | Elimination of activity | 26 |
| Ser19Ala | Elimination of activity | 26 |
| Ser19Thr | Elimination of activity | 26 |
| Tyr20Gly | Elimination of activity | 26 |
| ΔCys21 | Elimination of activity | 26 |
| ΔCys22 | Elimination of activity | 26 |
The general summary of the effect of each mutant is provided relative to its activity.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
Bacterial strains and plasmids used in this study are listed in Table 2. Escherichia coli DH5α (Invitrogen, Carlsbad, CA) was used in cloning and was cultured at 37°C on Luria-Bertani (LB) agar. THY broth (30 g/liter Todd-Hewitt broth, 3 g/liter yeast extract) or agar (30 g/liter Todd-Hewitt broth, 3 g/liter yeast extract, 15 g/liter agar) medium (Bacto, Sparks, MD) was used to culture Streptococcus pneumoniae ATCC 27336, Staphylococcus aureus ATCC 25923, Clostridium difficile UK1, S. mutans UA159, S. mutans JH1140 ATCC 55676, and Micrococcus luteus ATCC 10240 at 37°C. THY broth and THY agar media were used for antimicrobial activity assays.
Table 2.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristic(s)b | Source or reference(s) |
|---|---|---|
| Strains | ||
| E. coli DH5α | F− φ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rk−, mk+) phoA supE44 λ-thi-1 gyrA96 relA1 tonA | Invitrogen |
| M. luteus ATCC 10240 | Antibiotic indicator strain | ATCC |
| S. aureus ATCC 25923 | ATCC | |
| S. pneumoniae ATCC 27336 | ATCC | |
| C. difficile UK1 | 41, 42 | |
| S. mutans UA159 | 43 | |
| S. mutans ATCC 55676 | ATCC | |
| JH1140 | Wild-type strain | 31 |
| JH1140 Phe1Gly | mutA::Phe1Gly site-directed derivative | This study |
| JH1140 Phe1Ser | mutA::Phe1Ser site-directed derivative | This study |
| JH1140 Phe1Thr | mutA::Phe1Thr site-directed derivative | This study |
| JH1140 Phe1Ile | mutA::Phe1Ile site-directed derivative | This study |
| JH1140 Phe1Trp | mutA::Phe1Trp site-directed derivative | This study |
| JH1140 Trp4Ala | mutA::Trp4Ala site-directed derivative | This study |
| JH1140 Trp4insAla | mutA::Trp4insAla site-directed derivative | This study |
| JH1140 ΔTrp4 | mutA::ΔTrp4 site-directed derivative | This study |
| JH1140 DHA5Ala | mutA::Dha5Ala site-directed derivative | This study |
| JH1140 Alas7insAla | mutA::Alas7insAla site-directed derivative | This study |
| JH1140 Arg13Asp | mutA::Arg13Asp site-directed derivative | This study |
| JH1140 Phe17insAla | mutA::Phe17insAla site-directed derivative | This study |
| JH1140 Asn18Ala | mutA::Asn18Ala site-directed derivative | This study |
| JH1140 Trp4Ala-Arg13Asp | mutA::Trp4Ala Arg13Asp site-directed derivative | This study |
| JH1140 muta | mutA::heterodiploid derivatives; Ermr | This study |
| Plasmids | ||
| TOPO-TA | Cloning vector; 3′ T overhangs; lacZ for blue/white screening; Kanr, Ampr | Invitrogen |
| pVA891 | Suicide vector; Ermr | 27 |
| pVA891mutA | Suicide vector; Ermr; cloned mutA containing approx 500 bp of flanking DNA | Oragenics Inc. |
Site-directed heterodiploid mutants of S. mutans containing two copies of mutA and pVA891 suicide vector.
Ermr, erythromycin resistant; Kanr, kanamycin resistant; Ampr, ampicillin resistant.
Mutagenesis of mutacin 1140.
The S. mutans genome database and lan gene cluster (GenBank/EMBL accession number AF051560) were used to design primers for the mutagenesis and sequencing work. The open reading frame (ORF) of the native mutacin 1140 structural gene (mutA) plus 500 bp of 5′ and 3′ flanking DNA was cloned into the pVA891 plasmid (5.4 kb) (27, 28) to create pVA891mutA. The cloned insert in pVA891mutA was derived by PCR amplification of chromosomal DNA of S. mutans strain JH1140 using the primers SRWmutA_F and SRWmutA_R (see Table S1 in the supplemental material). Reagents and media were purchased from VWR (Radnor, PA), enzymes were purchased from New England BioLabs (Ipswich, MA), and primers were purchased from Integrated DNA Technologies (IDT; Coralville, IA). Mutations were introduced into the core peptide region of mutA using appropriately modified primers to create the variants of mutacin 1140 (Fig. 2). The pVA891mutA plasmid was used as a template, and the site-specific mutations were introduced using PCR. PCRs were performed using Taq polymerase (New England BioLabs) with appropriate primers (see Table S1 in the supplemental material). The cloned inserts from the confirmed TOPO-TA vector (Invitrogen, Carlsbad, CA) were then subcloned into the S. mutans suicide vector pVA891 using the EcoRI site. Purified pVA891 DNA containing confirmed inserts was transformed into S. mutans JH1140 with selection on medium containing erythromycin (15 μg/ml) to create a heterodiploid intermediate, as described by Li et al. (29). Clones containing the desired mutA mutations were obtained by spontaneous resolution of the heterodiploid state. The mutA region of erythromycin-sensitive clones was amplified by PCR and sequenced to identify colonies having the mutated mutA gene.
Fig 2.

Mutagenesis of mutacin 1140 core peptide. Changes in amino acid sequence are represented by the circles above and below the mutacin 1140 core peptide sequence. The arrows represent insertions or deletions in the core peptide. The circles are positioned in the region in which the substitution, insertions, and deletions occur. Active mutants are indicated by the circles above the core peptide sequence, while the inactive mutants are positioned below the core peptide sequence.
Bioactivity of mutants.
The bioactivities of the mutants were determined by a deferred antagonism assay. The parent S. mutans strain, JH1140, and the mutants were grown to an optical density at 600 nm (OD600) of 0.8 and diluted to an OD600 of 0.2. Samples (2 μl) of the cultures were spotted in triplicate on a prewarmed THY agar plate and allowed to air dry. This assay was performed in this manner to help ensure that each sample had the same colony size for comparing zones of inhibition. The plate was incubated for 24 h at 37°C and then placed in an oven at 65°C for 1 h to kill the bacteria and thereby stop further mutacin 1140 production. M. luteus was grown to an OD600 between 0.4 and 0.8 and diluted to an OD600 of 0.2. Four hundred microliters of cells was added to 10 ml of molten top agar (42°C) (30 g/liter Todd-Hewitt broth and 7.5 g/liter agar), which was then poured over the surface of each THY agar plate. The plates were allowed to solidify before being inverted and incubated overnight at 37°C. Each inhibitory zone radius (n = 10) was measured in millimeters from one edge of the colony to the farthest portion of the zone. The average area of the inhibitory zone was calculated for each zone and compared to the average zone area of the wild type. The statistical method used was Student's t test. The MIC of mutacin 1140 and variants of mutacin 1140 was determined against S. mutans UA159, S. pneumoniae ATCC 27336, S. aureus ATCC 25923, C. difficile UK1, and M. luteus ATCC 10240. The MIC was the lowest concentration of mutacin 1140 and variants that inhibited the visible growth of the bacteria after 24 h of incubation. Preparation of the antimicrobial agent and bacterial inoculum for MICs was performed by following the method described by the Clinical and Laboratory Standards Institute (CLSI) (30) with some minor modifications. S. mutans UA159 was tested overnight at 37°C in a shaking incubator at 175 rpm to maintain uniform dispersion of the bacteria, and C. difficile UK1 was tested in an anaerobic chamber at 37°C. The medium used for the MIC assays was THY.
Purification and mass spectrometry.
Purification of the mutacin 1140 variants Phe1Ile, Phe1Gly, Phe1Thr, Phe1Ser, Phe1Trp, Trp4Ala, Dha5Ala, and Arg13Asp was performed based on methods described by Hillman et al. (31), with some modifications (32). Isolation of the mutacin 1140 variants Phe1Gly, Phe1Thr, Phe1Ser, Phe1Trp, and Dha5Ala was performed on a small scale to enable mass identification by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS). Briefly, S. mutans strains were grown on a 150-mm petri dish in 50 ml of THY containing 0.3% agar supplemented with salts (5 mg/liter FeSO4·7H2O, 200 mg/liter K2HPO4, 1 g/liter KH2PO4, 0.7 g/liter MgSO4·7H2O, and 5 mg/liter MnSO4) for 72 h at 37°C. S. mutans JH1140 mutants F1I, Trp4Ala, and R13D, which had the most significant increases in bioactivity in the deferred antagonism assay, were cultured on a larger scale to provide enough material to enable determination of their specific activities using the CLSI MIC assay (30). The mutant strains of S. mutans JH1140 were grown in 0.5 liters of the THY medium containing the supplements described above in a 1-liter flask for 72 h at 37°C. The cultured samples were then frozen at −70°C and thawed quickly in a 65°C incubator. The culture was transferred into a centrifuge tube and spun for 30 min at 20,000 × g. The supernatant was extracted with an equal volume of chloroform. The emulsion at the chloroform-aqueous interface was collected. The dried pellet was resuspended in 35% acetonitrile (ACN)/water (vol/vol) containing 0.1% trifluoroacetic acid. The samples were fractionated using C18 columns (Grace-Vydac; catalog numbers 201TP54 and 201TP510) on a Bio-Rad BioLogic F10 Duo Flow with Quad Tech UV-visible (UV-Vis) detector system as previously described (33, 34). Reverse-phase high-performance liquid chromatography (RP-HPLC) fractions that tested positive for bioactivity against the indicator strain M. luteus were evaporated to dryness and dissolved in 100 μl of 35% acetonitrile containing 0.1% trifluoroacetic acid. From these resuspended fractions, 0.5 μl was mixed with 0.5 μl of α-cyano-4-hydroxycinnamic acid matrix (6 mg/ml in 50% acetonitrile) containing 0.1% trifluoroacetic acid and dried on the target plate. MALDI-TOF MS (Shimadzu/Kratos) operating in the positive linear mode was used to determine the mass of the major component in RP-HPLC fractions as previously described (33, 34). More than seven milligrams of mutacin 1140 Phe1Ile, Trp4Ala, and Arg13Asp variants was isolated, enabling accurate weight measurements for MIC assays. Representative chromatograms of each purified variant are shown in Fig. S2 in the supplemental material.
RESULTS AND DISCUSSION
Mutations in the structural gene (mutA) for mutacin 1140 were generated to determine the effect of the following amino acid alterations: Phe1Gly, Phe1Ser, Phe1Thr, Phe1Ile, Phe1Trp, Trp4Ala, Trp4insAla, ΔTrp4, Dha5Ala, Alas7insAla, Arg13Asp, Phe17insAla, Asn18Ala, and double mutant Trp4Ala Arg13Asp.
Bioactivity of N-terminal core peptide mutants of mutacin 1140.
Nisin, epidermin, and Pep5 lanthipeptides are the representative structures for each subgroup of type A(I) lanthipeptides (see Table S2 in the supplemental material). Given that there is a substantial amount of variability at the first position of the type A(I) lanthipeptides, we wanted to determine whether the first position of the core peptide was important for MutP activity. First, we replaced Phe1 with a glycine to determine whether the presence of a phenylalanine at the N-terminal end was essential for biological activity of mutacin 1140. This substitution did not significantly alter the bioactivity (Fig. 3; see also Table S3 in the supplemental material). The mass of the purified product matched the predicted mass of 2,176 Da (Table 3; see also Fig. S1 in the supplemental material). The nisin and epidermin groups of type A(I) lanthipeptides contain phenylalanine, valine, isoleucine, and tryptophan at the first position (see Table S2 in the supplemental material). We replaced mutacin 1140's N-terminal phenylalanine with isoleucine or tryptophan. Neither of these substitutions resulted in a significant reduction in bioactivity. On the contrary, the Phe1Ile substitution resulted in an 84% increase in the area of inhibitory activity (mm2), which was statistically significant (P < 0.05) (Fig. 3; see also Table S3 in the supplemental material). The masses of the purified products matched the predicted masses for the amino acid substitutions (Table 3; see also Fig. S1 in the supplemental material). The Pep5 group of type A(I) lanthipeptides contains serine and threonine at the first position (see Table S2 in the supplemental material), which are posttranslationally dehydrated and deaminated to pyruvate (2-oxopropionyl) and 2-oxobutyryl residues. We replaced Phe1 of mutacin 1140 with a serine or threonine with an expectation that they would be dehydrated. The variant with the serine substitution had activity similar to that of wild-type mutacin 1140, while the threonine substitution resulted in a 32% increase in the area of inhibitory activity. The latter result was statistically significant (P < 0.05) (Fig. 3; see also Table S3 in the supplemental material). The mass of these purified products showed that the serine and threonine residues were not dehydrated, suggesting that the dehydratase, MutB, does not function at the +1 position of mutacin 1140's core peptide (Table 3; see also Fig. S1 in the supplemental material). Our results demonstrate that the first position of the core peptide was amenable to amino acid substitutions, and the observation that bioactivity was unchanged or increased in these mutants indicates that the substitutions did not affect MutP substrate recognition.
Fig 3.
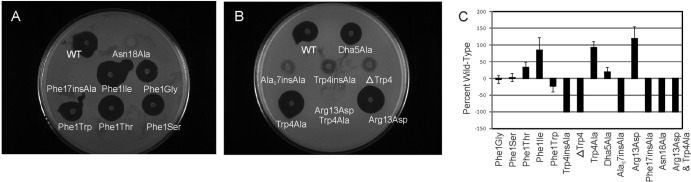
Bioactivity of mutacin 1140 variants. (A) Growth inhibition of the intertwined rings C and D and N-terminal mutants in comparison to the wild-type strain. (B) Growth inhibition of ring A and hinge region mutants in comparison to the wild-type strain. (C) Relative activity of each mutant with respect to the wild-type area (mm2) of inhibition. Micrococcus luteus ATCC 10240 was used as the indicator strain for antimicrobial activity.
Table 3.
MALDI-TOF spectrum masses
| Strain | Predicted mass (Da) | Actual mass (Da) |
|---|---|---|
| JH1140 | 2,266 | 2,266 |
| JH1140 Phe1Gly | 2,176 | 2,176 |
| JH1140 Phe1Ser | 2,188 | 2,206a |
| JH1140 Phe1Thr | 2,202 | 2,220a |
| JH1140 Phe1Ile | 2,232 | 2,232 |
| JH1140 Phe1Trp | 2,300 | 2,300 |
| JH1140 ΔTrp4 | 2,080 | 2,080 |
| JH1140 Trp4Ala | 2,151 | 2,151 |
| JH1140 Trp4insAla | 2,337 | 2,355a |
| JH1140 DHA5A | 2,268 | 2,268 |
| JH1140 AlaS7insAla | 2,337 | 2,355a |
| JH1140 R13D | 2,225 | 2,225 |
| Phe17insAla | 2,337 | 2,337 |
Mass is different from the predicted mass.
There is a great deal of diversity for the LanP proteases that are involved in removing the leader peptide of lanthipeptides. Some of these proteases are intracellular, while others are extracellular. Furthermore, the specificity for proteolysis varies among different lanthipeptides. Some lanthipeptide proteases have no apparent sequence-specific motifs for cleavage, suggesting that they may recognize a tertiary structure of the prepropeptide (8). Many lanthipeptide proteases belong to the serine peptidase S8 lantibiotic-specific protease family (35). The sequence of the S8 lantibiotic-specific protease domain of mutacin 1140's protease MutP was compared to other proteins in the BLASTP database. It was found that MutP had about the same amount of similarity to nisin's protease (NisP) and epidermin's protease (EpiP). The similarities are 38% and 36% for nisin and epidermin, respectively. MutP similarity to the Pep5 lanthipeptide protease PepP is only 19%.
Bioactivity of lipid II binding domain mutants of mutacin 1140.
The flexibility of ring A with respect to ring B is thought to be important for orientating the rings during lipid II binding. Insertion of an alanine after Trp4 (Trp4insAla) and deletion of Trp4 (ΔTrp4) were engineered into the mutacin 1140 peptide to determine the effect of altering the size of ring A. The Trp4Ala and Dha5Ala mutations were constructed to determine whether these residues are important for bioactivity. An Alas7insAla mutation results in the introduction of an alanine between rings A and B. Therefore, the Alas7insAla mutation was constructed to determine whether the proximity of ring A to ring B is essential for bioactivity. The results of inhibitory assays using these mutants showed that the Trp4insAla and ΔTrp4 substitutions had no measurable mutacin 1140 activity (Fig. 3; see also Table S3 in the supplemental material). The mass of the ΔTrp4 variant matched the predicted mass, suggesting that the variant has undergone all of the posttranslational modifications by MutB, MutD, and MutP (Table 3; see also Fig. S1 in the supplemental material). The mass of the Trp4insAla variant was 18 Da larger than the predicted mass, suggesting that the insertion of an alanine interferes with a dehydration of a serine or threonine residue by MutB (Table 3; see also Fig. S1 in the supplemental material). Ser5 may be the residue that is not dehydrated, given its proximity to the insertion. These results suggest that shortening or lengthening ring A has a significant effect on the inhibitory activity of the mutant strains, indicating that the size of ring A may be important for mutacin 1140's interaction with lipid II or for the formation of a thioether linkage. The Trp4Ala substitution resulted in a 94% increase in the area of inhibitory activity (P < 0.05). The Dha5Ala substitution resulted in a 19% increase in the area of inhibitory activity.
These results demonstrate that certain substitutions of residues in ring A can significantly increase the measurable bioactivity of mutacin 1140. A difference in bioactivity may be due to the size difference or the bioavailability of the two amino acids, or the Trp may impede the wild type's travel to its target by its interaction with various structures via its lone pair of electrons. The underlying mechanism for the increase in bioactivity will require further investigation. Replacement of Dha5 with alanine is potentially very useful in solid-phase peptide synthesis (SPPS). As these techniques improve, the incorporation of an alanine at this position will simplify the synthesis and should therefore ultimately impact the cost of production. When this same mutation was made in nisin, the product showed bioactivity similar to that of wild-type nisin (36).
Tryptophan residues have been shown to possess antiaggregation properties in lipid bilayers (37). Thus, it was hypothesized that the tryptophan may play a role in coordinating the lateral assembly of mutacin 1140 (20). In this model, the Trp4 side chain is directed away from the rest of molecule and was thought to be important for oligomerization specificity by preventing mutacin 1140 from forming random aggregates and promoting uniform assembly of the mutacin monomers into large complexes that would trap lipid II. From our results, it appears that this model is incorrect: the presence of the tryptophan is not essential for the bioactivity of mutacin 1140.
The Alas7insAla mutation had no measurable zone of inhibition. All of the mutations in the lipid II binding region that altered the size of ring A or proximity of ring A to ring B resulted in a loss of activity, signifying the importance of this region. The free rotation of the Ψ bond of SAla7 is presumably important for promoting the correct binding interaction of rings A and B to its molecular target lipid II (20). Having a single amino acid inserted after the SAla7 position between rings A and B destroys the bioactivity of the resultant peptide. The mass of the sAla7insAla variant was 18 Da larger than the predicted mass, suggesting that the insertion of an alanine interferes with a dehydration of a serine or threonine residue by MutB (Table 3; see Fig. S1 in the supplemental material). It is probable that the insertion prevents the dehydration of Thr8, leading to the disruption of ring B formation. This would prevent the interaction of mutacin 1140 with lipid II.
Bioactivity of hinge and C-terminal core peptide mutants of mutacin 1140.
The importance of arginine at position 13 in the hinge region of mutacin 1140 and the structural importance of the phenylalanine and asparagine residues at positions 17 and 18 of the intertwined rings C and D were investigated. Substitution of a negatively charged aspartate in place of the positively charged arginine resulted in the largest increase in bioactivity among the mutants tested in this study. The Arg13Asp substitution had a 119% increase in the area of activity (P < 0.05) (Fig. 3; see also Table S3 in the supplemental material). Surprisingly, the double mutant Trp4Ala Arg13Asp resulted in a complete loss of activity. There was no product isolated for the mutacin 1140 Trp4Ala Arg13Asp variant that would enable mass determination. The bioactivity of mutacin 1140 was not amenable to an insertion of an alanine after Phe17 or to the substitution of alanine at the Asn18 position in the intertwined rings C and D. Phe17insAla had no definable zones of inhibition. The mass of the Phe17insAla variant matched the predicted mass, suggesting that the variant has undergone all of the posttranslational modifications by MutB, MutD, and MutP (Table 3; see also Fig. S1 in the supplemental material). Asn18Ala also had no definable zones of inhibition, but there was a visible reduction in growth of the indicator bacterium (Fig. 3). However, there was no product isolated for the Asn18Ala variant that would enable mass determination.
The Arg13 was predicted to provide an anchor to the cell surface of the bacterium (20). The cell surfaces of bacteria are generally negatively charged, and this is believed to be important for attracting the cationic type A lanthipeptides. The three-dimensional structure of mutacin 1140 places the charged amino terminal, Lys2 side chain, and Arg13 side chain along a common plane (20), and this charge distribution was predicted to be important for promoting the proper orientation of mutacin 1140 in the lipid bilayer (20). Even though the replacement of the positive charge with a negative charge was tolerated and resulted in a mutant with increased activity, this prediction may still be accurate, given that the charge of the aspartic acid may still bind to a cationic lipid. Future substitution experiments for lysine in position 2 would be interesting to determine whether the positive charge is critical for mutacin 1140 function. Mutacin 1140 has demonstrated resistance to trypsin cleavage (21, 22). Nevertheless, the incorporation of an aspartic acid at this position may afford some additional protection against proteases. In addition, this mutation introduces a carboxyl group into the peptide which would enable labeling procedures to facilitate fluorescence microscopy studies, as described for nisin (10, 11, 13). It is noteworthy that replacement of arginine (AGA/AGG/CGT/CGC/CGA/CGG) with aspartic acid (GAT/GAC) is unlikely to occur in nature. The complete loss of activity of the double mutant Trp4Ala Arg13Asp was surprising given that each of these mutants contributed to the largest increase in bioactivity. Presumably, the loss in activity can be attributed to the bacterium's inability to synthesize the product and not to a loss in activity of the modified lanthipeptide. If the modified lanthipeptide is produced and has no bioactivity, this would suggest that the two distant regions are important for coordinating the bioactivity. Synthetic synthesis of this mutant should provide an answer to this question.
The insertion of an alanine in ring C after Phe17 resulted in no measurable zone of inhibition. Based on the determined mass of the product, the insertion of alanine did not interfere with the dehydration of the serine residues in rings C and D. However, it may interfere with the formation of the thioether linkages within intertwined rings C and D. The Asn18Ala mutant also had no measurable zone of inhibition. Rings C and D are believed to be important for the lateral assembly mechanism that has been shown to trap lipid II into large complexes (11, 13). Asparagine has been shown to be an important residue in lipid bilayers, serving as a hydrogen bond donor and acceptor (38, 39), and its presence may be necessary for lateral assembly. However, the loss of the asparagine residue at position 18 may interfere with the biosynthesis or transport of the peptide, given that no product was isolated. Based on what has been reported for the sequence motif needed for the decarboxylase EpiD, these C-terminal mutations should not interfere with C-terminal oxidative decarboxylation of mutacin 1140 (40). The enzymatic decarboxylation of peptides by EpiD was shown to occur when an asparagine at the same position was replaced by an alanine (40). Therefore, the observed loss of activity of these mutants is probably not due to a loss of function by MutD decarboxylase.
Specific activities for mutacin 1140 and variants Phe1Ile, Trp4Ala, and Arg13Asp.
The deferred antagonism activities described for the lanthipeptide variants of mutacin 1140 were not determined using purified products or impure products but have been characterized by the bacterial strains' abilities to inhibit the growth of the indicator strain M. luteus ATCC 10240. The reported bioactivity in these experiments depends on the biosynthesis of the active peptide, transport, or diffusion in the medium. It is conceivable that some mutations may perturb or enhance these activities, resulting in lower or higher yield of the molecule being produced. Therefore, the bioactivity in the deferred antagonism assay is not a direct indication of the specific activity of the antimicrobial peptide but is a direct measurement of the overall bioactivity produced by Streptococcus mutans JH1140. This approach provides a rapid screen to find mutants with increased activity and to identify regions that are important for production or bioactivity. S. mutans JH1140 mutacin 1140 variants Phe1Ile, Trp4Ala, and Arg13Asp had the most significant increases in activity in the M. luteus bioassay compared to the wild-type strain and were isolated for MIC assays.
The mutacin 1140 variants Phe1Ile, Trp4Ala, and Arg13Asp were each isolated and purified in a measurable quantity that would enable direct comparison of their antibacterial activities to that of wild-type mutacin 1140. Interestingly, the mutacin 1140 variant Arg13Asp (mu1140 Arg13Asp) had reduced activity against M. luteus ATCC 10240; the values observed were 0.125 μg/ml and 0.0625 μg/ml for the wild type and mu1140 Arg13Asp, respectively (Table 4). This mutant had the largest increase in activity against M. luteus ATCC 10240 in the plate assays described above. The variant also had reduced activity toward S. pneumoniae ATCC 27336 and S. aureus ATCC 25923 compared to that of wild-type mutacin 1140. Mu1140 Arg13Asp had the same level of activity as wild-type mutacin 1140 against S. mutans UA159 and C. difficile UK1. Mu1140 Trp4Ala had a 4-fold increase in activity compared to wild-type mutacin 1140 against S. pneumoniae ATCC 27336; the values observed were 0.125 μg/ml and 0.5 μg/ml for the wild type and mu1140 Trp4Ala, respectively (Table 4). The variant had a 2-fold increase in activity over that of wild-type mutacin 1140 against C. difficile UK1 and M. luteus ATCC 10240. Mu1140 Trp4Ala had the same level of activity as wild-type mutacin 1140 against S. mutans UA159 and S. aureus ATCC 25923. Mu1140 Phe1Ile had a 4-fold increase in activity over that of wild-type mutacin 1140 against M. luteus ATCC 10240; the values observed were 0.0156 μg/ml and 0.625 μg/ml for the wild type and mu1140 Phe1Ile, respectively (Table 4). The variant had a 2-fold increase in activity compared to that of wild-type mutacin 1140 against S. pneumoniae ATCC 27336 and S. aureus ATCC 25923 and the same level of activity as wild-type mutacin 1140 against S. mutans UA159 and C. difficile UK1.
Table 4.
MICs for mutacin 1140 and structural variants
| Mutacin 1140 variant | MIC (μg/ml) against: |
||||
|---|---|---|---|---|---|
| Streptococcus mutans UA159 | Streptococcus pneumoniae ATCC 27336 | Staphylococcus aureus ATCC 25923 | Clostridium difficile UK1 | Micrococcus luteus ATCC 10240 | |
| Mu1140 | 2 | 0.5 | 16 | 16 | 0.0625 |
| Mu1140_Phe1Ile | 2 | 0.25 | 8 | 16 | 0.0156 |
| Mu1140_Trp4Ala | 2 | 0.125 | 16 | 8 | 0.0312 |
| Mu1140_Arg13Asp | 2 | 4 | >16 | 16 | 0.125 |
The mutacin 1140 variants mu1140 Phe1Ile and mu1140 Trp4Ala demonstrated improvements in their bioactivity against three of the strains tested. Interestingly, the observed improvement in activity against the bacterial strains tested varied between the two mutacin 1140 variants. Presumably, the variations in activity are attributed to each variant's ability to navigate through the cell wall to reach its molecular target lipid II. The improvement in activity for mu1140 Arg13Asp observed in the deferred antagonism assay against M. luteus ATCC 10240 was not observed in the MIC assay. The observed increase in activity in the deferred antagonism assay may be attributed to an increase in biosynthesis and export efficiency of the mu1140 Arg13Asp variant or to the variant being less impeded by the agar used in this assay.
Conclusions.
Our studies examined amino acid substitutions in the N-terminal end of the mutacin 1140 core peptide as well as substitutions in residues comprising ring A, the hinge region, and the C-terminal intertwined rings C and D. The results obtained revealed mutants with increased, similar, or no bioactivity, all of which provide insight into the importance of the structural regions within the core peptide. Some of our engineered variants may have the potential to significantly improve the pharmacokinetics and pharmacodynamics of mutacin 1140. Our finding in this mutagenesis study will be important for future studies aimed at developing mutacin 1140 for the treatment of Gram-positive infections.
In summary, from this study, it was determined that the first residue and ring A are amenable to some degree of modification. The size of ring A and the distance between rings A and B are essential for maintaining bioactivity. Also, modifications of the highly conserved intertwined rings C and D resulted in a loss of bioactivity. Trp4Ala, Dha5Ala, and Arg13Asp substitutions all resulted in an increase in bioactivity in the deferred antagonism assay, but the mu1140 Arg13Asp variant had the same level of bioactivity or reduced bioactivity in the MIC assays. Interestingly, despite significant increases in bioactivities for the Trp4Ala and Arg13Asp mutations, a combination of Trp4Ala and Arg13Asp mutations resulted in a complete loss of bioactivity.
Supplementary Material
ACKNOWLEDGMENTS
Mass spectrometric data were collected at the Texas A&M University Protein Chemistry Lab. We thank Lawrence Dangott for his expert assistance. We also thank Robert A. Burne from the University of Florida for providing bacterial strains.
This research was supported by funds from Texas A&M University (to L.S.).
Jeffrey D. Hillman served as Chief Scientific Officer of Oragenics Inc. before his retirement in the fall of 2012. Leif Smith has consulted for Oragenics Inc. on their lanthipeptide projects.
Footnotes
Published ahead of print 19 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00704-13.
REFERENCES
- 1. Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian K-D, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl Schmidt H-GEW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang G-L, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. 2013. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30:108–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kodani S, Hudson ME, Durrant MC, Buttner MJ, Nodwell JR, Willey JM. 2004. The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor. Proc. Natl. Acad. Sci. U. S. A. 101:11448–11453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bierbaum G, Sahl HG. 2009. Lantibiotics: mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol. 10:2–18 [DOI] [PubMed] [Google Scholar]
- 4. Nagao J-I, Nishie M, Sonomoto K. 2011. Methodologies and strategies for the bioengineering of lantibiotics. Curr. Pharm. Biotechnol. 12:1221–1230 [DOI] [PubMed] [Google Scholar]
- 5. Smith L, Hillman JD. 2008. Therapeutic potential of type A (I) lantibiotics, a group of cationic peptide antibiotics. Curr. Opin. Microbiol. 11:401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilson-Stanford S, Smith L. 2011. Commercial development and application of type A lantibiotics. Recent Pat. Antiinfect. Drug Discov. 6:175–185 [DOI] [PubMed] [Google Scholar]
- 7. Bierbaum G, Sahl HG. 1993. Lantibiotics—unusually modified bacteriocin-like peptides from Gram-positive bacteria. Zentralbl. Bakteriol. 278:1–22 [DOI] [PubMed] [Google Scholar]
- 8. Chatterjee C, Xie PML, van der Donk W. 2005. Biosynthesis and mode of action of lantibiotics. Chem. Rev. 105:633–683 [DOI] [PubMed] [Google Scholar]
- 9. Smith L, Novak J, Rocca J, McClung S, Hillman JD, Edison AS. 2000. Covalent structure of mutacin 1140 and a novel method for the rapid identification of lantibiotics. Eur. J. Biochem. 267:6810–6816 [DOI] [PubMed] [Google Scholar]
- 10. Breukink E, van Heusden HE, Vollmerhaus PJ, Swiezewska E, Brunner L, Walker S, Heck AJR, de Kruijff B. 2003. Lipid II is an intrinsic component of the pore induced by nisin in bacterial membranes. J. Biol. Chem. 278:19898–19903 [DOI] [PubMed] [Google Scholar]
- 11. Hasper HE, Kramer NE, Smith JL, Hillman JD, Zachariah C, Kuipers OP, de Kruijff B, Breukink E. 2006. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 313:1636–1637 [DOI] [PubMed] [Google Scholar]
- 12. Smith L, Hasper H, Breukink E, Novak J, Cerkasov J, Hillman JD, Wilson-Stanford S, Orugunty RS. 2008. Elucidation of the antimicrobial mechanism of mutacin 1140. Biochemistry 47:3308–3314 [DOI] [PubMed] [Google Scholar]
- 13. Wilson-Stanford S, Kalli A, Hakansson K, Kastrantas J, Orugunty RS, Smith L. 2009. Oxidation of lanthionines renders the lantibiotic nisin inactive. Appl. Environ. Microbiol. 75:1381–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsu STD, Breukink E, Tischenko E, Lutters MAG, de Kruijff B, Kaptein R, Bonvin A, van Nuland NAJ. 2004. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat. Struct. Mol. Biol. 11:963–967 [DOI] [PubMed] [Google Scholar]
- 15. Hillman JD. 2001. Replacement therapy of dental caries. Oper. Dent. 6(Suppl):39–49 [Google Scholar]
- 16. Hillman JD. 2002. Genetically modified Streptococcus mutans for the prevention of dental caries. Antonie Van Leeuwenhoek 82:361–366 [PubMed] [Google Scholar]
- 17. Hillman JD, Brooks TA, Michalek SM, Harmon CC, Snoep JL, van der Weijden CC. 2000. Construction and characterization of an effector strain of Streptococcus mutans for replacement therapy of dental caries. Infect. Immun. 68:543–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghobrial O, Derendorf H, Hillman JD. 2010. Pharmacokinetic and pharmacodynamic evaluation of the lantibiotic MU1140. J. Pharm. Sci. 99:2521–2528 [DOI] [PubMed] [Google Scholar]
- 19. Ghobrial OG, Derendorf H, Hillman JD. 2009. Pharmacodynamic activity of the lantibiotic MU1140. Int. J. Antimicrob. Agents 33:70–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith L, Zachariah C, Thirumoorthy R, Rocca J, Novak J, Hillman JD, Edison AS. 2003. Structure and dynamics of the lantibiotic mutacin 1140. Biochemistry 42:10372–10384 [DOI] [PubMed] [Google Scholar]
- 21. Hillman JD, Johnson KP, Yaphe BI. 1984. Isolation of a Streptococcus mutans strain producing a novel bacteriocin. Infect. Immun. 44:141–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hillman JD, Johnson KP, Yaphe BI. 1983. Characterization of a Streptococcus mutans bacteriocin with novel properties. J. Dent. Res. 62:241–241 [Google Scholar]
- 23. Field D, Connor PMO, Cotter PD, Hill C, Ross RP. 2008. The generation of nisin variants with enhanced activity against specific Gram-positive pathogens. Mol. Microbiol. 69:218–230 [DOI] [PubMed] [Google Scholar]
- 24. Rink R, Wierenga J, Kuipers A, Muskens LD, Driessen AJM, Kuipers OP, Moll GN. 2007. Production of dehydroamino acid-containing peptides by Lactococcus lactis. Appl. Environ. Microbiol. 73:1792–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonelli RR, Schneider T, Sahl HG, Wiedemann I. 2006. Insights into in vivo activities of lantibiotics from gallidermin and epidermin mode-of-action studies. Antimicrob. Agents Chemother. 50:1449–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ottenwalder B, Kupke T, Brecht S, Gnau V, Metzger J, Jung G, Gotz F. 1995. Isolation and characterization of genetically-engineered gallidermin and epidermin analogs. Appl. Environ. Microbiol. 61:3894–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Macrina FL, Evans RP, Tobian JA, Hartley DL, Clewell DB, Jones KR. 1983. Novel shuttle plasmid vehicles for Escherichia-Streptococcus transgeneric cloning. Gene 25:145–150 [DOI] [PubMed] [Google Scholar]
- 28. Perry D, Nilsen LJ, Kuramitsu HK. 1985. Mapping of a cloned glucosyltransferase gene in Streptococcus mutans. Infect. Immun. 50:130–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y-H, Tang N, Aspiras MB, Lau PCY, Lee JH, Ellen RP, Cvitkovitch DG. 2002. A quorum-sensing signaling system essential for genetic competence in Streptococcus mutans is involved in biofilm formation. J. Bacteriol. 184:2699–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clinical and Laboratory Standards Institute 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—eighth edition. CLSI document M07-A8. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 31. Hillman JD, Novak J, Sagura E, Gutierrez JA, Brooks TA, Crowley PJ, Hess M, Azizi A, Leung KP, Cvitkovitch D, Bleiweis AS. 1998. Genetic and biochemical analysis of mutacin 1140, a lantibiotic from Streptococcus mutans. Infect. Immun. 66:2743–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qi FX, Chen P, Caufield PW. 1999. Purification of mutacin III from group III Streptococcus mutans UA787 and genetic analyses of mutacin III biosynthesis genes. Appl. Environ. Microbiol. 65:3880–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chaney N, Wilson-Stanford S, Kastrantas J, Dahal N, Smith L. 2009. Rapid method for extracting the antibiotic mutacin 1140 from complex fermentation medium yeast extract. Can. J. Microbiol. 55:1261–1266 [DOI] [PubMed] [Google Scholar]
- 34. Dahal N, Chaney N, Ellis D, Lu S-E, Smith L. 2010. Optimization of the production of the lantibiotic mutacin 1140 in modified M9 media. Process Biochem. 5:1187–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Siezen RJ, Leunissen JA. 1997. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 6:501–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chan WC, Dodd HM, Horn N, Maclean K, Lian LY, Bycroft BW, Gasson MJ, Roberts GCK. 1996. Structure-activity relationships in the peptide antibiotic nisin: role of dehydroalanine 5. Appl. Environ. Microbiol. 62:2966–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Killian JA, Salemink I, dePlanque MRR, Lindblom G, Koeppe RE, II, Greathouse DV. 1996. Induction of nonbilayer structures in diacylphosphatidylcholine model membranes by transmembrane alpha-helical peptides: importance of hydrophobic mismatch and proposed role of tryptophans. Biochemistry 35:1037–1045 [DOI] [PubMed] [Google Scholar]
- 38. Choma C, Gratkowski H, Lear JD, DeGrado WF. 2000. Asparagine-mediated self-association of a model transmembrane helix. Nat. Struct. Biol. 7:161–166 [DOI] [PubMed] [Google Scholar]
- 39. Zhou FX, Merianos HJ, Brunger AT, Engelman DM. 2001. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. U. S. A. 98:2250–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kupke T, Kempter C, Jung G, Gotz F. 1995. Oxidative decarboxylation of peptides catalyzed by flavoprotein EpiD—determination of substrate specificity using peptide libraries and neutral loss mass spectrometry. J. Biol. Chem. 270:11282–11289 [DOI] [PubMed] [Google Scholar]
- 41. Killgore G, Thompson A, Johnson S, Brazier J, Kuijper E, Pepin J, Frost EH, Savelkoul P, Nicholson B, van den Berg RJ, Kato H, Sambol SP, Zukowski W, Woods C, Limbago B, Gerding DN, McDonald LC. 2008. Comparison of seven techniques for typing international epidemic strains of Clostridium difficile: restriction endonuclease analysis, pulsed-field gel electrophoresis, PCR-ribotyping, multilocus sequence typing, multilocus variable-number tandem-repeat analysis, amplified fragment length polymorphism, and surface layer protein A gene sequence typing. J. Clin. Microbiol. 46:431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sorg JA, Sonenshein AL. 2010. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J. Bacteriol. 192:4983–4990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ajdić D, McShan WM, McLaughlin RE, Savić G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. U. S. A. 99:14434–14439 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.