

Abstract
Infectious diseases disproportionately affect indigent regions and are the greatest cause of childhood mortality in developing countries. Practical, low-cost vaccines for use in these countries are paramount to reducing disease burdens and concomitant poverty. Algae are a promising low-cost system for producing vaccines that can be orally delivered, thereby avoiding expensive purification and injectable delivery. We engineered the chloroplast of the eukaryotic alga Chlamydomonas reinhardtii to produce a chimeric protein consisting of the 25-kDa Plasmodium falciparum surface protein (Pfs25) fused to the β subunit of the cholera toxin (CtxB) to investigate an alga-based whole-cell oral vaccine. Pfs25 is a promising malaria transmission-blocking vaccine candidate that has been difficult to produce in traditional recombinant systems due to its structurally complex tandem repeats of epidermal growth factor-like domains. The noncatalytic CtxB domain of the cholera holotoxin assembles into a pentameric structure and acts as a mucosal adjuvant by binding GM1 ganglioside receptors on gut epithelial cells. We demonstrate that CtxB-Pfs25 accumulates as a soluble, properly folded and functional protein within algal chloroplasts, and it is stable in freeze-dried alga cells at ambient temperatures. In mice, oral vaccination using freeze-dried algae that produce CtxB-Pfs25 elicited CtxB-specific serum IgG antibodies and both CtxB- and Pfs25-specific secretory IgA antibodies. These data suggest that algae are a promising system for production and oral delivery of vaccine antigens, but as an orally delivered adjuvant, CtxB is best suited for eliciting secretory IgA antibodies for vaccine antigens against pathogens that invade mucosal surfaces using this strategy.
INTRODUCTION
There is an urgent need to develop vaccines for practical use in developing countries, both in terms of cost and delivery. Infectious and parasitic diseases killed more than 1.7 million children and adolescents in low-income countries in 2008 (1), which represents nearly half of all deaths in that age bracket. In contrast, infectious and parasitic diseases caused 4,500 deaths (4%) in high-income countries in the same age bracket (1). This disparity highlights one of the great challenges in human health today, which is to reduce the disease burden in the poorest regions of the world, especially for the young. Much of the inequality is the result of disparate vaccine coverage in the developing world (2), mainly brought about by the high cost and logistical difficulty of large-scale vaccination campaigns in countries with underdeveloped health infrastructures. Additionally, pathogens for which no traditional vaccine exists, like malaria, disproportionately affect the poorest countries (3), and some have argued that these countries are unable to climb out of poverty because of this disease burden (4). Thus, feasibility of implementation must be considered when developing vaccines for such regions of the world. Heat-stable oral vaccines could overcome the largest obstacles that deter widespread vaccination in low-income countries. Thermostability would eliminate the need for cold-chain storage, and oral delivery would be safer, simpler, and cheaper than injectable vaccines (5–8). Indeed, the National Institutes of Health, the World Health Organization, and others have emphasized the need for needle-free vaccination strategies.
Oral vaccines are not a new idea; indeed, oral vaccines for a few human pathogens, including Vibrio cholerae (Dukoral), Salmonella typhi (Vivotif), rotavirus (Rotarix and RotaTeq), and polio (Sabin vaccine), are commercially available. Despite these few examples, the potential of oral vaccines remains largely unrealized. The complexity of the mucosal immune system, which must discriminate between detrimental and innocuous antigens in the gut, has slowed the development of oral vaccine candidates (9, 10). This is especially true for subunit vaccines, because the response to most antigens at mucosal surfaces is one of nonresponsiveness or tolerance (11, 12). Recent studies suggest that this may be overcome by proteins that act as adjuvants at mucosal surfaces. The best-characterized mucosal adjuvants are the ADP-ribosylating enterotoxins from V. cholerae (cholera toxin [CT]) and Escherichia coli (heat labile toxin [LT]) (13, 14). Other potential mucosal adjuvants include cytokines, which have been investigated for intranasal vaccines (15), and Toll-like receptor (TLRs) or other pattern recognition receptor (PRR) agonists (16–18). Alternatively, antigens could be specifically targeted to microfold (M) cells, which overlay Payer's patches and are often the site of entry for pathogens or particles from the gut lumen to immune cells (19, 20).
One major challenge for oral vaccines is to overcome antigen degradation by commensal bacteria, proteases, and the acidic stomach environment in order to safely deliver intact antigens and mucosal adjuvants to the gut-associated lymphoid tissue. Plant delivery systems are particularly attractive for this purpose for two reasons. First, oral delivery is restricted to Food and Drug Administration (FDA)-approved organisms that are generally regarded as safe (GRAS) (21). Unlike conventional organisms used for recombinant protein production, GRAS organisms pose little risk of harmful viral, prion, toxin, or bacterial contaminants. Second, plant cells have a rigid cell wall that can protect encapsulated vaccine antigens, allowing ambient temperature storage. The cell wall can also provide a layer of protection from proteolysis in the gastrointestinal tract.
Advances in molecular genetics have made recombinant protein production possible in many plants (22); the first example of a plant-based oral vaccine was accomplished in tobacco and potatoes more than 15 years ago (23). Plants also afford an unprecedented economy of scale that results in a robust and inexpensive platform for producing recombinant proteins (24). Oral delivery via an edible plant also eliminates the need for antigen purification and drives costs down even further. Several experimental plant-based oral vaccines elicit an immune response, although not all demonstrate protective efficacy (25). There are inherent drawbacks of terrestrial plants, however, which undoubtedly contribute to the absence of commercially available plant-based vaccines despite a few that have been approved for use (26). First, terrestrial plants grow relatively slowly. It can take years from the initial transformation to validate an antigen from a stable plant line, although newer methods using transient transfection in tobacco allow a more rapid turnaround (27). Both methods often result in antigen production variability between plants. Second, the environmental impact of gene flow or lateral gene transfer remains unclear (28). Thus, from a time and regulatory perspective, transgenic food crops may not be ideal. Lastly, many plants require extensive processing to become palatable, which reduces antigen immunogenicity (29).
The unicellular eukaryotic green alga Chlamydomonas reinhardtii is an attractive alternative to terrestrial plants for producing oral vaccines. Eukaryotic green algae are considered GRAS organisms by the FDA (30). C. reinhardtii can be rapidly transformed into stable transgenic strains and grown in enclosed bioreactors or outdoor ponds. Importantly, several algae are already grown at commercial scales for consumption (e.g., Earthrise and Cyanotech), and the interest in alga-based biofuel is further driving innovation for low-cost biomass production. The impending industrial utility of algae is also accelerating the development of genetic tools for metabolic engineering and recombinant protein production as potential coproducts (31). Thus far, C. reinhardtii chloroplasts have been used to make vaccine antigens (32, 33), industrial enzymes (34), antibodies (35), human therapeutics (36), and immunotoxins (37). The versatility of the chloroplast is owed to the unique combination of eukaryotic chaperones (38), protein disulfide isomerases (39), and peptidylprolyl isomerases (40) coupled with prokaryotic-like ribosomal machinery (41).
Here, we describe the production and characterization of a complex chimeric protein consisting of the CT β subunit (CtxB), a mucosal adjuvant, and Pfs25, a malaria transmission-blocking vaccine (TBV) candidate (42), in C. reinhardtii chloroplasts. CtxB assembles into a pentameric structure and binds the GM1 ganglioside receptor found on gut epithelial cells, which facilitates an immune response via the gut mucosa but lacks the toxic ADP-ribosylating activity of the CT α subunit (CtxA) (43). Pfs25 is a structurally complex protein found on the surface of sexual stage P. falciparum parasites (44); antibodies to Pfs25 block malaria transmission (45). We previously demonstrated that alga-produced Pfs25 resembles native Pfs25 structure and elicits transmission-blocking antibodies (46). In these studies, we characterized the chimeric CtxB-Pfs25 fusion protein and the immune response in mice that were administered lyophilized whole algae that produce CtxB-Pfs25.
MATERIALS AND METHODS
Plasmid and C. reinhardtii strain construction.
The peptide sequence of CtxB(Thr22-Asn124; UniProt identifier P01556) was reverse translated using the Gene Designer software (DNA2.0, Menlo, CA) using the C. reinhardtii chloroplast codon table (www.kazusa.or.jp) as a reference table. The codon-optimized C. reinhardtii ctxB nucleotide sequence (Cr.ctxB) was validated by calculating the codon adaptation index (CAI; 0.94) (47) and synthesized by Integrated DNA technologies (San Diego, CA). C. reinhardtii codon-optimized Plasmodium falciparum surface protein 25 (Cr.pfs25) was previously described (46). Cr.ctxB and Cr.pfs25 were separately amplified and fused together with uracil-specific excision reagent (USER) cloning (48) such that it contained a linker composed of a flexible hinge (G-P-G-P), a furin protease recognition sequence (R-A-R-R) (49), and a BglII restriction site. Flanking Cr.ctxB-pfs25 are NdeI and AgeI restriction sites on the 5′ and 3′ ends, respectively. The chimeric gene (Cr.ctxB-pfs25) was digested with NdeI and AgeI and cloned into the chloroplast transformation vector pJAG15 (46), a pD1-KanR (50) derivative, also digested with NdeI and AgeI. The resulting plasmid, pJAG101, encodes Cr.CtxB-Pfs25 with a C-terminal FLAG affinity tag driven by the psbA promoter and flanked by the psbA 5′ and 3′ untranslated regions for message stabilization. Integration of pJAGA101 into the psbA locus of the chloroplast is directed by 5′ and 3′ psbA homology regions and confers kanamycin resistance. All constructs were verified by sequencing (Retrogen, San Diego, CA). Mid-log-phase C. reinhardtii strain W1.1 (51) cells were transformed with pJAG101 by particle bombardment using DNA-coated gold particles as previously described (46, 52) to make strain JAG101. Transformants were selected on Tris-acetate-phosphate (TAP) agar plates supplemented with 100 μg/ml kanamycin and propagated with 150 μg/ml kanamycin. Gene-specific primers were used to confirm integration of Cr.ctxB-Pfs25, and homoplasmicity was confirmed as previously described (36). A C. reinhardtii strain that is isogenic to JAG101 (Cr.ctxB-Pfs25), except that it lacks Cr.ctxB-Pfs25 and the kanamycin resistance cassette, was constructed (here referred to as the ΔpsbA strain) using previously described methods to remove the selectable marker (53). The ΔpsbA strain has a seamless deletion that extends from −300 bp to +1,525 bp, relative to the psbA start codon and stop codon, respectively.
Western blotting of alga lysates.
Initial screens for the Cr.CtxB-Pfs25 protein were performed on homoplasmic JAG101 strains. JAG101 and ΔpsbA cultures were grown in liquid TAP medium on a rotary shaker in low light. Cultures were shifted to high light (∼5,000 lx) at mid-log phase (∼1 × 106 cells/ml), and samples were harvested at various time points after light shift and stored at −20°C. Alga pellets were resuspended in lysis buffer (50 mM Tris [pH 8.0], 400 mM NaCl, 0.1% Tween 20) with protease inhibitor cocktail (Roche, Mannheim, Germany) and lysed by sonication. Lysates were cleared by centrifugation at 30,000 × g and quantitated with the DC Bio-Rad protein assay. Soluble protein samples were prepared in SDS buffer with urea and β-mercaptoethanol (50 mM Tris, 2% SDS, 10% glycerol, 2 M urea, and 10% β-mercaptoethanol), heated at 37°C for 10 min, resolved on RUNBLUE precast SDS-PAGE gels (Expedeon, San Diego, CA), and transferred to nitrocellulose membranes. The nitrocellulose membrane was probed with mouse anti-FLAG monoclonal antibodies (MAbs) (Sigma catalog no. F3165) and detected with goat anti-mouse IgG antibodies conjugated to alkaline phosphatase (Sigma catalog no. A1682) using nitro-blue tetrazolium (NBT) and 5-bromo-4-chloro-3′-indolylphosphate (BCIP) p-toluidine salt as the substrates in alkaline phosphatase buffer.
Affinity purification of alga-produced CtxB-Pfs25.
C. reinhardtii strain JAG101 (Cr.CtxB-Pfs25) and JAG9 (Cr.Pfs25) were grown in 20-liter photobioreactors constructed from Nalgene carboys (part number 2251-0050) fitted with Nunc bulkhead adapters (part number 6149-002). Photobioreactors were inoculated with 250 ml of stationary-phase JAG101 grown in TAP and grown to mid-log phase in the dark. Mixing was achieved by bubbling air through a 0.1-μm-pore-size filter. Cultures were then illuminated with 32-W 6500K T8 fluorescent light bulbs and harvested 24 h later using a Lavin L2 continuous-flow centrifuge (AML Industries, Hatboro, PA) fed by a peristaltic pump. Cells were snap-frozen with liquid nitrogen and stored at −80°C. Cells were resuspended and lysed as described above, followed by the addition of M2 anti-FLAG affinity resin (Sigma-Aldrich) and end-over-end mixing for 2 to 4 h at 4°C. Resin was washed twice with 20 column volumes of lysis buffer or until washes were no longer green, followed by one wash with lysis buffer without Tween 20. Bio-Rad Econo-pac columns were loaded with the washed resin, and bound protein was eluted using 100 mM glycine (pH 3.5) and 400 mM NaCl. Eluted fractions were neutralized with 1 M Tris (pH 8.0) to a final concentration of 50 mM Tris and tested for protein by Western blotting. Fractions containing protein were combined and buffer exchanged and concentrated into phosphate-buffered saline (PBS) using a 10-kDa cutoff Vivaspin centrifugal concentrator (GE Healthcare) and quantitated using the DC Bio-Rad protein assay. Pure protein was prepared with 4× native buffer (Expedeon) for nonreduced samples or SDS buffer with β-mercaptoethanol for reduced samples and resolved by SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were probed using rabbit anti-CT antibodies (Sigma catalog no. C3062), mouse anti-FLAG MAbs (Sigma catalog no. F3165), or anti-Pfs25 (4B7) MAbs and detected with goat anti-rabbit IgG (Sigma catalog no. A3687) or goat anti-mouse IgG (Sigma catalog no. A1682) secondary antibodies conjugated to alkaline phosphatase. Bands were visualized as described above. For gel staining, samples were resolved on 4 to 20% gradient SDS-PAGE precast gels (Bio-Rad) and stained using Coomassie blue.
Determination of alga-produced CtxB-Pfs25 accumulation.
The percent total soluble protein that constitutes the Cr.CtxB-Pfs25 fraction was determined by enzyme-linked immunosorbent assay (ELISA) as previously described (46). Briefly, soluble lysates from JAG101 and the ΔpsbA mutant were prepared as described above and diluted to 0.5 mg/ml in PBS. A standard curve was prepared by adding known amounts of purified CtxB (Sigma catalog no. C9903) to ΔpsbA lysates. JAG101 and ΔpsbA lysates with purified CtxB were applied to Maxisorp plates in triplicate, detected with rabbit anti-CT primary antibodies (Sigma catalog no. C3062) and goat anti-rabbit IgG secondary antibodies conjugated to alkaline phosphatase. Plates were then visualized with p-nitrophenyl phosphate disodium salt (PNPP) in 1 M diethanolamine buffer (pH 9.8; Thermo Scientific catalog no. 34047) and read with an Infinite M200 pro plate reader (Tecan, Switzerland) at 405 nm.
GM1 ganglioside binding ELISA.
MaxiSorp plates (Nunc, Rochester, NY) were coated with 150 ng/ml GM1 ganglioside (Sigma, G7641) in PBS, incubated overnight at 4°C, and blocked with 5% milk in PBS with 0.1% Tween 20 (PBS-T). Total soluble protein lysates from JAG101 and ΔpsbA algae were prepared as described above. Lysates were diluted to 20 μg/ml soluble protein in PBS and applied to the GM1-coated plate for 1 h at room temperature in triplicate. GM1 binding by Cr.CtxB-Pfs25 in lysates was detected with rabbit anti-CT antibodies followed by goat anti-rabbit IgG antibodies conjugated to horseradish peroxidase (HRP; Thermo Scientific). Binding was then visualized with the TMB substrate kit (Thermo Scientific) and read with an Infinite M200 Pro plate reader (Tecan, Switzerland) at 450 nm.
Stability of alga-produced CtxB-Pfs25 in lyophilized algae.
The stabilities of Cr.CtxB-Pfs25 in lyophilized algae stored at 4°C, 22°C (room temperature), and 37°C were determined by GM1 ganglioside binding as described above with minor modifications. The JAG101 and ΔpsbA strains were grown in 20-liter photobioreactors as described above and stored at −80°C in aliquots, several of which were lyophilized. Total soluble protein lysates from the JAG101 and ΔpsbA strains were prepared from frozen and lyophilized samples and diluted to 20 μg/ml. Two-fold serial dilutions were prepared in PBS and applied to GM1 ganglioside-coated plates in triplicate. Purified CtxB was used as a positive control. Plates were washed with PBS-T, and binding was detected with rabbit anti-CT primary antibodies and goat anti-rabbit secondary antibodies conjugated to horseradish peroxidase. Plates were visualized with the TMB substrate kit (Thermo Scientific). Endpoint titers of GM1 binding activity in JAGA101 lysates were defined as the last dilution that gave a reading that was three standard deviations above the signal from ΔpsbA lysates at a concentration of 10 μg/ml.
Laboratory mice and animal protocols.
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Diego, under protocol S-12026. Female BALB/c mice 4 to 6 weeks of age were purchased from Charles River Labs and housed in a UCSD vivarium. Mice were given ad libitum access to food and water. Experimental groups were composed of at least five mice.
Immunization regimes and blood and fecal sample collection.
For comparing the immunogenicity of Cr.CtxB-Pfs25 to that of Cr.Pfs25, mice were vaccinated by intraperitoneal injection with 20 μg of affinity-purified Cr.CtxB-Pfs25 or an equimolar amount of Cr.Pfs25 in a 1:1 mix of 2% Alhydrogel (IvivoGen). Three injections were administered at 3-week intervals, and blood was collected prior to each immunization and 1 week following the final injection.
Oral vaccinations were administered to naive and primed mice. Mice were primed by intraperitoneal injection with 40 μg of affinity-purified Cr.CtxB-Pfs25 using complete Freund's adjuvant; naive mice were not injected. Primed and naive mice were each orally immunized by intragastric gavage with 10 mg lyophilized JAG101 or the ΔpsbA strain for a total of four groups of five mice: (i) primed with ΔpsbA algae, (ii) primed with JAG101 algae, (iii) naive with ΔpsbA algae, and (iv) naive with JAG101 algae. Oral immunizations were prepared by resuspending lyophilized algae in PBS and were administered using a bulb-tipped gastric gavage needle every 7 days. In total, 12 oral immunizations were administered over a 12-week period. From each mouse, fecal pellets were collected weekly and blood was collected prior to vaccinations (prebleed) and periodically thereafter by submandibular bleed; final bleeds were performed by cardiac puncture at the completion of the immunization regime.
Determination of antigen-specific IgG and IgA endpoint titers.
Pfs25- and CtxB-specific serum IgG antibody titers were determined by ELISA as follows. Purified CtxB and Cr.Pfs25 were diluted to 1 μg/ml in PBS and applied to MaxiSorp or Immulon 2HB ELISA plates, which were incubated overnight at 4°C on a rocker, blocked with 5% milk in PBS-T for 2 h at room temperature, and washed three times with PBS-T. Serum samples from each mouse were prepared by 1:4 serial dilutions starting at 1:100 in PBS; diluted samples were added to the plate and incubated overnight at 4°C. After three washes with PBS-T, antigen-specific IgG antibodies were detected with goat anti-mouse IgG antibodies (Sigma; A3562) conjugated to alkaline phosphatase for 2 h at room temperature. Plates were visualized with PNPP and measured at 405 nm using an Infinite M200 Pro plate reader. Endpoint titers were defined as the lowest dilution with a signal that was three standard deviations above that of prebleed sera. Statistics were calculated in Excel using a paired Student t test, and figures were assembled in JMP version 9.02 (SAS, Cary, NC).
Pfs25- and CtxB-specific fecal IgA antibody levels were determined by ELISA as follows. ELISA plates were prepared in an identical manner to IgG antibody detection. Fecal extracts were prepared by pooling fecal pellets from each experimental group and resuspending in PBS with 0.01% Tween 20. Samples were homogenized with a 19-guage needle followed by sonication. Lysates were cleared by centrifugation and quantitated using the DC protein assay (Bio-Rad). Fecal extracts were added to antigen-coated ELISA plates at 0.5 mg/ml in triplicate and incubated overnight at 4°C on a rocker. Plates were washed three times with PBS-T, and IgA antibodies were detected with goat anti-mouse IgA antibodies (Sigma catalog no. A4937) conjugated to alkaline phosphatase at room temperature for 2 h. Following three washes with PBS-T, plates were visualized with PNPP and measured at 405 nm with an Infinite M200 Pro plate reader. Figures were assembled in JMP version 9.02.
RESULTS
Protein engineering and construction of transgenic algal chloroplasts expressing ctxB-pfs25.
We synthesized the β subunit of the Vibrio cholerae toxin (ctxB) gene using a C. reinhardtii chloroplast codon bias (here referred to as Cr.ctxB). Synthetic Cr.ctxB was fused to the 5′ end of a codon-optimized pfs25 (Cr.pfs25). We placed the Pfs25 domain on the carboxy side of the CtxB domain, because N-terminal peptide additions to CtxB reduce oral immunogenicity (54). Pfs25 is a malaria transmission-blocking vaccine candidate protein that lacks glycosylation and contains tandem repeats of structurally complex epidermal growth factor (EGF)-like domains. The codon adaptation index (CAI), which quantitates the codon bias of a transgene against a reference set, of Cr.ctxB-pfs25 is 0.909 (47). A CAI score of 1 indicates that every instance of an amino acid is encoded by the most prevalent codon of the reference table for that amino acid (55). CtxB and Pfs25 are separated by a flexible glycine linker and a furin protease recognition sequence, which facilitates antigen separation following endocytosis by gut epithelial cells. A nine-amino-acid flag epitope was placed on the carboxy end of the protein to facilitate purification, with a TEV protease site between the flag epitope and Pfs25 to allow for removal of the flag epitope following purification. Cr.ctxB-pfs25 was cloned into a chloroplast expression vector containing the light-dependent psbA promoter and 5′ and 3′ untranslated regions (Fig. 1A; pJAG101). C. reinhardtii strain W1.1 (51) was transformed with plasmid pJAG101 by DNA-coated gold particle bombardment; integration of Cr.ctxB-pfs25 and chloroplast homoplasmicity were confirmed by PCR as previously described (data not shown; 36), and protein accumulation was confirmed by Western blotting (Fig. 1B). Cr.CtxB-Pfs25 accumulates in chloroplasts and reaches levels of 0.09% total soluble protein as determined by ELISA (see Materials and Methods).
Fig 1.

Diagram of ctxB-pfs25 chloroplast transformation vector and Western blots of CtxB-Pfs25 accumulation in transformed C. reinhardtii. (A) The codon-optimized ctxB gene was cloned upstream and in-frame with codon-optimized pfs25, which contains four tandem epidermal growth factor-like domain repeats. The resulting ctxB-pfs25 fusion gene was integrated into the psbA locus by homologous recombination such that transgene expression is driven by the light-regulated psbA promoter. (B) Western blot analysis of soluble protein lysates at 0 and 24 h after light shift from C. reinhardtii expressing Cr.ctxB-pfs25 and control C. reinhardtii lacking psbA.
Characterization of purified CtxB-Pfs25.
Purified Cr.CtxB-Pfs25 was prepared using anti-FLAG M2 affinity resin (see Materials and Methods) and evaluated by Western blotting (Fig. 2A). As expected, Cr.CtxB-Pfs25 migrates more slowly than Cr.Pfs25 when analyzed by SDS-PAGE, near the predicted size of 34 kDa when probed with anti-Flag MAbs (Fig. 2A, arrow). There is also a slightly larger band that migrates around 40 kDa (Fig. 2A, asterisk), which is similar to the band in JAG101 algae lysates (Fig. 1B). The absence of the smaller band in lysates is unclear but could be due to incomplete reduction of Cr.CtxB-Pfs25, because both CtxB and Pfs25 contain multiple disulfide bonds.
Fig 2.

Western blot analysis of affinity-purified alga-produced CtxB-Pfs25. (A) Affinity-purified Cr.Pfs25 and Cr.CtxB-Pfs25 were reduced, separated by SDS-PAGE, transferred to nitrocellulose, and detected with anti-FLAG MAbs. (B and C) Reduced and nonreduced Cr.CtxB-Pfs25 were resolved by SDS-PAGE, transferred to nitrocellulose, and detected with anti-Pfs25 4B7 MAbs (B) and anti-cholera toxin Abs (C). (D) Five micrograms of nonreduced and reduced Cr.CtxB-Pfs25 and a BSA standard (2.5 μg, 1.25 μg, 1.125 μg, 0.56 μg) were resolved by SDS-PAGE (4 to 20% gradient) and stained with Coomassie blue. R, reduced; N, nonreduced.
We previously demonstrated that Cr.Pfs25 resembles native Pfs25 when produced in algal chloroplasts (46). To test whether Cr.Pfs25 retains its structure when produced as a fusion with CtxB, we probed both reduced and nonreduced Cr.CtxB-Pfs25. Reduced Cr.CtxB-Pfs25 lacks the necessary disulfide bridges that are essential to Pfs25 structure, while nonreduced Cr.CtxB-Pfs25 should maintain conformational epitopes. We first probed with conformation-specific anti-Pfs25 monoclonal antibodies (4B7 MAbs). 4B7 MAbs block malaria transmission and are specific to an epitope found on properly folded Pfs25 (56). Consistent with conformation-dependent binding of Pfs25, 4B7 MAbs recognized nonreduced but not reduced Cr.CtxB-Pfs25 (Fig. 2B). Thus, Cr.CtxB-Pfs25 contains the 4B7 epitope found only on native Pfs25, suggesting that CtxB does not disrupt Cr.Pfs25 folding. Interestingly, nonreduced Cr.CtxB-Pfs25 migrates as a larger-molecular-weight species. This could be due to Cr.Pfs25, which was previously shown to migrate at a larger molecular weight in native and nonreduced gels (46), or because native CtxB assembles into a pentameric structure (57). Similar to 4B7 MAbs, anti-cholera toxin (CT) antibodies recognize larger but less well-defined molecular weight species in nonreduced samples. Stronger recognition of nonreduced Cr.CtxB-Pfs25 by anti-CT antibodies suggests that Cr.CtxB is folding correctly when fused to Pfs25. Additionally, the wide distribution suggests that Cr.CtxB-Pfs25 is assembling into quaternary structures, including but not limited to pentamers. These data suggest that both Cr.Pfs25 and Cr.CtxB are folded correctly when produced together as a chimeric protein.
Purified Cr.CtxB-Pfs25 separated by SDS-PAGE and stained with Coomassie blue (Fig. 2D) is consistent with the Western blots. Reduced and nonreduced Cr.CtxB-Pfs25 migrate near 33 kDa (Fig. 2D, arrowhead) and 100 to 135 kDa (Fig. 2D, arrow), respectively. However, the quantitation of purified Cr.CtxB-Pfs25 determined by the DC Bio-Rad protein assay (see Materials and Methods) appears to overestimate the amount of recombinant protein recovered compared to bovine serum albumin (BSA) standards. Thus, where purified Cr.CtxB-Pfs25 was used in animal studies (described below), we likely used less than the indicated amounts.
Alga-produced CtxB-Pfs25 binds the GM1 ganglioside receptor.
The mucosal adjuvant properties of CtxB are dependent on GM1 ganglioside receptor binding and subsequent internalization into the gut-associated lymphoid tissue. GM1 binding was evaluated in soluble lysates prepared from Cr.CtxB-Pfs25-producing algae (strain JAG101) and an isogenic control strain lacking psbA (the ΔpsbA strain) collected at 0, 24, 48, and 72 h after light induction. Light induction is necessary because Cr.ctxB-pfs25 is controlled by the psbA promoter, which is light dependent. Lysates were applied to ELISA plates coated with GM1 ganglioside and tested for Cr.CtxB-mediated binding using anti-CT antibodies by ELISA (Fig. 3). Significant binding was detected in JAG101 samples collected at 24, 48, and 72 h after light induction compared to control algae lacking psbA. Thus, fusing Cr.Pfs25 to the C terminus of Cr.CtxB does not disrupt GM1 ganglioside binding by Cr.CtxB-Pfs25.
Fig 3.

GM1 ganglioside binding by alga-produced CtxB-Pfs25. Soluble protein extracts (20 μg/ml) from JAG101 and ΔpsbA C. reinhardtii strains collected at 0, 24, 48, and 72 h after light induction (JAG101 is driven by the light-dependent psbA promoter) were applied to GM1 ganglioside-coated ELISA plates in triplicate. Binding was detected with anti-CT antibodies. Error bars shown are 1 standard deviation.
Effect of lyophilization and storage temperature on GM1 ganglioside binding.
Cold-chain storage increases vaccine distribution complexity and costs, which could be avoided if antigens were temperature stable. We evaluated GM1 binding by Cr.CtxB-Pfs25 by ELISA from both pre- and postlyophilized JAG101 and ΔpsbA algae (Fig. 4A). Significant binding compared to that of controls was detected in both pre- and postlyophilized samples at below 2 μg/ml soluble protein. Importantly, no difference in binding could be detected after lyophilization. Thus, lyophilization does not impact GM1 ganglioside binding, suggesting that Cr.CtxB-Pfs25 structure is retained. We also assessed the stability of Cr.CtxB-Pfs25 within lyophilized algae stored at 4°C, 22°C (room temperature), and 37°C. The binding activity of Cr.CtxB-Pfs25 in lysates is reported as an endpoint titer using a cutoff of three standard deviations above that of controls (see Materials and Methods). GM1 binding was reduced in all samples after 6 months but remained relatively stable at 4°C and 22°C, ranging from 0.5 to 2.1 μg/ml and 0.6 to 5 μg/ml, respectively (Fig. 4B). In comparison, GM1 binding titers in samples stored at 37°C were reduced 10-fold compared to those of samples stored at 4°C. Thus, Cr.CtxB-Pfs25 is relatively stable in lyophilized algae at moderate temperatures.
Fig 4.

Stability of alga-produced CtxB-Pfs25 in lyophilized algae. (A) Soluble protein extracts from JAG101 (both pre- and postlyophilization) and ΔpsbA C. reinhardtii strains were serially diluted and applied to GM1 ganglioside-coated ELISA plates in triplicate. Binding was detected with anti-CT antibodies. (B) Lyophilized JAG101 C. reinhardtii was stored at 4°C, 22°C, and 37°C; lyophilized ΔpsbA C. reinhardtii was stored at 4°C. Soluble protein extracts were prepared on the indicated days, serially diluted, and applied to GM1 ganglioside-coated ELISA plates in triplicate. Binding was detected with anti-CT Abs. Error bars shown are 1 standard deviation. Binding endpoints were defined as the last dilution with an absorbance greater than that of the ΔpsbA strain at 10 μg/ml plus 3 standard deviations.
Immunogenicity of CtxB-Pfs25.
We previously demonstrated that injecting mice with Cr.Pfs25 elicits significant levels of Pfs25-specific IgG antibodies. To compare the immune response to Cr.CtxB-Pfs25 and Cr.Pfs25 alone, we vaccinated BALB/c mice via intraperitoneal injection with 20 μg of affinity-purified Cr.CtxB-Pfs25 and an equimolar amount of Cr.Pfs25 formulated with Alhydrogel. Injections were given at 3-week intervals, and blood samples were collected prior to each vaccination and 1 week after the last injection. Sera were then tested for Pfs25-specific IgG antibodies by ELISA (Fig. 5). Surprisingly, Pfs25-specific IgG titers were significantly higher in mice injected with Cr.Pfs25 alone after the first two injections, while both proteins had similar high titers after the third injection. Thus, Cr.CtxB-Pfs25 elicits Pfs25-specific antibodies, which reach levels that are similar to Cr.Pfs25 alone after two boosts using Alhydrogel.
Fig 5.

Immunogenicity of alga-produced CtxB-Pfs25 and Pfs5 using Alhydrogel. Mice were vaccinated by intraperitoneal injection with 20 μg of affinity-purified Cr.CtxB-Pfs25 or the molar equivalent of Cr.Pfs25 adsorbed to Alhydrogel. Pfs25-specific IgG antibodies were detected by ELISA. Endpoint titers were defined as the reciprocal of the last dilution that gave an absorbance greater than prebleed sera plus 3 standard deviations. Comparisons between groups were done using a two-tailed parametric Student t test (*, P < 0.05; **, P < 0.01).
Oral vaccination of primed mice with lyophilized algae.
We hypothesized that oral vaccination with whole algae that produce Ctxb-Pfs25 could act to boost antigen-specific serum IgG antibody levels of mice previously injected with a single antigen dose (58). We also wanted to determine whether oral vaccination with CtxB-Pfs25 elicits a secretory IgA response not normally associated with injectable vaccines (59). To test this hypothesis, BALB/c mice were injected with affinity-purified Cr.CtxB-Pfs25 using complete Freund's adjuvant followed by weekly oral vaccinations with 10 mg of JAG101 or ΔpsbA algae, starting 2 weeks after injection (Fig. 6; oral vaccinations are indicated by arrows). Soluble protein was measured as 23% of the total algal biomass, and of that 0.09% is Cr.CtxB-Pfs25, which results in an effective dose of approximately 31 μg of Cr.CtxB-Pfs25 for each immunization. Pfs25- and CtxB-specific IgG and IgA antibodies in sera and feces, respectively, were detected by ELISA. Pfs25- and CtxB-specific IgG antibodies were similar for mice fed with JAG101 or control ΔpsbA algae (Fig. 6A and B). Pfs25-specific IgG antibody titers were also similar when mice were injected with Cr.Pfs25 (rather than Cr.CtxB-Pfs25) followed by six doses of JAG101 or ΔpsbA algae over 3 days (see Fig. S1 in the supplemental material). Thus, oral vaccination appears to have no effect on serum IgG antibodies on a primed immune system when using relatively low doses of antigen (microgram quantities). However, CtxB- and Pfs25-specific IgA antibodies were significantly higher in fecal extracts of mice that were fed JAG101 algae than in those fed ΔpsbA algae (Fig. 6C and D). Significant levels of CtxB-specific IgA antibodies were seen after a single dose of JAG101 algae, whereas four doses were required for Pfs25-specific IgA antibodies. Together, these data suggest that lyophilized algae sufficiently protect Cr.CtxB-Pfs25 from degradation in the gastrointestinal tract and that Cr.CtxB-Pfs25 binds the GM1 ganglioside receptor and elicits an antigen-specific IgA immune response to both Cr.CtxB and its fusion partner, Cr.Pfs25.
Fig 6.
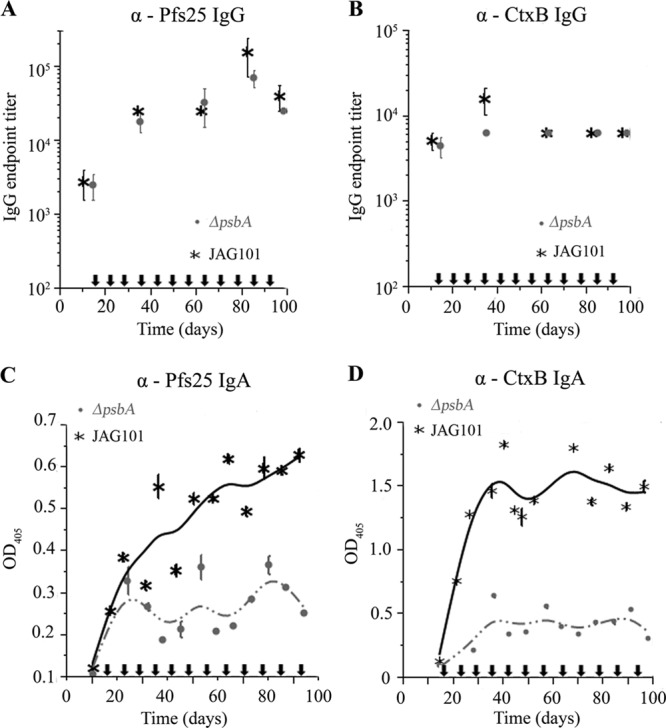
Immune response to whole-cell alga oral immunization in primed mice. Mice were injected with Cr.CtxB-Pfs25 using complete Freund's adjuvant followed by weekly doses of 10 mg of JAG101 or ΔpsbA C. reinhardtii starting 2 weeks postinjection. Serum IgG antibodies for Pfs25 (A) and CtxB (B) were detected by ELISA. Error bars indicate the standard errors of the means. Pfs25 (C)- and CtxB (D)-specific IgA antibodies in fecal extracts diluted to 0.5 mg/ml were detected by ELISA. Error bars indicate 1 standard deviation. Arrows indicate when oral doses were administered.
Oral vaccination of naive mice with lyophilized algae.
We characterized the immune response of naive BALB/c mice during a 12-week oral vaccination schedule to evaluate whether antigen-specific IgA antibodies were dependent on prior injections of the antigen. Lyophilized JAG101 or ΔpsbA algae were administered once per week starting at day 0 by oral gavage in PBS; blood and fecal samples were collected periodically and tested for antigen-specific IgG and IgA antibodies, respectively, by ELISA. Pfs25-specific IgG antibodies in sera were not detected at any point during the vaccination regime (Fig. 7A). CtxB-specific IgG antibodies in sera increased after three immunizations (∼21 days) and reached significant levels after five immunizations (35 days) relative to those in preimmune sera (Fig. 7B). Similar to primed mice, significant levels of CtxB- and Pfs25-specific IgA antibodies were detected in mice fed JAG101 algae compared to those in mice fed ΔpsbA algae (Fig. 7C and D). Antigen-specific IgA antibodies appeared to increase more slowly in naive mice than in primed mice, which indicates that priming by injection may enhance the IgA antibody response to oral vaccinations. Thus, oral vaccination using Cr.CtxB-Pfs25-producing algae elicits both CtxB-specific IgG and IgA antibodies and Pfs25-specific IgA antibodies.
Fig 7.
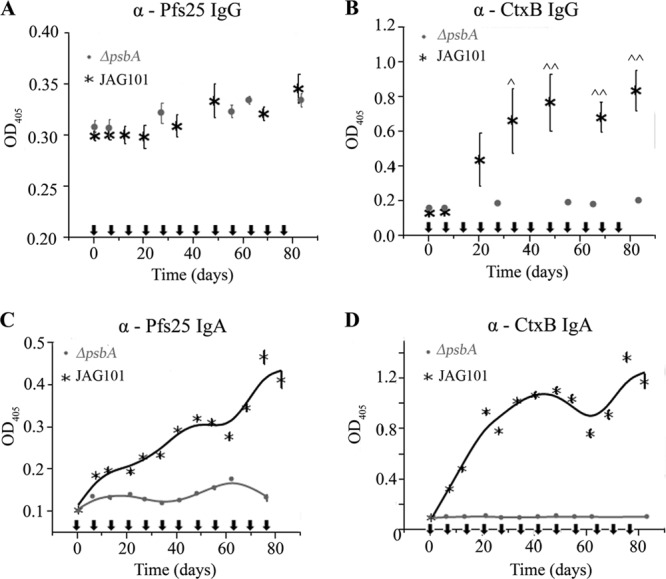
Immune response to whole-cell alga oral immunization in naive mice. Mice were administered weekly doses of JAG101 algae or ΔpsbA C. reinhardtii. Serum IgG antibodies for Pfs25 (A) and CtxB (B) were detected by ELISA. Error bars indicate standard errors of the means. Pfs25 (C)- and CtxB (D)-specific IgA antibodies in fecal extracts diluted to 0.5 mg/ml were detected by ELISA. Error bars indicate 1 standard deviation. Arrows indicate when oral doses were administered. ^, P < 0.05; ^^, P < 0.01.
DISCUSSION
In this study, we demonstrated that C. reinhardtii chloroplasts can produce a structurally complex chimeric protein consisting of CtxB, the β subunit from the V. cholerae toxin, and Pfs25, a eukaryotic protein from P. falciparum and promising malaria TBV candidate. To date, algae are the only recombinant system to produce an unmodified aglycosylated Pfs25 protein that resembles native Pfs25 structure (46). Like alga-produced Pfs25, the Cr.CtxB-Pfs25 chimera is recognized by conformation-specific anti-Pfs25 transmission-blocking monoclonal antibodies, suggesting that Cr.Pfs25 is similar in structure to native Pfs25 even when produced as a chimeric protein. Furthermore, GM1 ganglioside binding by Cr.CtxB-Pfs25 in soluble alga extracts indicates that Cr.CtxB also resembles its native counterpart from V. cholerae. Thus, Cr.CtxB-Pfs25 accumulates as a soluble, correctly folded, and functional protein within algal chloroplasts.
Bifunctional chimeric proteins, like the CtxB-Pfs25 fusion produced here, are becoming increasingly useful for vaccine formulation, cancer therapeutics, immunotherapy (60, 61), and industrial enzymes (62). Ultimately, algae could be an inexpensive platform for producing many of these complex proteins, because they can be grown photosynthetically and have minimal growth requirements. Economic manufacturing of recombinant proteins using algae will further improve as new methods to increase yields are identified. In the near term, proteins that are recalcitrant to production in conventional systems or otherwise biologically suited for production in algae will help drive development of the algae platform. Indeed, algae were recently used to make immunotoxins in a single step (37). Immunotoxins are currently made by chemically conjugating a human antibody, made and purified from CHO cells, to a toxin molecule or protein, followed by a second purification of the chimeric molecule. Another significant advantage for an alga platform is oral delivery of a protein, thereby eliminating all purification steps. Unlike bacterial and mammalian expression platforms, eukaryotic green algae are considered safe and can therefore be consumed, making oral delivery of unpurified protein a possibility.
In this work, we demonstrated that Cr.CtxB-Pfs25 is protected from degradation in the gastrointestinal tract within lyophilized algae. Cr.CtxB-Pfs25 is eventually released from the alga cell in the gut, whereupon it binds the GM1 ganglioside receptor found on gut epithelial cells and elicits an immune response. Thus, lyophilized algae could be a cost-effective and efficient means of storing and delivering proteins to the gut. In this case, Cr.CtxB-Pfs25 did not elicit the prerequisite IgG antibodies that would block malaria transmission. However, a similar strategy using the D2 fibronectin-binding domain from Staphylococcus aureus fused to CtxB did elicit IgG antibodies (63); eliciting an antigen-specific IgG response therefore appears to be dependent on the antigen itself and not just on CtxB. Hence, CtxB is not the appropriate mucosal adjuvant for an oral Pfs25-based transmission-blocking vaccine. The predominantly IgA antibody response in this study may be more appropriate for Helicobacter pylori, Shigella spp., rotaviruses, or pathogens that invade host mucosal surfaces (64).
Whole-cell oral vaccines would greatly simplify vaccine delivery and eliminate costly purification steps, both of which would enable affordable vaccination in poor countries. Eukaryotic green algae are devoid of endotoxins, human pathogens, or other known toxic compounds and provide a stable storage and delivery mechanism. Furthermore, lyophilized whole cells are a significant cost advantage over injectable vaccines and alternative oral vaccination strategies that use formulated nanoparticles (65, 66). For example, algal biomass can be produced for about $3/kg (67), meaning that a therapeutic dose could cost just a few cents. A more thorough understanding of mucosal adjuvant proteins is paramount to developing oral vaccines moving forward. Ideally, antigens could be paired with mucosal adjuvants that elicit an appropriate immune response that confers long-lived protection from disease. If this can be achieved, the gap in vaccine coverage between wealthy and poor regions of the world could be closed, and the disease burdens could be reduced in areas of the world where this is needed most.
Supplementary Material
ACKNOWLEDGMENTS
We thank Cecilia Berin and Zivko Nikolov for helpful discussions and comments on the manuscript and Javier Gimpel for the psbA knockout strain.
This research was supported by funding from the San Diego Foundation, the California Energy Commission (500-10-039), and the National Science Foundation (CBET-1160184).
J.A.G. and S.M. conceived and designed the experiments; J.A.G., A.B.T., and D.Z.D. performed the experiments; J.A.G. analyzed the data and wrote the paper.
Footnotes
Published ahead of print 19 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00714-13.
REFERENCES
- 1. WHO 2008. Global health observatory data repository, cause-specific mortality. WHO, Geneva, Switzerland [Google Scholar]
- 2. WHO/UNICEF 2012. Estimates of vaccine coverage 1980–2011: 2012 update. WHO, Geneva, Switzerland [Google Scholar]
- 3. WHO 2012. The world health statistics. WHO, Geneva, Switzerland [Google Scholar]
- 4. Ingstad B, Munthali AC, Braathen SH, Grut L. 2012. The evil circle of poverty: a qualitative study of malaria and disability. Malar. J. 11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ekwueme DU, Weniger BG, Chen RT. 2002. Model-based estimates of risks of disease transmission and economic costs of seven injection devices in sub-Saharan Africa. Bull. World Health Organ. 80:859–870 [PMC free article] [PubMed] [Google Scholar]
- 6. Giudice EL, Campbell JD. 2006. Needle-free vaccine delivery. Adv. Drug Deliv. Rev. 58:68–89 [DOI] [PubMed] [Google Scholar]
- 7. Levine MM. 2011. “IDEAL” vaccines for resource poor settings. Vaccine 29(Suppl 4):D116–D125 [DOI] [PubMed] [Google Scholar]
- 8. Mitragotri S. 2005. Immunization without needles. Nat. Rev. Immunol. 5:905–916 [DOI] [PubMed] [Google Scholar]
- 9. Bilsborough J, Viney JL. 2004. Gastrointestinal dendritic cells play a role in immunity, tolerance, and disease. Gastroenterology 127:300–309 [DOI] [PubMed] [Google Scholar]
- 10. Mowat AM. 2003. Anatomical basis of tolerance and immunity to intestinal antigens. Nat. Rev. Immunol. 3:331–341 [DOI] [PubMed] [Google Scholar]
- 11. Chase MW. 1946. Inhibition of experimental drug allergy by prior feeding of the sensitizing agent. Proc. Soc. Exp. Biol. Med. 61:257–259 [DOI] [PubMed] [Google Scholar]
- 12. Mason KL, Huffnagle GB, Noverr MC, Kao JY. 2008. Overview of gut immunology. Adv. Exp. Med. Biol. 635:1–14 [DOI] [PubMed] [Google Scholar]
- 13. da Hora VP, Conceicao FR, Dellagostin OA, Doolan DL. 2011. Nontoxic derivatives of LT as potent adjuvants. Vaccine 29:1538–1544 [DOI] [PubMed] [Google Scholar]
- 14. Sanchez J, Holmgren J. 2011. Cholera toxin—a foe & a friend. Indian J. Med. Res. 133:153–163 [PMC free article] [PubMed] [Google Scholar]
- 15. Thompson AL, Staats HF. 2011. Cytokines: the future of intranasal vaccine adjuvants. Clin. Dev. Immunol. 2011:289597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103 [DOI] [PubMed] [Google Scholar]
- 17. Lahiri A, Das P, Chakravortty D. 2008. Engagement of TLR signaling as adjuvant: towards smarter vaccine and beyond. Vaccine 26:6777–6783 [DOI] [PubMed] [Google Scholar]
- 18. McCluskie MJ, Weeratna RD, Payette PJ, Davis HL. 2001. The potential of CpG oligodeoxynucleotides as mucosal adjuvants. Crit. Rev. Immunol. 21:103–120 [PubMed] [Google Scholar]
- 19. Azizi A, Kumar A, Diaz-Mitoma F, Mestecky J. 2010. Enhancing oral vaccine potency by targeting intestinal M cells. PLoS Pathog. 6:e1001147. 10.1371/journal.ppat.1001147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clark MA, Jepson MA, Simmons NL, Hirst BH. 1994. Preferential interaction of Salmonella typhimurium with mouse Peyer's patch M cells. Res. Microbiol. 145:543–552 [DOI] [PubMed] [Google Scholar]
- 21. U.S. Food and Drug Administration 2012, posting date GRAS notice inventory. FDA, Silver Spring, MD: http://www.accessdata.fda.gov/scripts/fcn/fcnNavigation.cfm?rpt=graslisting [Google Scholar]
- 22. Ma JK, Drake PM, Christou P. 2003. The production of recombinant pharmaceutical proteins in plants. Nat. Rev. Genet. 4:794–805 [DOI] [PubMed] [Google Scholar]
- 23. Haq TA, Mason HS, Clements JD, Arntzen CJ. 1995. Oral immunization with a recombinant bacterial antigen produced in transgenic plants. Science 268:714–716 [DOI] [PubMed] [Google Scholar]
- 24. Dove A. 2002. Uncorking the biomanufacturing bottleneck. Nat. Biotechnol. 20:777–779 [DOI] [PubMed] [Google Scholar]
- 25. Streatfield SJ, Howard JA. 2003. Plant-based vaccines. Int. J. Parasitol. 33:479–493 [DOI] [PubMed] [Google Scholar]
- 26. Kaiser J. 2008. Is the drought over for pharming? Science 320:473–475 [DOI] [PubMed] [Google Scholar]
- 27. Giritch A, Marillonnet S, Engler C, van Eldik G, Botterman J, Klimyuk V, Gleba Y. 2006. Rapid high-yield expression of full-size IgG antibodies in plants coinfected with noncompeting viral vectors. Proc. Natl. Acad. Sci. U. S. A. 103:14701–14706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ellstrand NC. 2001. When transgenes wander, should we worry? Plant Physiol. 125:1543–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kong Q, Richter L, Yang YF, Arntzen CJ, Mason HS, Thanavala Y. 2001. Oral immunization with hepatitis B surface antigen expressed in transgenic plants. Proc. Natl. Acad. Sci. U. S. A. 98:11539–11544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rosenberg JN, Oyler GA, Wilkinson L, Betenbaugh MJ. 2008. A green light for engineered algae: redirecting metabolism to fuel a biotechnology revolution. Curr. Opin. Biotechnol. 19:430–436 [DOI] [PubMed] [Google Scholar]
- 31. DOE 2010. National algal biofuels technology roadmap. Department of Energy, Biomass Program, Washington, DC [Google Scholar]
- 32. Jones CS, Luong T, Hannon M, Tran M, Gregory JA, Shen Z, Briggs SP, Mayfield SP. 2012. Heterologous expression of the C-terminal antigenic domain of the malaria vaccine candidate Pfs48/45 in the green algae Chlamydomonas reinhardtii. Appl. Microbiol. Biotechnol. 97:1987–1995 [DOI] [PubMed] [Google Scholar]
- 33. Surzycki R, Greenham K, Kitayama K, Dibal F, Wagner R, Rochaix JD, Ajam T, Surzycki S. 2009. Factors effecting expression of vaccines in microalgae. Biologicals 37:133–138 [DOI] [PubMed] [Google Scholar]
- 34. Rasala BA, Lee PA, Shen Z, Briggs SP, Mendez M, Mayfield SP. 2012. Robust expression and secretion of Xylanase1 in Chlamydomonas reinhardtii by fusion to a selection gene and processing with the FMDV 2A peptide. PLoS One 7:e43349. 10.1371/journal.pone.0043349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mayfield SP, Franklin SE, Lerner RA. 2003. Expression and assembly of a fully active antibody in algae. Proc. Natl. Acad. Sci. U. S. A. 100:438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rasala BA, Muto M, Lee PA, Jager M, Cardoso RM, Behnke CA, Kirk P, Hokanson CA, Crea R, Mendez M, Mayfield SP. 2010. Production of therapeutic proteins in algae, analysis of expression of seven human proteins in the chloroplast of Chlamydomonas reinhardtii. Plant Biotechnol. J. 8:719–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tran M, Van C, Barrera DJ, Pettersson PL, Peinado CD, Bui J, Mayfield SP. 2013. Production of unique immunotoxin cancer therapeutics in algal chloroplasts. Proc. Natl. Acad. Sci. U. S. A. 110:E15–E22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schroda M. 2004. The Chlamydomonas genome reveals its secrets: chaperone genes and the potential roles of their gene products in the chloroplast. Photosynth. Res. 82:221–240 [DOI] [PubMed] [Google Scholar]
- 39. Kim J, Mayfield SP. 1997. Protein disulfide isomerase as a regulator of chloroplast translational activation. Science 278:1954–1957 [DOI] [PubMed] [Google Scholar]
- 40. Breiman A, Fawcett TW, Ghirardi ML, Mattoo AK. 1992. Plant organelles contain distinct peptidylprolyl cis,trans-isomerases. J. Biol. Chem. 267:21293–21296 [PubMed] [Google Scholar]
- 41. Manuell AL, Quispe J, Mayfield SP. 2007. Structure of the chloroplast ribosome: novel domains for translation regulation. PLoS Biol. 5:e209. 10.1371/journal.pbio.0050209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaslow DC, Quakyi IA, Syin C, Raum MG, Keister DB, Coligan JE, McCutchan TF, Miller LH. 1988. A vaccine candidate from the sexual stage of human malaria that contains EGF-like domains. Nature 333:74–76 [DOI] [PubMed] [Google Scholar]
- 43. Lonnroth I, Holmgren J. 1973. Subunit structure of cholera toxin. J. Gen. Microbiol. 76:417–427 [DOI] [PubMed] [Google Scholar]
- 44. Sharma B. 2008. Structure and mechanism of a transmission blocking vaccine candidate protein Pfs25 from P. falciparum: a molecular modeling and docking study. In Silico Biol. 8:193–206 [PubMed] [Google Scholar]
- 45. Gozar MM, Price VL, Kaslow DC. 1998. Saccharomyces cerevisiae-secreted fusion proteins Pfs25 and Pfs28 elicit potent Plasmodium falciparum transmission-blocking antibodies in mice. Infect. Immun. 66:59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gregory JA, Li F, Tomosada LM, Cox CJ, Topol AB, Vinetz JM, Mayfield S. 2012. Algae-produced Pfs25 elicits antibodies that inhibit malaria transmission. PLoS One 7:e37179. 10.1371/journal.pone.0037179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Puigbo P, Bravo IG, Garcia-Vallve S. 2008. CAIcal: a combined set of tools to assess codon usage adaptation. Biol. Direct 3:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nour-Eldin HH, Hansen BG, Norholm MH, Jensen JK, Halkier BA. 2006. Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res. 34:e122. 10.1093/nar/gkl635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou A, Webb G, Zhu X, Steiner DF. 1999. Proteolytic processing in the secretory pathway. J. Biol. Chem. 274:20745–20748 [DOI] [PubMed] [Google Scholar]
- 50. Rasala BA, Mayfield SP. 2011. The microalga Chlamydomonas reinhardtii as a platform for the production of human protein therapeutics. Bioeng. Bugs 2:50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Manuell AL, Beligni MV, Elder JH, Siefker DT, Tran M, Weber A, McDonald TL, Mayfield SP. 2007. Robust expression of a bioactive mammalian protein in Chlamydomonas chloroplast. Plant Biotechnol. J. 5:402–412 [DOI] [PubMed] [Google Scholar]
- 52. Boynton JE, Gillham NW, Harris EH, Hosler JP, Johnson AM, Jones AR, Randolph-Anderson BL, Robertson D, Klein TM, Shark KB. 1988. Chloroplast transformation in Chlamydomonas with high velocity microprojectiles. Science 240:1534–1538 [DOI] [PubMed] [Google Scholar]
- 53. Fischer N, Stampacchia O, Redding K, Rochaix JD. 1996. Selectable marker recycling in the chloroplast. Mol. Gen. Genet. 251:373–380 [DOI] [PubMed] [Google Scholar]
- 54. Elson CO. 1992. Cholera toxin as a mucosal adjuvant: effects of H-2 major histocompatibility complex and lps genes. Infect. Immun. 60:2874–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sharp PM, Li WH. 1987. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15:1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barr PJ, Green KM, Gibson HL, Bathurst IC, Quakyi IA, Kaslow DC. 1991. Recombinant Pfs25 protein of Plasmodium falciparum elicits malaria transmission-blocking immunity in experimental animals. J. Exp. Med. 174:1203–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang RG, Westbrook ML, Westbrook EM, Scott DL, Otwinowski Z, Maulik PR, Reed RA, Shipley GG. 1995. The 2.4 A crystal structure of cholera toxin B subunit pentamer: choleragenoid. J. Mol. Biol. 251:550–562 [DOI] [PubMed] [Google Scholar]
- 58. Doherty TM, Olsen AW, van Pinxteren L, Andersen P. 2002. Oral vaccination with subunit vaccines protects animals against aerosol infection with Mycobacterium tuberculosis. Infect. Immun. 70:3111–3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meeusen EN. 2011. Exploiting mucosal surfaces for the development of mucosal vaccines. Vaccine 29:8506–8511 [DOI] [PubMed] [Google Scholar]
- 60. Zhu D, Kepley CL, Zhang K, Terada T, Yamada T, Saxon A. 2005. A chimeric human-cat fusion protein blocks cat-induced allergy. Nat. Med. 11:446–449 [DOI] [PubMed] [Google Scholar]
- 61. Zhu D, Kepley CL, Zhang M, Zhang K, Saxon A. 2002. A novel human immunoglobulin Fc gamma Fc epsilon bifunctional fusion protein inhibits Fc epsilon RI-mediated degranulation. Nat. Med. 8:518–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nahalka J, Nidetzky B. 2007. Fusion to a pull-down domain: a novel approach of producing Trigonopsis variabilisD-amino acid oxidase as insoluble enzyme aggregates. Biotechnol. Bioeng. 97:454–461 [DOI] [PubMed] [Google Scholar]
- 63. Dreesen IA, Charpin-El Hamri G, Fussenegger M. 2010. Heat-stable oral alga-based vaccine protects mice from Staphylococcus aureus infection. J. Biotechnol. 145:273–280 [DOI] [PubMed] [Google Scholar]
- 64. Holmgren J, Svennerholm AM. 2012. Vaccines against mucosal infections. Curr. Opin. Immunol. 24:343–353 [DOI] [PubMed] [Google Scholar]
- 65. Chadwick S, Kriegel C, Amiji M. 2010. Nanotechnology solutions for mucosal immunization. Adv. Drug Deliv. Rev. 62:394–407 [DOI] [PubMed] [Google Scholar]
- 66. Mahapatro A, Singh DK. 2011. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J. Nanobiotechnol. 9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Amer L, Adhikari B, Pellegrino J. 2011. Technoeconomic analysis of five microalgae-to-biofuels processes of varying complexity. Bioresour. Technol. 102:9350–9359 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.