

Abstract
The infectious metacyclic promastigotes of Leishmania protozoa establish infection in a mammalian host after they are deposited into the dermis by a sand fly vector. Several Leishmania virulence factors promote infection, including the glycosylphosphatidylinositol membrane-anchored major surface protease (MSP). Metacyclic Leishmania infantum chagasi promastigotes were treated with methyl-beta-cyclodextrin (MβCD), a sterol-chelating reagent, causing a 3-fold reduction in total cellular sterols as well as enhancing MSP release without affecting parasite viability in vitro. MβCD-treated promastigotes were more susceptible to complement-mediated lysis than untreated controls and reduced the parasite load 3-fold when inoculated into BALB/c mice. Paradoxically, MβCD-treated promastigotes caused a higher initial in vitro infection rate in human or murine macrophages than untreated controls, although their intracellular multiplication was hindered upon infection establishment. There was a corresponding larger amount of covalently bound C3b than iC3b on the parasite surfaces of MβCD-treated promastigotes exposed to healthy human serum in vitro, as well as loss of MSP, a protease that enhances C3b cleavage to iC3b. Mass spectrometry showed that MβCD promotes the release of proteins into the extracellular medium, including both MSP and MSP-like protein (MLP), from virulent metacyclic promastigotes. These data support the hypothesis that plasma membrane sterols are important for the virulence of Leishmania protozoa at least in part through retention of membrane virulence proteins.
INTRODUCTION
Leishmania protozoa shuttle between a mammalian host as intracellular amastigotes and a sand fly vector as flagellated promastigotes. The sand fly acquires amastigote-laden cells during a blood meal. In the sand fly midgut, procyclic promastigotes derived from transformation of amastigotes undergo multiplication and development via intermediate stages, eventually yielding metacyclic promastigotes (1). The infectious metacyclic promastigote is inoculated into a pool of blood in the dermis of a mammalian host formed during a sand fly bite. Parasites are phagocytized by host macrophages, where they transform to amastigotes and multiply in parasitophorous vacuoles. Amastigotes spread to new macrophages at local or disseminated sites, perpetuating the infection and ultimately resulting in asymptomatic infections or symptomatic leishmaniasis (2). Various forms of leishmaniasis are endemic in 88 countries on four continents, leading to approximately two million new cases and 59,000 deaths annually (3).
Both host and parasite factors contribute to the success of infection. On one hand, mammalian host environmental risk factors and genetic background influence the clinical manifestations of infection (4). On the other hand, parasite virulence determinants required for disease development include, but are not limited to, the major surface protease (MSP) (also called GP63 or leishmanolysin) and lipophosphoglycan (LPG) (5–8). These two molecules play both overlapping and unique roles in pathogenesis. Both are attached to the exoplasmic leaflet of the plasma membranes of promastigotes by a glycosylphosphatidylinositol (GPI) membrane anchor and are localized in lipid-enriched microdomains called lipid rafts (9–11).
We hypothesized that plasma membrane lipids of metacyclic Leishmania promastigotes are important for proper display of virulence-associated proteins, including MSP, and are necessary for establishing infection of mammalian hosts. Because these parasites lack the enzymes for cholesterol biosynthesis, it was not clear which lipids would actually be most important for maintaining the intact membrane structure of virulent parasites. We used the chelating reagent methyl-beta-cyclodextrin (MβCD), which has been used in other studies of membrane cholesterol, to deplete membrane sterols from metacyclic promastigotes. The resultant parasites contained greatly reduced levels of sterols, including ergosterol and cholesterol. Resistance of sterol-depleted parasites to complement-mediated lysis in vitro was remarkably diminished, and in vivo virulence was significantly decreased in a BALB/c mouse model. Concomitantly, sterol-depleted promastigotes had larger amounts of covalently bound C3b than iC3b on their surfaces when exposed to healthy human sera. Consistently, release of MSP, the major protease responsible for cleavage of C3b to iC3b (12), into extracellular medium was enhanced by sterol perturbation. These data support the hypothesis that molecules localized at the plasma membrane and associated with membrane sterols in metacyclic promastigotes provide critical interactions with the host environment.
MATERIALS AND METHODS
Ethics statement.
Procedures with human subjects were approved by the Institutional Review Board (IRB) of the University of Iowa and the Iowa City VA Medical Center R&D Committee. Written informed consent was provided by all study participants.
This study was carried out in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Protocols were approved by the Animal Care and Use Committee of the Iowa City Veterans' Affairs Medical Center (protocol numbers 1190301 and 1190302). All efforts were taken to minimize numbers and suffering of animals.
Parasites and sterol depletion.
A Brazilian strain of Leishmania infantum chagasi (MHOM/BR/00/1669) was continuously passaged in golden hamsters to maintain virulence as previously described (13–15). Amastigotes isolated from the spleens of infected hamsters spontaneously transformed in vitro into promastigotes at 26°C in hemoflagellate-modified minimal essential medium (HOMEM), which was prepared from reagents from GIBCO (Rockville, MD) supplemented with 10% heat-inactivated fetal calf serum (FCS) (GIBCO) according to a published formula (16). Virulent promastigotes were passed weekly in HOMEM and used for experiments within three passages. Metacyclic promastigotes were isolated from the stationary-phase growth cultures using discontinuous Ficoll gradients as previously described (17, 18). Membrane sterols were depleted from stationary-phase or metacyclic promastigotes by incubation of 2 × 108 cells/ml for 1 h in freshly prepared MβCD (Sigma, St. Louis, MO) in RPMI 1640 (GIBCO), ranging in concentration from 0 to 50 mM. Treated cells were collected by centrifugation (3,000 × g, 10 min, 4°C), washed twice by centrifugation in Hanks' balanced salt solution (GIBCO) to remove MβCD, and used immediately. Supernatants were collected, filtered through a 0.22-μm filter to remove cell debris, and assayed for released proteins in some experiments. Untreated control and MβCD-treated conditions were coded prior to assessing parasite loads.
SDS-PAGE and Western blotting.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and silver staining were conducted as described previously (13, 19). SDS-polyacrylamide gels were silver stained using the SilverQuest staining kit (Invitrogen, Carlsbad, CA), spots containing proteins of interest were sliced from gels, and proteins were extracted and digested with trypsin in preparation for identification by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as previously described (20). For Western blots, proteins from metacyclic promastigotes incubated with or without MβCD and/or human serum were separated on 10% Tris-HCl SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and blocked in Tris-buffered saline with 5% milk, 3% bovine serum albumin (BSA), and 0.02% Tween 20. In some experiments, samples were treated prior to separation with 1 M hydroxylamine (pH 10.5) in 1% SDS for 30 min at 37°C to remove ester-bound C3 fragments. C3b and iC3b were from Complement Technology, Inc. (Tyler, TX). Immunoblotting was performed with 1:10,000 goat antiserum to human C3 (Quidel Corp., San Diego, CA), 1:10,000 sheep polyclonal antiserum to MSP (21), or a monoclonal antibody against α-tubulin (AB-1, 0.1 μg/ml; Oncogene, San Diego, CA) followed by peroxidase-conjugated affinity purified anti-goat (1:25,000), anti-sheep (1:10,000), or anti-mouse (1:10,000) IgG (Kirkegaard & Perry Laboratories, Gaithersburg, MD). Blots were developed using an enhanced chemiluminescence kit (Amersham Pharmacia, Arlington Heights, IL), and proteins were detected with Fuji Imager (Fuji Photo Film Co., Ltd., Valhalla, NY) or X-ray films (Kodak, Rochester, NY). Quantitation of bands was performed using Image Gauge software (Fuji Photo Film Co., Ltd.).
GC-MS analysis of sterols.
Sterol extraction from L. infantum chagasi promastigotes was performed according to a published protocol (22). Briefly, 1 × 109 promastigotes were washed three times by centrifugation in phosphate-buffered saline (PBS) (GIBCO) and rotated for 24 h at 4°C in a mixture of dicholoromethane and methanol (2:1 ratio [vol/vol]; Sigma) at a cell density of 2 × 108 cells/ml. Extracted sterols were recovered from the supernatants of two sequential centrifugations at 1,000 × g for 10 min at 4°C followed by 11,000 × g for 1 h at 4°C. After evaporation of the organic solvents, aliquots were stored at −20°C until use. Sterols were saponified by incubation in 0.5 ml of 30% KOH in methanol at 80°C for 2 h and extracted twice with an equal volume of petroleum ether (Sigma), followed by evaporation of solvents. Dichloromethane (50 μl) and two volumes of bis(trimethylsilyl)trifluroacetamide (BSTFA) (Sigma) were added to the residue in a Reacti-Vial Small Reaction vial (Pierce, Rockford, IL) and incubated at 80°C for 1 h. The resultant derivatized trimethylsilyl derivatives were dried under nitrogen in an N-EVAP analytical evaporator (Associates Inc., South Berlin, MA). The solvent decane was added, and the samples were analyzed by gas chromatography-mass spectrometry (GC-MS).
A ThermoFinnigan Voyager single quadruple mass spectrometer interfaced with a Trace2000 GC was used for the GC-MS analysis of sterols. A DB-5ht capillary column (inner diameter, 0.25 mm; length 30 m) (JW Scientific Inc., Folsom, CA) was used. The GC temperature program settings were 175°C for 1 min, ramp up at 10°C/min to 280°C, and hold at 280°C for 30 min. The GC inlet temperature was set at 280°C. The mass spectrometer was set to detect a mass range 45 to 700 Da. Electron ionization at 70 eV was used to ionize the GC eluent. ThermoFinnigan's Xcalibur software was used for both data acquisition and processing.
For sterol identification, two methods were utilized. First, the GC retention times of sample peaks were compared with those of the known standards cholesterol and ergosterol of chromatograph grade (Sigma). Second, the mass spectrum of each GC peak was searched against the small-molecule library of the National Institute of Standards and Technology library (NIST). The library was the NIST/EPA/NIH Mass Spectral Library (2001 version) that was purchased with the GC-MS, and the library search program was part of its operating software, Xcalibur. A sterol was positively identified if its mass spectrum fairly matched up with one in the library (score of SI and RSI matching factors of ≥800) with a probability score of ≥70. Details about SI, RSI, and probability may be found in the Library Search Results window of Xcalibur. For absolute quantification of sterols by MS, unknowns were compared to a standard curve generated with various amounts of cholesterol deuterated at six positions ([2H]cholesterol; Sigma) and various amounts of chromatograph-grade ergosterol (Sigma) with each experiment. Known amounts of [2H]cholesterol were also spiked into each sample, and samples were analyzed at three different cell equivalents to facilitate comparison to the standard curve. The quality of each GC-MS analysis was closely monitored, and samples and standards were rerun if a correlation factor of the standard curve in an individual run was less than 0.98. Due to a lack of available deuterated standards for ergosterol, it was not feasible to directly measure absolute amounts of ergosterols identified in Leishmania spp. Instead, a combination of an absolute amount of cholesterol and the relative amount of each positively identified sterol from the same sample was used to extrapolate the absolute amounts of ergosterol sterols. The peak areas were used to generate these extrapolated values.
Complement-mediated lysis.
Peripheral blood was collected from three healthy adult human donors who had not been exposed to Leishmania infections, and serum was separated by centrifugation. Sera were pooled in aliquots and stored at −80°C until a single use. Metacyclic promastigotes (1 × 107) were incubated in 75 μl of serial 1:2 dilutions of pooled sera in PBS, ranging from 3.1% to 50.0% serum, for 30 min at 37°C. Controls were incubated under parallel conditions with PBS alone. After incubations, each batch of cells was diluted in 1 ml PBS, followed by incubation in propidium iodide (Sigma) at a final concentration of 1 μg/ml. Viable cells that excluded propidium iodide were quantified by flow cytometry on a Becton Dickinson FACScan (BD, Franklin Lakes, NJ). Positive control cells were obtained by incubation in 100 mM hydrogen peroxide (H2O2) for 30 min at 37°C (23). Hydrogen peroxide-treated cells had an average of 5.6% ± 2.7% viable promastigotes (n = 4) under these conditions.
Macrophage infections.
Monocyte-derived macrophages (MDMs) were derived from the peripheral blood of healthy human donors. Peripheral blood mononuclear cells were isolated by centrifugation on Ficoll-Hypaque (Sigma), washed twice in RPMI 1640, and resuspended in RPMI 1640 supplemented with 10% FCS, 50 μg streptomycin/ml, and 100 U penicillin/ml (24). Monocytes were allowed to differentiate to MDMs by cultivation on petri dishes at 37°C in 5% CO2 for 5 to 7 days.
Bone marrow-derived macrophages (BMMs) were generated and cultured as previously described (25). Briefly, bone marrow cells derived from femurs of female BALB/c mice were cultured at 37°C with 5% CO2 in RPMI 1640 supplemented with 10% heat-inactivated FCS, 2 mM l-glutamine, 100 U of penicillin/ml, and 50 μg of streptomycin/ml. Twenty percent of cell culture supernatant from L929 cells (American Type Culture Collection, Manassas, VA) was added as a source of macrophage colony-stimulating factor. The cells were allowed to differentiate in 100-mm by 20-mm polystyrene-treated tissue culture dishes (Corning, Lowell, MA) for 7 to 9 days at 37°C.
Prior to experiments, nonadherent cells were removed by rinsing in PBS, and adherent MDMs or BMMs were released from the plates with 2.5 mg trypsin/ml plus 1 mM EDTA (GIBCO). After 2 washes, 5 × 105 macrophages were allowed to adhere to coverslips in 24-well plates overnight at 37°C with 5% CO2, followed by infection with opsonized metacyclic promastigotes at a multiplicity of infection (MOI) of 5:1. Parasites were opsonized either in 5% fresh C5-deficient human serum (GIBCO) or in 5% murine AJ serum for 30 min at 37°C for experiments with human or murine cells, respectively. Synchronized infections were achieved by centrifugation (330 × g, 4°C, 3 min) followed by incubation at 37°C with 5% CO2 for 30 min. Extracellular parasites were removed by rinsing twice with PBS. Cells were incubated in fresh RP-10 until the appropriate time points. Three replicate coverslips per condition were fixed and stained with Diff-Quik at each time point (Fisher Scientific, Pittsburgh, PA).
Mouse infections.
Four- to 6-week-old female BALB/c mice (Harlan Laboratories, Indianapolis, IN) were infected by tail vein injection of 1 × 106 control or MβCD-treated metacyclic promastigotes. They were euthanized 4 or 8 weeks later, and the parasite loads in livers and spleens were determined by microscopic enumeration of infected macrophages in Giemsa-stained touch preparations and by organ weights as described previously (26, 27). Parasite loads were also quantified by extraction of total tissue DNA, using a TaqMan assay amplifying Leishmania kinetoplastid DNA (kDNA). Primers and probes for the single-copy mouse gene encoding tumor necrosis factor alpha (TNF-α) served as validation that the PCR was working. Threshold cycle (CT) values were compared to a standard curve prepared from promastigote DNA extracted from the same L. infantum chagasi strain to calculate parasite equivalents as described previously (28). DNA was extracted from livers and spleens using a DNA extraction kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. To maximize DNA yields, an overnight incubation in cell lysis buffer with protease K was performed before the purification steps. DNA was eluted in 50 μl of distilled water and stored at −80°C until use.
RESULTS
Increased sensitivity to dose-dependent complement-mediated lysis after sterol depletion.
L. infantum chagasi metacyclic promastigotes were isolated from late-stationary-phase promastigote cultures using discontinuous Ficoll gradients as previously described (17, 18) and were left untreated (control) or partially depleted of membrane sterols by 60 min of incubation in increasing concentrations of MβCD (10 to 50 mM). Subsequent measure of viability with propidium iodide and flow cytometry suggested that control promastigotes were highly resistant to complement-mediated lysis. Consistent with our earlier report, 87.4% ± 0.1% of the cells were viable after exposure to 50% fresh human serum for 30 min at 37°C (Fig. 1A) (18). Cells treated with 10 mM MβCD were nearly the same as controls, exhibiting 85.3% ± 4.9% viability in 50% serum. However, cells exposed to 25 or 50 mM MβCD were more sensitive to complement, with 44.5% ± 7.7% (P < 0.05) or 27.0% ± 2.3% (P < 0.001) viability in 50% serum, respectively (Fig. 1A). Furthermore, variance of cells treated with the maximal 50 mM MβCD seemed to decrease dramatically in comparison with that of cells treated with lower concentrations. It is plausible that untreated cells with a range of sterol amounts as shown later effectively result in a heterogeneity of the response to serum among a population of cells, whereas treatment with the high MβCD concentrations eliminates that variable; i.e., the cell population is more homogeneous with regard to sterol concentration.
Fig 1.

Sterol depletion affects complement resistance but not viability of L. infantum chagasi metacyclic promastigotes. (A) Dose-dependent sensitivity to complement-mediated lysis. Metacyclic promastigotes were treated for 1 h with 0 (control), 10, 25, or 50 mM MβCD, followed by incubation in 1:2 serially diluted fresh pooled human sera. Cell viability was assayed by exclusion of membrane-impermeative propidium iodine measured by flow cytometry. Averages and standard deviations along with P values significant by the Student t test (n = 4) between untreated control and MβCD treatment are presented. (B) Similar cell density at the onset of logarithmic and stationary phases. Control or MβCD (25 mM, 1 h)-treated metacyclic promastigotes were incubated in cell culture medium at an initial density of 2 × 106 cells/ml (day 0). Cells were microscopically enumerated daily. Averages and standard deviations from four independent experiments are shown. Logarithmic growth is defined as the beginning of a 24-h period in which the cells increase >2-fold in the next 24 h. Stationary phase is defined as the beginning of a 24-h period in which cultures either increase by less than 25% or decrease in density over 24 h. No statistical differences were found between the cell densities of control or MβCD-treated parasites at beginning of the logarithmic (P > 0.2) or stationary (P > 0.6) phase of growth. (C) Growth curves of untreated and MβCD (25 mM)-treated metacyclic promastigotes. Results from two of four independent experiments are presented. (D) Cholesterol supplementation does not reverse the MβCD effect. Metacyclic promastigotes were incubated as described for panel A in 0 (control) or 25 mM MβCD in the absence (MβCD) or presence of various concentrations of water-soluble cholesterol (Cho) as indicated. Results of a representative of three independent experiments are shown.
Several lines of evidence suggested that the sensitivity of MβCD-treated promastigotes to complement-mediated lysis was not due to reduced cell survival caused by sterol depletion. First, MβCD-treated cells remained similarly capable of excluding the membrane-impermeative dye propidium iodide as controls did, suggesting little change in membrane integrity (Fig. 1A and D, 0% serum). Second, MβCD-treated promastigotes survived as well in low concentrations, up to 6.3%, of normal human serum as did control cells (Fig. 1A and D). Third, in vitro, control or MβCD-treated cells in HOMEM, monitored daily by microscopic enumeration, reached similar cell densities measured at the beginning of logarithmic or stationary phase of growth (P > 0.2) (Fig. 1B). A lag phase of 2 days was observed prior to the onset of logarithmic growth of stationary-phase promastigotes cultures (see Fig. S1 in the supplemental material), which is consistent with our previous data (13, 14). Interestingly, the usual logarithmic phase of 3 to 5 days observed prior to the onset of logarithmic growth of metacyclic promastigote cultures was shortened by 1 full day in MβCD-treated metacyclic promastigotes (Fig. 1C). The reasons for this interesting observation are not known, but the finding could imply that the membrane sterol content affects the maintenance of parasites in the virulent metacyclic form. Finally, we have previously shown that the rates of protein synthesis, as measured by incorporation of [35S]methionine into newly synthesized proteins, are very similar for MβCD-treated cells and untreated controls (19).
Incubation in an excess of water-soluble cholesterol (stoichiometrically chelated to MβCD) has been shown to replenish and restore function to cholesterol-depleted rat hepatoma cells, Chinese hamster ovary cells, the murine macrophage cell line J774A.1, and BMMs (25, 29, 30). We therefore investigated whether exogenous cholesterol would restore complement resistance of MβCD-treated Leishmania spp. Metacyclic L. infantum chagasi promastigotes were treated with 25 mM MβCD in the absence or presence of 100, 200, or 400 μg water-soluble cholesterol/ml, after which they were assessed for viability in the presence of complement. Unexpectedly, all MβCD-treated cells remained equally sensitive to complement lysis despite incubation in water-soluble cholesterol (Fig. 1D), suggesting that cholesterol repletion was insufficient to reverse the effects of sterol depletion from Leishmania promastigotes. Leishmania spp. are more closely related to yeast than to mammalian cells in their membrane sterol composition. Similar to yeast cells, Leishmania contains more ergosterol-like sterols (∼60 to 80%) than cholesterol (∼10 to 30%). Ergosterol is found in lipid rafts (31–34). This led us to hypothesize that the physiological effects of MβCD may be in large part caused by extraction of ergosterol-like sterols as opposed to cholesterol from metacyclic promastigote membranes. Unfortunately water-soluble ergosterol could not be prepared to rigorously test this hypothesis by ergosterol repletion.
Sterol identification by GC-MS.
Cauchetier and colleagues successfully identified more than a dozen sterols in logarithmic-phase L. infantum promastigotes using GC-MS to analyze sterols extracted with dichloromethane-methanol (22). The compounds identified included ergosterol and its biosynthetic pathway precursors. We applied the same technique to extract, identify, and quantify sterols of control or MβCD-treated L. infantum chagasi promastigotes. Five sterols were consistently identified in metacyclic promastigotes during repeated measurements (n = 25). Comparison with known standards showed that these compounds corresponded to cholesterol, two ergosterol isoform stereoisomers termed 1 and 2, ergosta-7,22-dien-3β-ol, and stigmasta-7-24(28)-dien-3β-ol (see Fig. S2 in the supplemental material), and they are indicated as peaks I to V, respectively, on the chromatogram (Fig. 2A).
Fig 2.

Cholesterol depletion of L. infantum chagasi metacyclic promastigotes by MβCD treatment. (A) Representative full GC outputs of sterols extracted from untreated control metacyclic promastigotes with a load of 2 × 105 cell equivalents. The five peaks labeled I to V were identified as sterols. The retention time (RT) and area (AA) of each sterol are shown. (B) MβCD treatment decreases cholesterol levels in metacyclic promastigotes. The absolute amount of cholesterol (ng/1 × 107 cell equivalents) of each sample was directly measured, whereas the relative amount was set as one arbitrary unit for controls and was calculated as ratios to corresponding controls for MβCD (25 mM)-treated samples. P values were obtained using the Student t test (n = 5).
Significant reduction in cellular sterol levels by sterol depletion.
As described above, MβCD treatment rendered L. infantum chagasi metacyclic promastigotes sensitive to complement-mediated lysis with little effect on cell viability in vitro. We therefore felt it important to document the exact sterol contents before and after MβCD treatment. First, we tested whether [2H]cholesterol could be used as an internal standard (IS) for quantification. In four independent analyses, [2H]cholesterol had a maximum of 0.7% overlap with chromatography-grade [1H]cholesterol. These data showed that [2H]cholesterol could be used as a reliable IS. Furthermore, an almost perfect correlation (r2 > 0.99) between the cholesterol amount/IS amount ratio and of cholesterol area/IS area ratio was achieved in each experiment, which convincingly showed that the area of each sterol in the GC-MS spectrum precisely correlates with amount of sterols.
GC-MS analysis was performed on 3 different dilutions of each unknown sample, and sterols per cell equivalent were calculated independently from each dilution. Because of the anticipated higher sterol concentration in untreated cells than in MβCD-treated cells, the numbers of cells in each run were adjusted to obtain results in the range of the standard curve. Data are expressed per 107 cell equivalents, and the average measured amounts of cholesterol are shown in Fig. 2B. The cholesterol content and the extrapolated levels of ergosterol and ergosterol-like sterols are summarized in Table 1. As expected, MβCD treatment significantly reduced sterols in L. infantum chagasi metacyclic promastigotes. Total cholesterol was reduced by 63.9% (P < 0.05). Total ergosterol (both steroisomers) was reduced by 46.5% (P < 0.05), and the other two ergosterol-like sterols were reduced between 60.9% and 74.7% (P < 0.05). Total cellular sterols were reduced by 67.3%, considering all five sterols (P < 0.01) (Table 1).
Table 1.
Sterol contents in control (n = 5) and MβCD-treated (n = 5, 25 mM) L. infantum chagasi metacyclic promastigotes
| Sterol | Peak(s) in Fig. 2A | Mean ng/1 × 107 cells (SD) in: |
Mean % reduction (SD)a | |
|---|---|---|---|---|
| Control cells | MβCD-treated cells | |||
| Cholesterol | I | 248.9 (120.8) | 91.0 (45.0) | 63.9 (10.1)* |
| Total ergosterol | II + III | 98.5 (34.4) | 49.8 (31.6) | 46.5 (32.6)* |
| Ergosterol isoform 1 | II | 68.4 (13.1) | 39.9 (24.9) | 44.5 (31.9)* |
| Ergosterol isoform 2 | III | 30.0 (27.8) | 10.0 (9.5) | 60.4 (17.8) |
| Ergosta-7,22-dien-3β-ol | IV | 318.9 (137.8) | 83.6 (41.5) | 74.7 (6.5)** |
| Stigmasta-7,24(28)-dien-3β-ol | V | 50.4 (8.3) | 21.1 (6.3) | 60.9 (25.3)* |
| Total | 704.0 (281.2) | 241.4 (121.6) | 67.3 (12.3)** | |
*, P < 0.05; **, P < 0.01.
Reduced infectivity of promastigotes for BALB/c mice after sterol depletion.
Well-studied virulence factors of Leishmania include MSP and LPG, both of which are GPI anchored and are reported to localize in promastigote membrane rafts. We therefore hypothesized that membrane sterols are important for maintaining the virulence of Leishmania promastigotes. To test this hypothesis, we extracted membrane sterols with MβCD, leaving the promastigotes with transiently lowered membrane cholesterol and ergosterol levels. BALB/c mice were infected intravenously (i.v.) either with L. infantum chagasi metacyclic promastigotes that had been treated with 25 mM MβCD or with untreated controls. Four weeks after parasite inoculation, mice were euthanized and parasite loads were determined by quantitative PCR (qPCR) or by microscopic examination of Wright-Giemsa-stained liver/spleen impression smears (Fig. 3A and B). The qPCR data showed that sterol depletion caused a two-thirds decrease in parasite loads, which was significant in spleens and approached significance in livers. Microscopic examination verified that significantly fewer parasites were observed in liver impression smears from mice that were treated with MβCD (1.6 × 107 ± 0.7 × 107/organ) than in those from untreated controls (4.0 × 107 ± 0.5 × 107/organ), and this difference reached statistical significance (P < 0.001). Parasites were below the microscopic detection level in spleens. The same pattern was observed after 8 weeks of infection. Mice infected for 8 weeks with MβCD-treated L. infantum chagasi harbored 8.7 × 108 ± 2.3 × 108 and 8.0 × 109 ± 4.5 × 109 parasites in their spleens and livers, respectively, compared to 1.1 × 109 ± 0.3 × 109 and 4.5 × 1010 ± 2.9 × 1010 parasites in the spleens and livers, respectively, of mice infected with control parasites. Thus, murine infection with sterol-depleted promastigotes resulted in a sustained lower parasite burden than that with untreated promastigotes.
Fig 3.
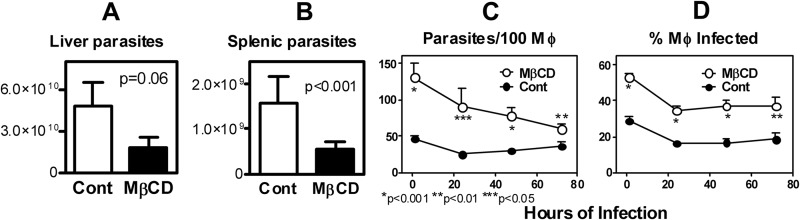
Sterol depletion of metacyclic promastigotes affects the outcome of infection. (A and B) Parasite loads in livers and spleens of BALB/c mice infected with metacyclic promastigotes with no treatment (open bars) or treated with 25 mM MβCD (filled bars). Parasite loads per organ were determined by qPCR at 4 weeks postinfection. (C and D) Opsonized metacyclic promastigotes of untreated control or MβCD treatment (25 mM, 1 h) groups were used to infect human MDMs at an MOI of 5. Infections of macrophages by control (filled) or treated (open) parasites were microscopically quantified daily until 72 h postinfection. (C) Number of parasites per 100 macrophages. (D) Percentage of macrophage infection. Results from one representative of three independent experiments are shown.
Infection of macrophages with sterol-depleted promastigotes.
The above intriguing in vivo observations were followed by studies of cultured macrophages to determine whether the sterol-dependent virulence characteristics of promastigotes are evident in isolated host phagocytes. Human MDMs were infected either with MβCD-treated, sterol-depleted promastigotes or with untreated control parasites (Fig. 3C and D). Unexpectedly, both the number of promastigotes associated with macrophages and the percentage of macrophages infected with MβCD-treated metacyclic promastigotes were significantly higher than those for MDMs incubated with untreated promastigotes. This was observed at all time points up to 72 h postinfection, although the kinetics suggested that the major difference occurred at the step of attachment (Fig. 3C and D). This difference at attachment was confirmed in parallel experiments using cytochalasin D to inhibit phagocytosis (25). The cytochalasin D experiment revealed that approximately twice the number of MβCD-treated promastigotes as untreated parasites attached to the surface of macrophages (61.5 ± 12.7 and 31.6 ± 5.0, respectively [n = 3]; P < 0.05) (Fig. 4, MβCD versus control at 0.5 h). Furthermore, after the initial killing usually observed after 24 h in control parasites, untreated parasites survived and began to replicate over 72 h of MDM infection, whereas MβCD-treated parasites progressively declined (Fig. 3 and 4, 24 h versus 72 h). These data suggest the MβCD effect on macrophage infections occurs at the step of macrophage attachment. Studies using murine BMMs showed similar results (n = 2; data not shown).
Fig 4.
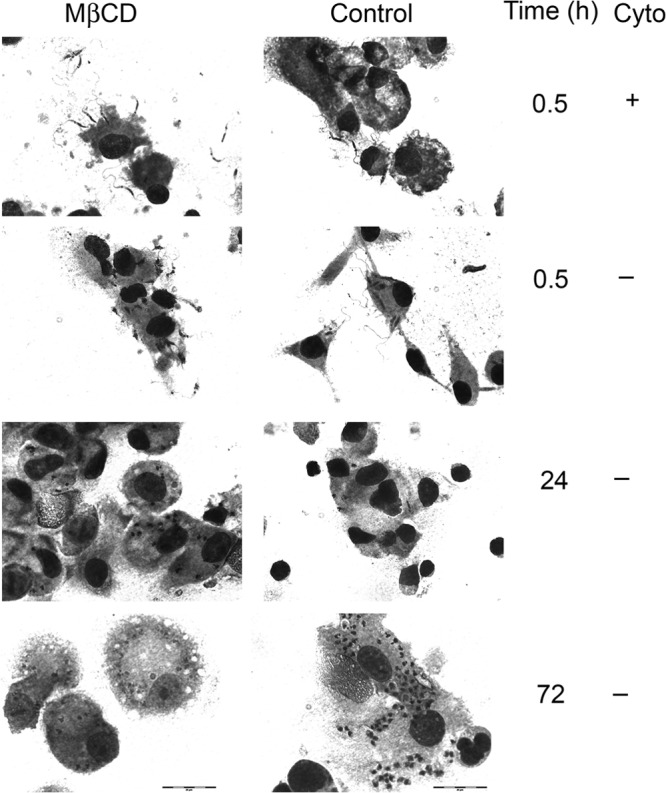
Effect of MβCD on L. infantum chagasi metacyclic promastigote attachment to and survival in macrophages. Human MDMs were infected at an MOI of 5 with opsonized metacyclic promastigotes of untreated control or MβCD-treated (25 mM, 1 h) groups in the absence (−) or presence (+) of cytochalasin D (Cyto) and fixed 30 min later to assess attachment or 24 and 72 h later to assess survival. Slides stained with Wright-Giemsa stain demonstrate a lack of internalized parasites in the presence of cytochalasin D. Scale bar, 20 μM.
The studies with the results shown in Fig. 3 and 4 were done with complement-opsonized metacyclic promastigotes that were either treated or not with MβCD. Because promastigotes are known to ligate macrophage complement receptors, we addressed whether the increased MDM entry by sterol-depleted promastigotes might be due to a differential complement opsonization following MβCD treatment. To this end, we compared whether MβCD treatment still augmented entry of nonopsonized metacyclic promastigotes into MDMs. Consistent with Fig. 3, a higher proportion of MDMs internalized promastigotes that had been treated with MβCD and opsonized than promastigotes that were opsonized without MβCD treatment at 30 min postinfection (51.8% ± 7.4% versus 31.9% ± 5.3% [mean ± standard deviations], respectively; P = 0.05). In contrast, significant differences were not observed between MβCD-treated and control nonopsonized parasites (40.7 ± 7.0 versus 46.3 ± 12.3, respectively; P = 0.58). These data are consistent with a model in which sterol depletion with MβCD augments or alters subsequent opsonization with at least some complement components, such that promastigote attachment to and entry into MDMs are enhanced.
Complement deposition on the surface of sterol-depleted or control parasites.
Serum-opsonized Leishmania promastigotes have been found to ligate two complement receptors on the surface of macrophages: CR3, which binds the inactive C3b derivative iC3b, and CR1, which binds C3b but also has a low affinity for iC3b (35–37). Phagocytosis stimulated through ligation of different receptors can lead to different rates or pathways of intracellular trafficking and consequent parasite survival (38). As such, if sterol content affects the type or amount of complement opsonization, this could influence parasite entry and survival.
Incubation of control or MβCD-treated parasites in 50% fresh human serum led to different patterns of immunoreactive C3 deposition (Fig. 5). Control parasites bound C3 as indicated by the 75-kDa β chain that is common to both C3b and iC3b and by the slowly migrating high-molecular-weight adducts of C3 covalently bound to their targets (lane 3). In contrast, MβCD-treated parasites demonstrated a paucity of high-molecular-weight adducts and demonstrated a prominent 110-kDa band consistent with the presence of the α′ chain of C3b (lane 5). To better understand the composition of C3 fragments bound to the parasites, we treated the samples with hydroxylamine to release ester-bound α-chain fragments. Consistent with the successful removal of these C3 fragments, the signals of the α′ and α1′ chains (110 kDa and 67 kDa, respectively) were increased by hydroxylamine treatment (lanes 4 and 6). To estimate the proportion of bound C3 fragments present as iC3b, we quantified the ratio of the 67-kDa α1′ chain (present in iC3b) to the 110-kDa α′ chain of C3b. Comparison of these ratios in lanes 4 and 6 revealed a greater proportion of iC3b on the control parasites (α1′/α′ ratios of 4.8 and 0.4 on the control and MβCD-treated parasites, respectively). The presence of bands that ran more slowly than 110 kDa in lane 4 suggests that hydroxylamine was unable to remove all α1′ and α2′ fragments from the control parasites. This observation is consistent with the data of Puentes et al., who noted that a majority of C3 bound to L. donovani promastigotes was iC3b and was bound via an amide linkage, which is not sensitive to cleavage by hydroxylamine (39). Unlike the 67-kDa and 110-kDa fragments of the α chain, the 75-kDa β chain is not covalently bound to its target, and thus its detection does not depend on hydroxylamine treatment. Comparison of the ratios of the 67-kDa α1′ chain to the 75-kDa β chain for lanes 4 and 6 (2.7 versus 1.2) was similarly consistent with a greater proportion of C3 present as iC3b on the control parasites. The ratios of the 110-kDa α′ chain of C3b to the β chain were 0.6 and 3.0 for the control and MβCD-treated parasites, respectively, consistent with a corresponding predominance of C3b on the MβCD-treated parasites. Taken together, these observations suggest that MβCD treatment alters the surface of L. infantum chagasi such that the parasite is less able to inactivate bound C3b to iC3b.
Fig 5.

C3 Western blotting of MβCD-treated cells versus untreated controls. One hundred nanograms of C3b and iC3b were loaded in lanes 1 and 2, respectively. Metacyclic promastigotes were incubated in a 50% concentration of the indicated sera for 30 min at 37°C and washed. Lanes 3, 4, and 7 were loaded with the untreated control, whereas lanes 5, 6, and 8 were loaded with metacyclic promastigotes that had been pretreated with 50 mM MβCD for 1 h. The membrane was probed with anti-C3 (C3) followed by α-tubulin (α-tub) as a loading control for samples prepared from metacyclic promastigotes (lanes 3 to 8). +HA, hydroxylamine treatment; PHS, pooled healthy human sera; HI, heat inactivated. Results from a representative of two independent experiments are shown. Quantification of C3 is shown at the bottom of each lane and is normalized to the 75-kDa band in lane 3. The C3 fragments present in each control complement protein are also indicated.
Proteins released from L. infantum chagasi metacyclic promastigotes after sterol depletion.
Surface-localized MSPs of Leishmania spp. are anchored to the plasma membrane through a GPI anchor. Other studies have found that GPI-anchored proteins partition mainly in membrane lipid subdomains enriched in cholesterol (9, 11, 40). We previously showed that stationary-growth-phase promastigotes depleted of sterols with MβCD displayed enhanced release of surface-localized MSP, whereas internal MSP stores remained unchanged (19). This enhanced release of the surface MSP was not due to a detrimental effect on cell viability, since both untreated and treated stationary-phase promastigotes incorporated similar levels of 35S-labeled radioisotopes into newly synthesized proteins (19). MβCD is known to extract cholesterol from membrane lipid bilayers by chelating sterols at its hydrophobic core (29, 41). We have demonstrated in Table 1 that MβCD also extracts ergosterol-like sterols from L. infantum chagasi promastigotes. We hypothesized that metacyclic promastigotes from which surface sterols were depleted by MβCD would release lipid-associated virulence determinants, including MSP, partially explaining the parasite attenuation observed in Fig. 3A and B. To test this hypothesis, cell-associated and released MSPs after MβCD treatment were investigated by Western blotting. There was a dose-dependent enhanced release of cell-associated MSP into the extracellular medium after promastigotes were exposed to MβCD. Specifically, fewer than 0.1% of MSPs were found in the supernatants of untreated stationary-phase promastigotes, whereas 37%, 44%, 52%, or 77% were found in the extracellular medium after the cells were exposed to 4, 6, 8, or 10 mM MβCD for 48 h, respectively (Fig. 6A).
Fig 6.

Released proteins from sterol-depleted promastigotes. (A) Stationary-phase promastigotes were treated with the indicated concentration of MβCD (0 to 10 mM) for 48 h. Filtered supernatants (S) and cells (C) were subjected to SDS-PAGE. MSP was detected by Western blotting (top panel). The same filter was reprobed with monoclonal antibody to α-tubulin (bottom panel) for a loading control. A total of 1 × 107 cell equivalents were loaded. The percentage of MSP in the supernatants of each treatment is shown. Results from a representative of two experiments are presented. (B) Released proteins were directly collected from the extracellular medium of metacyclic promastigotes that were untreated (lane 2) or subjected to 25 mM MβCD treatment (lane 1) for 1 h. A silver-stained SDS-polyacrylamide gel is shown. A total of 8 × 108 cell equivalents were loaded. There is an empty lane between lanes 1 and 2. Gel slices were excised for LC-MS/MS analysis (arrowhead), and the results are summarized in Table 2.
We have previously shown that stationary-phase promastigotes treated with 15 mM MβCD for 3 h release 34.6% more surface-located MSP than untreated control cells in experiments utilizing surface biotinylation followed by streptavidin affinity purification and immunoprecipitation (19). Here, released proteins were concomitantly collected from the cell-free supernatants of metacyclic promastigotes treated with buffer or MβCD and subjected to SDS-PAGE and silver staining (Fig. 6B). Proteins extracted from gel slices at 60 to 63 kDa were subjected to LC-MS/MS for protein identification to discover whether more MSP isoforms are released and to identify released MSP isoforms. In total, three MSPs, one MSP-like protein (MLP), and six other proteins with predicted masses ranging from 49.5 to 75.5 kDa were identified (Table 2). Dihydrolipoamide dehydrogenase and paraflagellar rod protein 1D were found to be released only from MβCD-treated and not from nontreated cells, indicating that at least some proteins were additionally released by sterol extraction. Other proteins released were present in both supernatants, although the method does not quantify amounts. We have already published the fact that MSPs are released from wild-type cells into medium (13, 19), and it was thus not surprising to discover three MSPs among the proteins released from both nontreated and MβCD-treated metacyclic cells. Sterol depletion did not result in release of additional MSP isoforms, suggesting that our immunoblots (Fig. 6A) reflected merely enhanced quantities, but not more isoforms, of released MSP. Several unanticipated proteins were identified in the mixture. These included MLP, confirming first that it is expressed as protein and second that it is released from infective metacyclic promastigotes. Since MLP is a novel secreted potential virulence-related protein, more information about MLP and its sequence is included in Table 3 and in Fig. S3 in the supplemental material.
Table 2.
Released proteins identified by LC-MS/MS of the gel slices excised at 60- to 63-kDa fractions of SDS-polyacrylamide gelsa
| Protein | Accession no. | Expected mass (Da) | pI | GPI anchor | Coverage (% AA) |
|
|---|---|---|---|---|---|---|
| MβCD-treated cells | Control cells | |||||
| ATPase β subunit | A4HE61 | 56,362.9 | 5.07 | No | 17.9 | 14.7 |
| Carboxypeptidase | A4HVE5 | 57,101.9 | 5.37 | No | 15.1 | 9.7 |
| Dihydrolipoamide dehydrogenase | Q6S4V7 | 50,586.7 | 6.42 | No | 6.7 | |
| Glucose-6-phosphate isomerase | P42861 | 67,245.8 | 6.19 | No | 4.8 | 11.6 |
| MSPL | P15706 | 63,808.6 | 6.83 | Yes | 18.2 | 21.2 |
| MSPS2 | A4HUF6 | 63,517.4 | 6.96 | Yes | 11.5 | 7.0 |
| MSPS4 | A4HUG0 | 75,528.9 | 8.88 | Yes | 6.0 | 5.0 |
| MLP | A4I3D1 | 60,546.0 | 7.41 | No | 10.1 | 9.2 |
| Paraflagellar rod protein 1D | A4HIY0 | 69,219.2 | 5.29 | No | 4.5 | |
| Surface antigen protein 2 | A4HV45 | 43,941.8 | 5.31 | Yes | 7.7 | 12.5 |
Protein bands in the gels were visualized by silver staining. Loaded in the gels were concentrated extracellular proteins released from L. infantum chagasi metacyclic promastigotes that were either treated with 25 mM MβCD or incubated in buffer.
Table 3.
Unique peptides of L. infantum chagasi metacyclic promastigotes found in MLP and MSP by LC-MS/MS
| Protein | Peptidesa |
|---|---|
| MLP | VTCSAADVLTR, VLLELLIPSAVQLHQER, ENGNIVVSPFIK, SNLPTYFQYFGDPRLGGPDPLMDFCPFVR |
| MSPL | DICTAEDILTD, ILVKHLIPQALQLHTER, VQDKWKVTGMG-, D-LPPYWQYFTDPSLAGISAFMDCCPVVE |
| MSPS2 | DICTAEDILTD, ILVKHLIPQALQLHTER, VQDKWKVTGMG-, S-LPPYWQYFTDPSLAGISAFMDCCPVVV |
| MSPS4 | TTCTAEDILD, ILVKHLIPQALQLHTER, VQDKWKVTGMD-, D-LPPYWQYFTDPSLAGISAFMDCCPVVE |
Dashes were inserted for maximizing alignment.
DISCUSSION
The sterol composition and sterol biosynthetic pathways of the Trypanosomatid protozoa, including Leishmania spp. and Trypanosoma spp., are similar to those of fungi (33, 42). In particular, these genera do not possess enzymes for cholesterol synthesis. Fungi instead synthesize and incorporate ergosterol into cell membranes (43, 44). Because many Leishmania virulence factors associate with the membranes through a GPI anchor and GPI-anchored proteins are often present in sterol-containing membrane domains, we explored what disruption of the unique membrane sterol content would do to parasite virulence. Using GC-MS, we documented the presence of five sterols in the surface membranes of L. infantum chagasi metacyclic promastigotes. Ironically, parasites do not contain the biosynthetic enzymes to generate cholesterol, ergosta-7,22-dien-3β-ol, or stigmasta-7-24(28)-dien-3β-ol (33), three sterols present in replicate assays. These sterols must therefore be derived by adsorption from the tissue culture environment. Ergosta-7,22-dien-3β-ol has been previously detected by GC-MS in Leishmania donovani and Leishmania amazonensis (45–47) as well as in the fungi Saccharomyces cerevisiae and Ganoderma pfeifferi (48, 49), whereas stigmasta-7-24(28)-dien-3β-ol has been found in the fungus Gymnosporangium juniper (50). Importantly, our data documented the fact that MβCD successfully reduced the content of all sterols by 67.3% and reduced ergosterol in particular by 46.5%. Therefore, we used MβCD-treated promastigotes to explore the functional effects of sterol variations on parasite infectivity.
Two of the GPI-anchored Leishmania virulence factors are LPG and MSP, both of which have been found in specialized lipid-enriched membrane microdomains (9–11). We have found that transient incubation with the cholesterol-chelating reagent MβCD depleted sterols of metacyclic promastigotes by two-thirds and released several lipid-associated proteins, including MSPs. Our data revealed that sterol-depleted metacyclic promastigotes were attenuated as defined by reduced parasite loads in a murine model and enhanced susceptibility to complement-mediated lysis. Unexpectedly, disruption of parasite membrane sterols enhanced the rate of promastigote binding to macrophages in the presence of serum complement, although parasite replication within macrophages was defective.
C3 immunoblots suggested that promastigotes partially depleted of membrane sterols with MβCD were readily opsonized with C3b but that inactivation of C3b to iC3b was less than that seen on control parasites. C3b forms an integral part of the C5 convertase and thus can contribute to assembly of the membrane attack complex and subsequent parasite lysis (51). In contrast, iC3b, although it retains opsonic activity, cannot participate in the formation of complement convertases. Thus, our observations may explain the greater degree of promastigote complement sensitivity after sterol depletion. Furthermore, a decreased abundance of ligand for the macrophage CR3 receptor (iC3b) on the promastigote surface would likely decrease the proportion of promastigotes ligating that receptor and possibly direct parasites to enter through different phagocytic receptors such as the mannose receptor. Phagocytosis through alternate receptors can alter the kinetics and efficiency of uptake and affect the ultimate survival of intracellular parasites, as illustrated in our recent report comparing avirulent to virulent parasite entry into macrophages (38). Similar to those in the prior study, our current observations may provide another example in which enhanced macrophage entry does not translate into enhanced intracellular survival (38). We hypothesize that the decreased abundance of MSP, a protease that cleaves C3b to iC3b, results in a greater proportion of C3b on treated parasites (12). We additionally hypothesize that MSP loss from the cell surface leads to the changes in complement opsonization that we observed in sterol-depleted parasites.
MSPs play a role in macrophage phagocytosis of promastigotes, possibly by serving as a ligand promoting attachment to macrophage receptor (52–54). One consequence of MβCD treatment of promastigotes is significantly diminished cellular MSPs (Fig. 6A). Rather than decreasing the rate of parasite binding to and uptake by macrophages, the binding was observed to increase (Fig. 3C and D and 4). One plausible explanation is that MβCD-treated cells bind to alternate macrophage receptors via ligands exposed by sterol depletion or via the enhanced opsonic C3, such as CR1 or mannose receptor (55). A parallel observation is made in the case of L. infantum chagasi logarithmic-phase promastigotes, which contain only one-fourteenth the level of steady-state MSPs of stationary-phase promastigotes (13) but contain only one-third of the MSPs of metacyclic promastigotes (18), yet their phagocytosis by MDMs is more efficient than that of metacyclic promastigotes (38).
Because MβCD works by chelating surface cholesterol/ergosterol and does not perturb sterol synthesis in the organism, one would anticipate that disruption of membrane lipids would be transient in parasites. As such, it is not inherently obvious why in vivo infection is lower throughout 8 weeks after inoculation of MβCD-treated compared to control promastigotes. Two possible mechanisms could contribute. First, MβCD-treated parasites would likely be killed more rapidly than controls when inoculated into mice by complement-mediated lysis, prior to entry into macrophages. This would not only lower the inoculation dose but also expose the host to extracellular dead or dying parasites early in the infection. Second, although the rate of macrophage entry is greater, the rate of killing seems to be enhanced over 72 h. Thus, it is conceivable that even those parasites that enter macrophages do not survive well intracellularly and are killed within host macrophages. Either mechanism would expose the host to products of dead or dying parasites. The sustained lower infection level after inoculation of MβCD-treated parasites may provide clues to events occurring in the earliest days and hours of infection that influence the ultimate immune response and outcome of infection.
It has been reported that most Leishmania spp., including L. infantum chagasi, L. donovani, L. amazonensis, L. panamensis, and L. major, become more resistant to complement during in vitro growth from logarithmic to stationary phase in cell culture, a process that reflects some of the changes that occur during parasite development to the virulent metacyclic form in the sand fly vector (56–59). The importance of MSP for promastigote resistance to complement (12) led us to investigate whether the isoforms, in addition to the abundance, of MSP are changed by sterol depletion. Mass spectrometric data of the 60- to 63-kDa fraction of released proteins showed that MSPs were indeed present in the mixture of proteins released from control or MβCD-treated promastigotes but that the isoforms did not differ. In addition to MSPs, two other promastigote surface proteins, MLP and surface antigen protein 2, were released by both untreated and MβCD-treated promastigotes. There were additional and more surprising released proteins in this fraction of released proteins, which were not predicted to be membrane associated. In particular, paraflagellar rod protein 1D released uniquely by promastigotes treated with MβCD does not have predicted GPI membrane anchors or transmembrane domains. Its enhanced release may implicate membrane sterol content in the directing of proteins toward nonclassical secretory pathways, such as the recently described release of Leishmania proteins through exosomes (60).
The release of MLP with MSPs raises the possibility that they too promote parasite virulence. Two MLP genes have been annotated on chromosome 28 of L. infantum and are 100% conserved in the coding region (LinJ28_V3.600 and LinJ28_V3.610), although significant differences exist in their untranslated regions (UTRs) (61). There are homologues of L. infantum MLP in L. donovani, L. major, and L. mexicana according to the annotated genomes (62–64). Interestingly, no homologous gene is found in the genome of the non-human-pathogenic species L. tarentolae (65). These MLPs do not have a putative C-terminal sequence for GPI anchor attachment and thus would not be anticipated in membrane or released fractions. MLPs bear minimum similarity to the MSPs, although they all contain the conserved HEXXHXXGXXH catalytic domain of the zinc metalloproteases (61, 63). Together these data confirm MLP expression and location in the plasma membrane of the infective metacyclic promastigotes, suggesting that it might be a virulence factor similar to MSPs.
Fungicidal chemicals such as ketoconazole, azasterol, and 2′,6′-dihydrooxy-4′methoxychalcone that target the ergosterol biosynthesis pathway have been shown to cause a depletion of normal sterol and an accumulation of abnormal sterol precursors in L. donovani, L. infantum, L. major, and L. amazonensis (31, 47, 66–70). It was further shown that depletion of membrane sterols by MβCD impaired action of the antileishmanial drug miltefosine on L. donovani promastigotes (71). The mechanisms of miltefosine action against Leishmania spp. have not well been defined, although its interference with phospholipid and sterol metabolism is proposed (71, 72). The current study accomplished sterol depletion by chelation rather than effects on sterol biosynthesis. The transient nature of this effect has enabled us to establish that sterol content is critical during the initial stages of Leishmania infection in host cells in vitro and live experimental animals. These data collectively lay a foundation for the use of sterol-depleted parasites to explore localized events at the parasite membrane that are critical for pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Melissa Miller, Bayan Sudan, and Yani Chen for maintaining parasites and infecting mice, to Michael Li for microscopic enumeration of slides of mouse and macrophage infections, and to Yalan Li for LC-MS/MS.
This work was supported by grants AI32135 and AI059451 (J.E.D. and M.E.W.) and AI45540 and AI048822 (M.E.W.) from the National Institutes of Health, by two Merit Review grants (C.Y. and M.E.W.), and by an MERP (C.Y.) and a Persian Gulf RFP (M.E.W.) from the Department of Veterans' Affairs.
Footnotes
Published ahead of print 29 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00214-13.
REFERENCES
- 1. Bates PA. 2007. Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int. J. Parasitol. 37:1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garcia LS. 2007. Diagnostic medical parasitology, 5th ed ASM Press, Washington, DC [Google Scholar]
- 3. Desjeux P. 2004. Leishmaniasis: current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 27:305–318 [DOI] [PubMed] [Google Scholar]
- 4. Blackwell JM, Fakiola M, Ibrahim ME, Jamieson SE, Jeronimo SB, Miller EN, Mishra A, Mohamed HS, Peacock CS, Raju M, Sundar S, Wilson ME. 2009. Genetics and visceral leishmaniasis: of mice and man. Parasite Immunol. 31:254–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beverley SM, Turco SJ. 1998. Lipophosphoglycan (LPG) and the identification of virulence genes in the protozoan parasite Leishmania. Trends Microbiol. 6:35–40 [DOI] [PubMed] [Google Scholar]
- 6. Chang KP, McGwire BS. 2002. Molecular determinants and regulation of Leishmania virulence. Kinetoplastid Biol. Dis. 1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yao C. 2010. Major surface protease of trypanosomatids: one size fits all? Infect. Immun. 78:22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yao C, Donelson JE, Wilson ME. 2003. The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol. Biochem. Parasitol. 132:1–16 [DOI] [PubMed] [Google Scholar]
- 9. Denny PW, Field MC, Smith DF. 2001. GPI-anchored proteins and glycoconjugates segregate into lipid rafts in Kinetoplastida. FEBS Lett. 491:148–153 [DOI] [PubMed] [Google Scholar]
- 10. Denny PW, Goulding D, Ferguson MA, Smith DF. 2004. Sphingolipid-free Leishmania are defective in membrane trafficking, differentiation and infectivity. Mol. Microbiol. 52:313–327 [DOI] [PubMed] [Google Scholar]
- 11. Zufferey R, Allen S, Barron T, Sullivan DR, Denny PW, Almeida IC, Smith DF, Turco SJ, Ferguson MA, Beverley SM. 2003. Ether phospholipids and glycosylinositolphospholipids are not required for amastigote virulence or for inhibition of macrophage activation by Leishmania major. J. Biol. Chem. 278:44708–44718 [DOI] [PubMed] [Google Scholar]
- 12. Brittingham A, Morrison CJ, McMaster WR, McGwire BS, Chang KP, Mosser DM. 1995. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J. Immunol. 155:3102–3111 [PubMed] [Google Scholar]
- 13. Yao C, Leidal KG, Brittingham A, Tarr DE, Donelson JE, Wilson ME. 2002. Biosynthesis of the major surface protease GP63 of Leishmania chagasi. Mol. Biochem. Parasitol. 121:119–128 [DOI] [PubMed] [Google Scholar]
- 14. Yao C, Luo J, Hsiao C, Donelson JE, Wilson ME. 2005. Internal and surface subpopulations of the major surface protease (MSP) of Leishmania chagasi. Mol. Biochem. Parasitol. 139:173–183 [DOI] [PubMed] [Google Scholar]
- 15. Yao C, Luo J, Storlie P, Donelson JE, Wilson ME. 2004. Multiple products of the Leishmania chagasi major surface protease (MSP or GP63) gene family. Mol. Biochem. Parasitol. 135:171–183 [DOI] [PubMed] [Google Scholar]
- 16. Berens RL, Brun R, Krassner SM. 1976. A simple monophasic medium for axenic culture of hemoflagellates. J. Parasitol. 62:360–365 [PubMed] [Google Scholar]
- 17. Spath GF, Beverley SM. 2001. A lipophosphoglycan-independent method for isolation of infective Leishmania metacyclic promastigotes by density gradient centrifugation. Exp. Parasitol. 99:97–103 [DOI] [PubMed] [Google Scholar]
- 18. Yao C, Chen Y, Sudan B, Donelson JE, Wilson ME. 2008. Leishmania chagasi: homogenous metacyclic promastigotes isolated by buoyant density are highly virulent in a mouse model. Exp. Parasitol. 118:129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao C, Donelson JE, Wilson ME. 2007. Internal and surface-localized MSP of Leishmania and their differential release from promastigotes. Eukaryot. Cell 6:1905–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yao C, Li Y, Donelson JE, Wilson ME. 2010. Proteomic examination of Leishmania chagasi plasma membrane proteins: contrast between avirulent and virulent (metacyclic) parasite forms. Proteomics Clin. Appl. 4:4–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilson ME, Hardin KK, Donelson JE. 1989. Expression of the major surface glycoprotein of Leishmania donovani chagasi in virulent and attenuated promastigotes. J. Immunol. 143:678–684 [PubMed] [Google Scholar]
- 22. Cauchetier E, Loiseau PM, Lehman J, Rivollet D, Fleury J, Astier A, Deniau M, Paul M. 2002. Characterisation of atovaquone resistance in Leishmania infantum promastigotes. Int. J. Parasitol. 32:1043–1051 [DOI] [PubMed] [Google Scholar]
- 23. Wilson ME, Andersen KA, Britigan BE. 1994. Response of Leishmania chagasi promastigotes to oxidant stress. Infect. Immun. 62:5133–5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang HK, Thalhofer C, Duerkop BA, Mehling JS, Verma S, Gollob KJ, Almeida R, Wilson ME. 2007. Oxidant generation by single infected monocytes after short-term fluorescence labeling of a protozoan parasite. Infect. Immun. 75:1017–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodriguez NE, Gaur U, Wilson ME. 2006. Role of caveolae in Leishmania chagasi phagocytosis and intracellular survival in macrophages. Cell. Microbiol. 8:1106–1120 [DOI] [PubMed] [Google Scholar]
- 26. Stauber LA. 1958. Host resistance to the Khartoum strain of Leishmania donovani. Rice Inst. Pamphlets 45:80–90 [Google Scholar]
- 27. Wilson ME, Innes DJ, Sousa AD, Pearson RD. 1987. Early histopathology of experimental infection with Leishmania donovani in hamsters. J. Parasitol. 73:55–63 [PubMed] [Google Scholar]
- 28. Weirather JL, Jeronimo SM, Gautam S, Sundar S, Kang M, Kurtz MA, Haque R, Schriefer A, Talhari S, Carvalho EM, Donelson JE, Wilson ME. 2011. Serial quantitative PCR assay for detection, species discrimination, and quantification of Leishmania spp. in human samples. J. Clin. Microbiol. 49:3892–3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Christian AE, Haynes MP, Phillips MC, Rothblat GH. 1997. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid. Res. 38:2264–2272 [PubMed] [Google Scholar]
- 30. Pucadyil TJ, Tewary P, Madhubala R, Chattopadhyay A. 2004. Cholesterol is required for Leishmania donovani infection: implications in leishmaniasis. Mol. Biochem. Parasitol. 133:145–152 [DOI] [PubMed] [Google Scholar]
- 31. Magaraci F, Jimenez CJ, Rodrigues C, Rodrigues JC, Braga MV, Yardley V, de Luca-Fradley K, Croft SL, de Souza W, Ruiz-Perez LM, Urbina J, Gonzalez Pacanowska D, Gilbert IH. 2003. Azasterols as inhibitors of sterol 24-methyltransferase in Leishmania species and Trypanosoma cruzi. J. Med. Chem. 46:4714–4727 [DOI] [PubMed] [Google Scholar]
- 32. Mbongo N, Loiseau PM, Billion MA, Robert-Gero M. 1998. Mechanism of amphotericin B resistance in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 42:352–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roberts CW, McLeod R, Rice DW, Ginger M, Chance ML, Goad LJ. 2003. Fatty acid and sterol metabolism: potential antimicrobial targets in apicomplexan and trypanosomatid parasitic protozoa. Mol. Biochem. Parasitol. 126:129–142 [DOI] [PubMed] [Google Scholar]
- 34. Urbina JA, Concepcion JL, Rangel S, Visbal G, Lira R. 2002. Squalene synthase as a chemotherapeutic target in Trypanosoma cruzi and Leishmania mexicana. Mol. Biochem. Parasitol. 125:35–45 [DOI] [PubMed] [Google Scholar]
- 35. Blackwell JM. 1985. Role of macrophage complement and lectin-like receptors in binding Leishmania parasites to host macrophages. Immunol. Lett. 11:227–232 [DOI] [PubMed] [Google Scholar]
- 36. Da Silva RP, Hall BF, Joiner KA, Sacks DL. 1989. CR1, the C3b receptor, mediates binding of infective Leishmania major metacyclic promastigotes to human macrophages. J. Immunol. 143:617–622 [PubMed] [Google Scholar]
- 37. Mosser DM, Edelson PJ. 1985. The mouse macrophage receptor for C3bi (CR3) is a major mechanism in the phagocytosis of Leishmania promastigotes. J. Immunol. 135:2785–2789 [PubMed] [Google Scholar]
- 38. Ueno N, Bratt CL, Rodriguez NE, Wilson ME. 2009. Differences in human macrophage receptor usage, lysosomal fusion kinetics and survival between logarithmic and metacyclic Leishmania infantum chagasi promastigotes. Cell. Microbiol. 11:1827–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Puentes SM, Dwyer DM, Bates PA, Joiner KA. 1989. Binding and release of C3 from Leishmania donovani promastigotes during incubation in normal human serum. J. Immunol. 143:3743–3749 [PubMed] [Google Scholar]
- 40. Ralton JE, Mullin KA, McConville MJ. 2002. Intracellular trafficking of glycosylphosphatidylinositol (GPI)-anchored proteins and free GPIs in Leishmania mexicana. Biochem. J. 363:365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goluszko P, Nowicki B. 2005. Membrane cholesterol: a crucial molecule affecting interactions of microbial pathogens with mammalian cells. Infect. Immun. 73:7791–7796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Urbina JA. 1997. Lipid biosynthesis pathways as chemotherapeutic targets in kinetoplastid parasites. Parasitology 114:91–99 [PubMed] [Google Scholar]
- 43. Jacquier N, Schneiter R. 2012. Mechanisms of sterol uptake and transport in yeast. J. Steroid Biochem. Mol. Biol. 129:70–78 [DOI] [PubMed] [Google Scholar]
- 44. Sturley SL. 2000. Conservation of eukaryotic sterol homeostasis: new insights from studies in budding yeast. Biochim. Biophys. Acta 1529:155–163 [DOI] [PubMed] [Google Scholar]
- 45. Rakotomanga M, Blanc S, Gaudin K, Chaminade P, Loiseau PM. 2007. Miltefosine affects lipid metabolism in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 51:1425–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rakotomanga M, Saint-Pierre-Chazalet M, Loiseau PM. 2005. Alteration of fatty acid and sterol metabolism in miltefosine-resistant Leishmania donovani promastigotes and consequences for drug-membrane interactions. Antimicrob. Agents Chemother. 49:2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Torres-Santos EC, Sampaio-Santos MI, Buckner FS, Yokoyama K, Gelb M, Urbina JA, Rossi-Bergmann B. 2009. Altered sterol profile induced in Leishmania amazonensis by a natural dihydroxymethoxylated chalcone. J. Antimicrob. Chemother. 63:469–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niedermeyer TH, Lindequist U, Mentel R, Gordes D, Schmidt E, Thurow K, Lalk M. 2005. Antiviral terpenoid constituents of Ganoderma pfeifferi. J. Nat. Prod. 68:1728–1731 [DOI] [PubMed] [Google Scholar]
- 49. Osumi T, Taketani S, Katsuki H, Kuhara T, Matsumoto I. 1978. Ergosterol biosynthesis in yeast. Pathways in the late stages and their variation under various conditions. J. Biochem. 83:681–691 [DOI] [PubMed] [Google Scholar]
- 50. Giner JL, Zhao H. 2004. Detailed sterol compositions of two pathogenic rust fungi. Lipids 39:763–767 [DOI] [PubMed] [Google Scholar]
- 51. Mosser DM, Brittingham A. 1997. Leishmania, macrophages and complement: a tale of subversion and exploitation. Parasitology 115:S9–23 [DOI] [PubMed] [Google Scholar]
- 52. Chang CS, Chang KP. 1986. Monoclonal antibody affinity purification of a Leishmania membrane glycoprotein and its inhibition of leishmania-macrophage binding. Proc. Natl. Acad. Sci. U. S. A. 83:100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Russell DG, Wilhelm H. 1986. The involvement of the major surface glycoprotein (gp63) of Leishmania promastigotes in attachment to macrophages. J. Immunol. 136:2613–2620 [PubMed] [Google Scholar]
- 54. Wilson ME, Hardin KK. 1988. The major concanavalin A-binding surface glycoprotein of Leishmania donovani chagasi promastigotes is involved in attachment to human macrophages. J. Immunol. 141:265–272 [PubMed] [Google Scholar]
- 55. Ueno N, Wilson ME. 2012. Receptor-mediated phagocytosis of Leishmania: implications for intracellular survival. Trends Parasitol. 28:335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dahlin-Laborde RR, Yu TP, Beetham JK. 2005. Genetic complementation to identify DNA elements that influence complement resistance in Leishmania chagasi. J. Parasitol. 91:1058–1063 [DOI] [PubMed] [Google Scholar]
- 57. Franke ED, McGreevy PB, Katz SP, Sacks DL. 1985. Growth cycle-dependent generation of complement-resistant Leishmania promastigotes. J. Immunol. 134:2713–2718 [PubMed] [Google Scholar]
- 58. Nunes AC, Almeida-Campos FR, Horta MF, Ramalho-Pinto FJ. 1997. Leishmania amazonensis promastigotes evade complement killing by interfering with the late steps of the cascade. Parasitology 115:601–609 [DOI] [PubMed] [Google Scholar]
- 59. Puentes SM, Sacks DL, da Silva RP, Joiner KA. 1988. Complement binding by two developmental stages of Leishmania major promastigotes varying in expression of a surface lipophosphoglycan. J. Exp. Med. 167:887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Silverman JM, Clos J, De'oliveira CC, Shirvani O, Fang Y, Wang C, Foster LJ, Reiner NE. 2010. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J. Cell Sci. 123:842–852 [DOI] [PubMed] [Google Scholar]
- 61. Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, Peters N, Adlem E, Tivey A, Aslett M, Kerhornou A, Ivens A, Fraser A, Rajandream MA, Carver T, Norbertczak H, Chillingworth T, Hance Z, Jagels K, Moule S, Ormond D, Rutter S, Squares R, Whitehead S, Rabbinowitsch E, Arrowsmith C, White B, Thurston S, Bringaud F, Baldauf SL, Faulconbridge A, Jeffares D, Depledge DP, Oyola SO, Hilley JD, Brito LO, Tosi LR, Barrell B, Cruz AK, Mottram JC, Smith DF, Berriman M. 2007. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 39:839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, Hilley JD, de Doncker S, Maes I, Mottram JC, Quail MA, Rijal S, Sanders M, Schonian G, Stark O, Sundar S, Vanaerschot M, Hertz-Fowler C, Dujardin JC, Berriman M. 2011. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 21:2143–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, et al. 2005. The genome of the kinetoplastid parasite, Leishmania major. Science 309:436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, Harris D, Her Y, Herzyk P, Imamura H, Otto TD, Sanders M, Seeger K, Dujardin JC, Berriman M, Smith DF, Hertz-Fowler C, Mottram JC. 2011. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 21:2129–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Raymond F, Boisvert S, Roy G, Ritt JF, Legare D, Isnard A, Stanke M, Olivier M, Tremblay MJ, Papadopoulou B, Ouellette M, Corbeil J. 2012. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 40:1131–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gangneux J-P, Dullin M, Sulahian A, Garin YJ-F, Derouin F. 1999. Experimental evaluation of second-line oral treatments of visceral leishmaniasis caused by Leishmania infantum. Antimicrob. Agents Chemother. 43:172–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Haughan PA, Chance ML, Goad LJ. 1995. Effects of an azasterol inhibitor of sterol 24-transmethylation on sterol biosynthesis and growth of Leishmania donovani promastigotes. Biochem. J. 308:31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lorente SO, Rodrigues JC, Jimenez Jimenez C, Joyce-Menekse M, Rodrigues C, Croft SL, Yardley V, de Luca-Fradley K, Ruiz-Perez LM, Urbina J, de Souza W, Gonzalez Pacanowska D, Gilbert IH. 2004. Novel azasterols as potential agents for treatment of leishmaniasis and trypanosomiasis. Antimicrob. Agents Chemother. 48:2937–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ramos H, Saint-Pierre-Chazalet M, Bolard J, Cohen BE. 1994. Effect of ketoconazole on lethal action of amphotericin B on Leishmania mexicana promastigotes. Antimicrob. Agents Chemother. 38:1079–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rodrigues JCF, Attias M, Rodriguez C, Urbina JA, Souza Wd. 2002. Ultrastructural and Biochemical Alterations Induced by 22,26-Azasterol, a delta(24(25))-sterol methyltransferase inhibitor, on promastigote and amastigote forms of Leishmania amazonensis. Antimicrob. Agents Chemother. 46:487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Saint-Pierre-Chazalet M, Ben Brahim M, Le Moyec L, Bories C, Rakotomanga M, Loiseau PM. 2009. Membrane sterol depletion impairs miltefosine action in wild-type and miltefosine-resistant Leishmania donovani promastigotes. J. Antimicrob. Chemother. 64:993–1001 [DOI] [PubMed] [Google Scholar]
- 72. Mishra BB, Kale RR, Singh RK, Tiwari VK. 2009. Alkaloids: future prospective to combat leishmaniasis. Fitoterapia 80:81–90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.