

Abstract
Heme oxygenases (HO) catalyze the rate-limiting step of heme degradation. The cytoprotective action of the inducible HO-1 isoform, encoded by the Hmox1 gene, is required for host protection against systemic infections. Here we report that upregulation of HO-1 expression in macrophages (Mϕ) is strictly required for protection against mycobacterial infection in mice. HO-1-deficient (Hmox1−/−) mice are more susceptible to intravenous Mycobacterium avium infection, failing to mount a protective granulomatous response and developing higher pathogen loads, than infected wild-type (Hmox1+/+) controls. Furthermore, Hmox1−/− mice also develop higher pathogen loads and ultimately succumb when challenged with a low-dose aerosol infection with Mycobacterium tuberculosis. The protective effect of HO-1 acts independently of adaptive immunity, as revealed in M. avium-infected Hmox1−/− versus Hmox1+/+ SCID mice lacking mature B and T cells. In the absence of HO-1, heme accumulation acts as a cytotoxic pro-oxidant in infected Mϕ, an effect mimicked by exogenous heme administration to M. avium-infected wild-type Mϕ in vitro or to mice in vivo. In conclusion, HO-1 prevents the cytotoxic effect of heme in Mϕ, contributing critically to host resistance to Mycobacterium infection.
INTRODUCTION
Mycobacterial infections remain a major global burden on society. Mycobacterium tuberculosis, the causative agent of tuberculosis, infects as much as one-third of the world population and accounts for up to 2 million human deaths per year (1). The advent of the AIDS epidemic and the introduction of immunosuppressive therapies have dramatically increased the number of individuals at risk of infection not only with M. tuberculosis but also with other Mycobacterium species, which would otherwise not cause disease, such as Mycobacterium avium, an ubiquitous bacterium that acts as an opportunistic pathogen in immunocompromised individuals (2).
Given the tropism of mycobacteria toward macrophages (Mϕ), host resistance to infection should operate within this specific cell compartment to confer protection against infection (3). In keeping with this notion, known factors in resistance to Mycobacterium infection include a series of mechanisms operating in Mϕ, including those regulating lysosome fusion, intracellular iron availability, and production of antimicrobial peptides or reactive oxygen and nitrogen species (4, 5). Mϕ antimicrobial capacity is also regulated by cytokines, including tumor necrosis factor (TNF) produced by Mϕ and gamma interferon (IFN-γ) produced by natural killer (NK) and T cells (4, 6).
One of the hallmarks of Mycobacterium infection is the formation of large multicellular structures composed of infected as well as noninfected Mϕ, i.e., granulomas. These confine mycobacteria within the site of infection, restraining pathogen dissemination to other organs (7, 8). Granulomas also play a critical role in the recruitment of effector immune cells, including NK and T cells that are critical for host resistance to infection. However, at late (chronic) stages of Mycobacterium infection, granulomas can undergo central necrosis, promoting pathogen dissemination (9) and causing irreversible tissue damage in the lung, a hallmark of lethal forms of the disease. On the other hand, studies on the zebra fish embryo model of granuloma formation induced by Mycobacterium marinum suggest that the dynamics of granuloma formation actually favor the initial expansion and dissemination of mycobacterium infection (62).
Programmed cell death of infected Mϕ can impact granuloma integrity and hence host resistance to Mycobacterium infection. While apoptosis promotes T cell cross-priming and as such restrains Mycobacterium growth, necrosis is used by mycobacteria to evade adaptive immunity and promote bacterial growth (10). This suggests that cytoprotective mechanisms that prevent Mϕ necrosis, but not apoptosis, should favor granuloma integrity and as such limit Mycobacterium growth (11). We hypothesized that the cytoprotective enzyme heme oxygenase-1 (HO-1) (12, 13) might play such a role when expressed by infected Mϕ.
Recognition of pathogen-associated molecular patterns by Mϕ is associated with the induction of HO-1 expression, which catabolizes free heme into biliverdin, iron, and the gasotransmitter carbon monoxide (14). The cytoprotective action of HO-1 (reviewed in reference 15) confers host protection against systemic infections in mice, as demonstrated for malaria (16–18) or severe sepsis (19). The protective effect of HO-1 against systemic infections relies to a large extent on its ability to negate the cytotoxic effects of free heme, that is, heme released from hemoproteins such as hemoglobin or myoglobin (18, 19; reviewed in reference 15). In the context of severe malaria or sepsis, this protective effect acts without regard to pathogen load to promote host survival (16–19) and as such is said to confer disease tolerance (3). Here we demonstrate that the cytoprotective effect of HO-1, when exerted in Mϕ, is essential to ensure granuloma integrity and to confer resistance, rather than tolerance, to mycobacterial infections.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations of the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (ETS 123) and Directive 86/609/EEC and Portuguese rules (DL 129/92). The animal experimental protocols were approved by the competent national authority, Direcção Geral de Veterinária (DGV) (protocol permit numbers 520/000/000/2006 and 0420/000/000/2011). All animal experiments were planned in order to minimize mouse suffering.
Mice.
We generated Hmox1−/− mice by mating Hmox1+/− mice, as previously described (20). Mice were bred at the pathogen-free facilities of the Instituto Gulbenkian de Ciência (IGC), housed at the Institute for Molecular and Cell Biology (IBMC) animal facility in high-efficiency particulate air (HEPA) filter-bearing cages, and fed sterilized food and water ad libitum. Genomic DNA was isolated from the tail, and the Hmox1 genotype was determined by PCR using the primers 5′-TCTTGACGAGTTCTTCTGAG-3′ with 5′-ACGAAGTGACGCCATCTGT-3′ and 5′-GGTGACAGAAGAGGCTAAG-3′ with 5′-CTGTAACTCCACCTCCAAC-3′.
Bacteria.
Mycobacterium avium strain 2447, forming smooth transparent (SmT) colonies, was isolated from an AIDS patient and was a gift from F. Portaels (Institute of Tropical Medicine, Antwerp, Belgium). Mycobacteria were grown to mid-log phase in Middlebrook 7H9 medium (Difco) containing 0.05% Tween 80 (Sigma) at 37°C. Bacteria were harvested by centrifugation, suspended in a small volume of saline containing 0.05% Tween 80, briefly sonicated to disrupt bacterial clumps, diluted, and stored in aliquots at −80°C until use. M. tuberculosis strain H37Rv, kindly provided by António Gil Castro (Life and Health Science Research Institute, Universidade do Minho, Portugal), was grown in Proskauer-Beck medium containing 0.05% Tween 80 to mid-log phase, harvested by centrifugation, and frozen at −80°C until use.
Mouse infection and quantification of bacterial loads in organs.
Mice were infected with M. avium intravenously (i.v.), through a lateral tail vein, with 106 CFU. Control animals received the same volume of saline. Mice were infected with M. tuberculosis via the aerosol route, using an airborne infection apparatus (Glas-Col Inc., Terre Haute, IN), resulting in the implantation, on average, of 30 bacilli in the lung of each mouse. At different time points, mice were sacrificed, and organs were aseptically collected and homogenized in a 0.05% Tween 80 solution in distilled water. Serial dilutions were plated into Middlebrook 7H10 (M. avium) or 7H11 (M. tuberculosis) agar medium (Gibco), the plates were incubated at 37°C for 1 week or 3 weeks, respectively, and the number of colonies was counted.
Gene expression analysis.
Tissue samples were collected and frozen at −80°C until use. Total RNA was extracted using the Micro-to-Midi total RNA purification system (Invitrogen) according to the manufacturer's specifications. Two micrograms of total RNA was transcribed into cDNA with Moloney murine leukemia virus reverse transcriptase (Fermentas), using an oligo(dT)18 primer. The primers used for amplification of cDNA were as follows: hprt1 (housekeeping), 5′-GTAATGATCAGTCAACGGGGGAC-3′ (forward) and 5′-CCAGCAAGCTTGCAACCTTAACCA-3′ (reverse); hmox1, 5′-GCCACCAAGGAGGTACACAT-3′ (forward) and 5′-GCTTGTTGCGCTCTATCTCC-3′ (reverse). The primers were shown not to coamplify genomic DNA. All reactions were performed in a total reaction volume of 20 μl with iQ SYBR Green Supermix (Bio-Rad) and carried out in the iQ5 instrument (Bio-Rad). Baseline thresholds were calculated by the Bio-Rad iQ5 program, and the threshold cycles (CT) were used in the REST software (21), where CT values for target genes were normalized to expression levels of hprt1. Values are reported as fold difference relative to those for the control samples.
Western blot.
Tissue samples were homogenized in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 8.0], 150 mM sodium chloride, 1% Igepal, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA) containing protease inhibitors (Sigma). Fifty micrograms of liver or 20 μg of spleen in electrophoresis sample buffer (50 mM Tris-HCl [pH 8.8], 2% SDS, 0.017% bromophenol blue, 10% glycerol, 2 mM EDTA, 100 mM dithiothreitol [DTT]) was subjected to electrophoresis in a 10% SDS-polyacrylamide gel and transferred to a 0.45-μm nitrocellulose membrane (Amersham Biosciences). The membrane was blocked with 5% fat-free milk and incubated with rabbit anti-HO-1 antibody (Ab) (Proteintech) or rabbit anti-β-actin (Abcam), followed by a horseradish peroxidase-conjugated sheep anti-rabbit IgG (The Binding Site). Immunoreactivity was visualized using an enhanced chemiluminescence (ECL) reagent (Pierce) according to the manufacturer's instructions, and the signal was recorded on a ChemiDoc XRS system (Bio-Rad). Band densitometry was performed with the Quantity One program (Bio-Rad).
Histopathology.
Samples of liver or lung were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and embedded in paraffin. Five-micrometer sections were stained with hematoxylin-eosin and the Masson-trichrome method using standard procedures. For immunofluorescence staining, slides were deparaffinized in Histoclear (National Diagnostics) and hydrated by passage through a grade of alcohols. Tissues were blocked with 4% bovine serum albumin (BSA) in PBS 0.05% Tween and incubated with purified Abs (rabbit anti-HO-1 [Proteintech] and rat anti-F4/80 [Serotec]) followed by secondary Abs (anti-rabbit IgG Alexa Fluor 568 and anti-rat IgG Alexa Fluor 634, both from Molecular Probes). Slides were analyzed in a Leica TCS SP2 AOBS confocal microscope (Leica Microsystems). Fragmented DNA was detected through terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining, according to the manufacturer's instructions (Roche). Slides were analyzed in a Zeiss AxioSkop wide-field microscope (Carl Zeiss, Germany). HO-1 was also detected by immunohistochemistry, using the Impress anti-rabbit Ig reagent (Vector). 8-OHdG was detected by immunohistochemistry using a monoclonal antibody (clone E2E; TrevigenUSA) and the M.O.M. immunodetection kit (Vector), according to the manufacturer's instructions.
Detection of cytokines in plasma and tissues.
Blood was harvested into EDTA tubes from mice anesthetized with isoflurane (Abbott Laboratories) by retro-orbital bleeding. The plasma was collected after centrifugation and frozen at −80°C until use. At the time of sacrifice, portions of the organs were stored at −80°C. Later, these portions were homogenized and protein extracts were prepared according to Bio-Rad's Bio-Plex recommended protocol. Cytokines were measured with the Bio-Plex multiplex cytokine assay system from Bio-Rad, according to the manufacturer's instructions. Total protein content was determined in parallel. Data analysis was performed with Bio-Plex Manager software (Bio-Rad).
Detection of plasma hemoglobin and heme.
Plasma hemoglobin was determined by spectroscopy at λ = 577 nm. Total plasma heme was measured with the 3,3′,5,5′ tetramethylbenzidine (TMB) peroxidase assay (BD Bioscience) at λ = 655 nm. Purified hemoglobin was used as a standard for hemoglobin and heme measurements.
Tissue iron measurements.
Tissue samples were weighed and desiccated in a microwave oven (MDS-2000). The dried samples were weighed and mineralized by acid digestion and heating at 65°C for 20 h. The sample supernatant was collected, and iron was complexed to the bathophenantroline sulfonate chromogen as described by Torrence and Bothwell (22). The nonheme iron concentration was measured spectrophotometrically at λ = 535 nm. Iron content in tissues was expressed as micrograms of nonheme iron per gram of dry tissue.
Protoporphyrins.
Heme (Frontier Scientific Inc.) was dissolved in 0.2 M NaOH, the pH was adjusted to 7.4 with 0.1 M HCl, and the volume was completed with distilled water. The solution was then filter sterilized and stored protected from light at −80°C until use. Mice received 0.2 ml of the heme solution every other day by a lateral tail vein, starting 1 day before the infection with M. avium. Control mice received an equal volume of vehicle.
Cell culture.
Bone marrow-derived Mϕ (BMMϕ) were obtained by culturing bone marrow cells that were flushed from the femurs of the mice with Hanks' balanced salt solution (HBSS) (Gibco). The resulting cell suspension was centrifuged and the cells resuspended in Dulbecco's modified essential medium (DMEM) (Gibco) supplemented with 10 mM glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 10% fetal bovine serum (FBS) (Gibco), and 10% L929 cell-conditioned medium (LCCM) as a source of macrophage colony-stimulating factor (M-CSF). To remove fibroblasts, the cells were cultured overnight, at 37°C in a 7% CO2 atmosphere on cell culture dishes. The nonadherent cells were collected with warm HBSS, distributed in 24-well plates (5 × 105 cells/well) or 96-well plates (1 × 105 cells/well), and incubated at 37°C in a 7% CO2 atmosphere. After 3 days, 10% LCCM was added. On day 7, the medium was renewed. On day 10, when the cells were fully differentiated into Mϕ, they were infected with M. avium, with 106 CFU of M. avium (24-well plates) or 2 × 105 CFU (96-well plates) added to each well. Cells were incubated for 4 h at 37°C in a CO2 atmosphere and then washed with warm HBSS to remove noninternalized bacteria and reincubated in DMEM with 10% FBS and 10% LCCM. To quantify the mycobacteria, Mϕ from triplicate wells were lysed at different time points with 0.1% saponin (Sigma). Serial dilutions were plated into Middlebrook 7H10 agar medium, and the plates were incubated at 37°C for 1 week, when the colonies were counted. For cytotoxicity assays, BMMϕ were washed with HBSS and exposed to heme diluted in HBSS for 1 h. After the removal of heme, the cells were infected with M. avium as described above, and 24 h later, resazurin (Sigma) was added to the wells (125 μM) and incubated at 37°C for 3 h. The conversion of resazurin to the highly fluorescent resorufin by viable cells was evaluated by measuring the relative fluorescence units (RFU) in a SpectraMAX GeminiXS instrument (Molecular Devices).
Statistical analysis.
Statistically significant differences between groups were determined using the unpaired Student t test. Significance is indicated as follows: *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
RESULTS
M. avium infection is associated with induction of HO-1 expression in Mϕ.
Expression of HO-1 supports the viability of activated Mϕ (23, 24), a cell type that is critically involved in providing host resistance to mycobacterial infections (5). Therefore, we investigated whether M. avium infection modulates the expression of HO-1. We found that M. avium infection was associated with the induction of HO-1 in the livers of BALB/c mice, with the highest levels of expression at day 30 postinfection, i.e., 5.2 ± 2.7- and 9.4 ± 5.8-fold increases at the mRNA and protein levels versus noninfected controls, respectively (Fig. 1A). HO-1 expression in the liver was restricted to F4/80+ Mϕ, as assessed by immunofluorescence (Fig. 1B). Expression of HO-1 in the spleen was not significantly induced in response to M. avium infection (see Fig. S1 in the supplemental material). This was not unexpected, as the basal level of HO-1 expression in this organ is very high due to its function in erythrocyte recycling.
Fig 1.
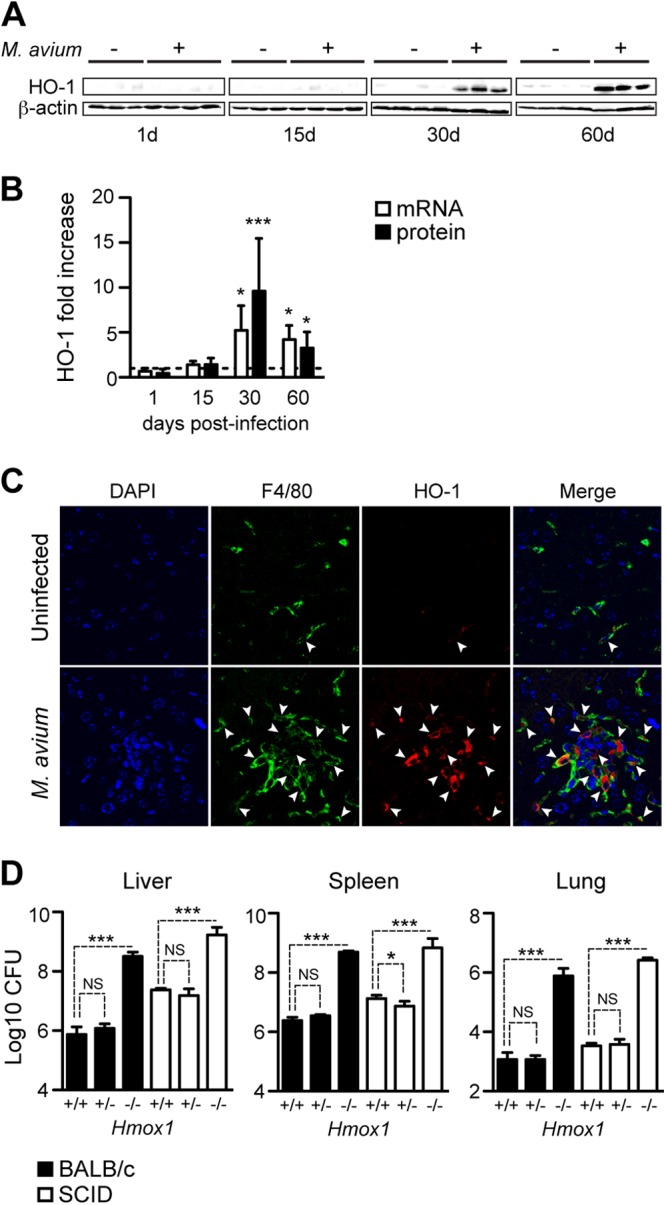
Expression of HO-1 is required for host resistance to M. avium infection. BALB/c mice were infected Mycobacterium avium 2447 SmT (106 CFU, i.v.). (A) Expression of HO-1 protein in the liver, detected by Western blotting at 1, 15, 30, and 60 days after infection. (B) Hmox1 mRNA expression quantified by quantitative reverse transcription-PCR (qRT-PCR) in the livers of BALB/c mice and densitometry of the Western blot shown in panel A. Data are shown as fold change in infected relative to noninfected mice (dashed line) (n = 3 to 5 mice per time point). (C) HO-1 expression detected by immunofluorescence in a representative liver section from BALB/c mice at 30 days after M. avium infection. Arrows indicate F4/80+ cells expressing HO-1. (D) Bacterial loads in the livers, spleens, and lungs of M. avium-infected Hmox1+/+, Hmox1+/−, and Hmox1−/− BALB/c (n = 3 per genotype) or SCID (n = 4 or 5 per genotype) mice. Bars represents mean + standard deviation of log10 CFU/organ. *, P < 0.05; ***, P < 0.001; NS, not significant.
HO-1 confers resistance to M. avium infection.
To assess whether host resistance to M. avium infection operates via a mechanism involving HO-1, we used a loss-of-function approach, comparing the outcomes of M. avium infection in wild-type (Hmox1+/+) versus heterozygous (Hmox1+/−) or Hmox1-deficient (Hmox1−/−) BALB/c mice (20). The bacterial load was about 100-fold higher in Hmox1−/− than in Hmox1+/+ mice, as assessed in the liver, spleen, and lungs, at 60 days postinfection (Fig. 1D). This reveals that host resistance to M. avium relies on a mechanism involving the expression of HO-1, presumably in Mϕ. The bacterial load in heterozygous (Hmox1+/−) mice was similar to that in wild-type (Hmox1+/+) mice (Fig. 1D).
Given that host resistance to Mycobacterium infection relies on interleukin-12 (IL-12)-driven IFN-γ-production by CD4+ T helper (TH) cells (25), we asked whether impaired resistance to M. avium infection in Hmox1-deficient mice is associated with inhibition of this antimicrobial response. To test this hypothesis, we compared the outcomes of M. avium infection in Hmox1−/− mice and Hmox1+/+ BALB/cscid (SCID) mice lacking peripheral T and B cells. As expected (26), SCID mice were more permissive to M. avium growth than immunocompetent mice, as revealed by a 5- to 10-fold increase in bacterial load (Fig. 1D). The bacterial load was about 100-fold higher in Hmox1−/− than in Hmox1+/+ SCID mice, revealing that the mechanism via which HO-1 contributes to host resistance to M. avium acts independently of adaptive immunity (Fig. 1D). This increase in bacterial growth in different organs of Hmox1−/− mice was also observed at earlier infection time points, namely, at 15 and 30 days of infection (see Fig. S2A in the supplemental material).
Given that induction of HO-1 expression in response to M. avium infection is restricted to Mϕ (Fig. 1C), we compared the capacities of Mϕ derived from the bone marrow of wild-type and Hmox1-deficient mice to control M. avium growth in vitro. There was no difference in M. avium growth in Hmox1+/+ and Hmox1−/− Mϕ, as assessed at 4 or 7 days after infection (see Table S1 in the supplemental material). This suggests that HO-1 controls M. avium growth in vivo via a mechanism that is not driven by a cell intrinsic effect of this enzyme on bacterial growth within Mϕ, as suggested by data obtained in vitro for Mycobacterium tuberculosis (27, 28).
The protective effect of HO-1 against Mycobacterium infection acts independently of the regulation of key cytokines.
Resistance to Mycobacterium infection relies on the production of several proinflammatory cytokines (26, 29), while anti-inflammatory cytokines, e.g., IL-10, can increase host susceptibility (30). To assess whether enhanced susceptibility of Hmox1-deficient mice to M. avium infection was associated with deregulated cytokine production, the concentrations of several cytokines functionally involved in host protection against M. avium infection were compared in the plasma, liver, and spleen of infected Hmox1−/− versus Hmox1+/+ SCID mice. Expression of IFN-γ, TNF, IL-17, and IL-10 was similar in Hmox1−/− and Hmox1+/+ SCID mice, as assessed at 30 days postinfection (Fig. 2). Expression of IL-12p40 was higher in infected Hmox1−/− mice than in wild-type controls (Fig. 2), which is at first inconsistent with the impaired resistance of Hmox1−/− mice to M. avium infection (Fig. 2). Expression of IL-1β and IL-6 was also higher in the spleen (but not in the plasma or liver) of infected Hmox1−/− versus Hmox1+/+ mice (Fig. 2). This indicates that the protective effect of HO-1 against M. avium infection acts via a mechanism that is probably not based on the regulation of cytokines involved in bacterial clearance, such as IL-12, TNF, IFN-γ, or IL-10 (29), instead being associated with a downregulation of the proinflammatory cytokines IL-1β and IL-6.
Fig 2.

Hmox1-deficient mice produce normal or elevated levels of inflammatory cytokines and chemokines in response to M. avium infection. Cytokine and chemokine concentrations in the plasma, livers, and spleens of SCID.BALB/c Hmox1+/+ and Hmox1−/− mice at 30 days after M. avium infection were assessed by a bead-based multiplex immune assay. Data are shown as mean + standard deviation for a group of 3 mice per genotype. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Expression of several chemokines functionally involved in host protection against Mycobacterium infection (31) was also compared in Hmox1−/− and Hmox1+/+ mice, 30 days after M. avium infection. The MCP-1/CCL2 concentration in plasma, liver, and spleen was higher in Hmox1−/− than in Hmox1+/+ mice (Fig. 2), which is in keeping with a recent report suggesting that HO-1 controls MCP-1/CCL2 during Mycobacterium infection (32). This was also the case for CCL3, but only in the spleen (Fig. 2). Expression of CCL4 was similar in Hmox1−/− and Hmox1+/+ mice (Fig. 2). Similar results were observed at 15 days of infection (see Fig. S2B in the supplemental material). Hmox1-deficient mice also showed higher hepatic expression of IL-12p40, Ccl2, Ccl5, and Icam-1 mRNAs than Hmox1+/+ mice (see Fig. S2C in the supplemental material).
Hmox1-deficient mice lack granulomas and develop tissue damage in response to M. avium infection.
To gain further understanding of the mechanisms underlying the susceptibility of Hmox1-deficient mice to Mycobacterium infection, histological analysis of the livers of M. avium-infected mice was performed. As expected (33), intravenous M. avium inoculation induced the formation of granulomas in the livers of wild-type BALB/c mice. These multicellular structures are composed of a core of epithelioid Mϕ surrounded by a lymphocytic cuff (Fig. 3A and B), with accumulation of collagen fibers (Fig. 3C). Granulomas were absent in Hmox1−/− mice (Fig. 3D to F). Although collagen fibers were detected in the liver, these were dispersed without apparent organization (Fig. 3F). Likewise, leukocyte infiltrates, composed mostly of granulocytes, were observed, but without granuloma-like organization (Fig. 3D and E). Several alterations in the overall architecture of the liver parenchyma were found in infected Hmox1-deficient versus wild-type mice. These include extensive vascular damage associated with smooth muscle cell proliferation (Fig. 3D), collagen deposition (Fig. 3F), and enlarged sinusoids and extensive cell infiltrations (Fig. 3E). Noninfected age-matched Hmox1-deficient mice did not show significant pathological alterations compared to wild-type controls (see Fig. S3A and E in the supplemental material). Similar pathological features, including the absence of granulomas, were also observed in M. avium-infected Hmox1−/− versus Hmox1+/+ SCID mice (see Fig. S3B to D and F to H in the supplemental material). This demonstrates that expression of HO-1 is required for granuloma formation and/or maintenance, independently of any effect on T or B lymphocytes.
Fig 3.
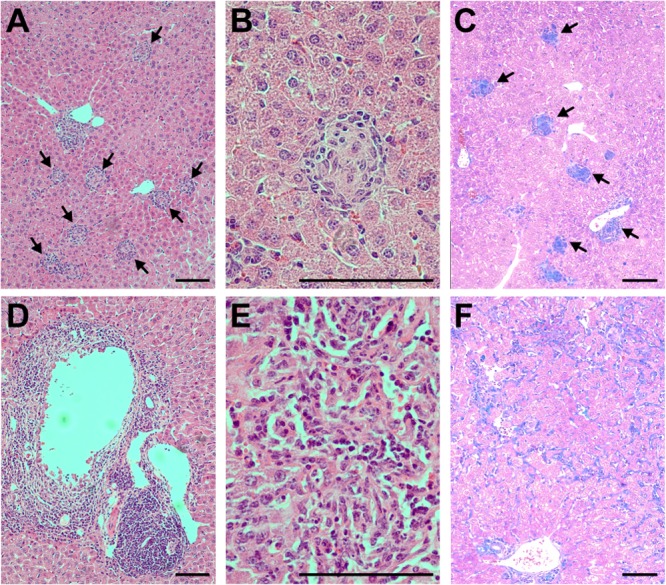
Hmox1-deficient mice do not form granulomas. Liver sections from BALB/c Hmox1+/+ (A, B, and C) and Hmox1−/− (D, E, and F) mice stained with hematoxylin and eosin (A, B, D, and E) or Masson's trichrome (collagen fibers stain blue) (C and F) at 60 days after M. avium infection are shown. Arrows indicate granulomas. Bar, 100 μm. Notice the lack of granulomas in Hmox1-deficient mice (D to F) versus wild-type controls (A to C).
When infected with M. avium, Hmox1−/− mice had twice as much iron accumulation in the liver as did wild-type mice (Fig. 4A). Moreover, Hmox1−/− mice accumulated cell-free hemoglobin and heme in plasma at levels that were 4-fold higher than those in wild-type mice (Fig. 4A). Given that heme and labile iron can act in a pro-oxidant manner during systemic infections (34), we asked whether infected Hmox1−/− developed oxidative stress, presumably leading to programmed cell death and tissue damage. We found that this is the case, as revealed by the higher accumulation of oxidized guanine in the livers of infected Hmox1−/− versus Hmox1+/+ mice, a hallmark of oxidative stress-induced nucleic acid damage (Fig. 4B).
Fig 4.
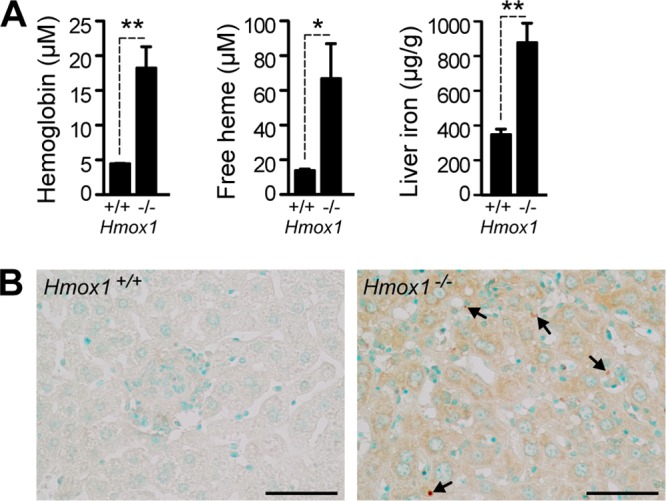
Hmox1-deficient mice develop oxidative stress. (A) Quantification of hemoglobin and free heme in the plasma and total iron in the livers of BALB/c Hmox1+/+ and Hmox1−/− mice at 60 days after M. avium infection (n = 3 to 5). Bars represent the average + standard deviation for each group. *, P < 0.05; **, P < 0.01. (B) Oxidative stress in the livers of BALB/c mice was monitored by detecting oxidized nucleosides 8-OHdG and 8-OHG, which are markers of oxidative damage to DNA and RNA, respectively, by immunohistochemistry. Note the increased DAB (brown) straining in the cytoplasm (RNA) and nuclei (DNA, arrows) of Hmox1−/− mice. The sections were counterstained with methyl green. Bar = 50 μm.
Heme causes oxidative stress leading to programmed cell death in response to several agonists, including TNF (18). In the case of Mϕ, heme promotes TNF-mediated necroptosis (24), a programmed form of necrosis (35). To investigate whether Mϕ programmed cell death occurs during M. avium infection in vivo, we detected cell death by TUNEL assay in the livers of infected mice. We found that M. avium infection was associated with increased cell death in Hmox1−/− versus Hmox1+/+ mice (Fig. 5A and C). We did not find a corresponding increase in cleaved caspase-3-positive cells (Fig. 5B and C), which is consistent with the notion, although does not prove, that cells are dying by necroptosis (24, 35, 36). This increased cell death in the tissues of Hmox1−/− mice is observed as soon as 15 days after infection (see Fig. S2D in the supplemental material). We conclude that expression of HO-1, presumably in Mϕ, acts in a cytoprotective manner to prevent programmed cell death during M. avium infection.
Fig 5.
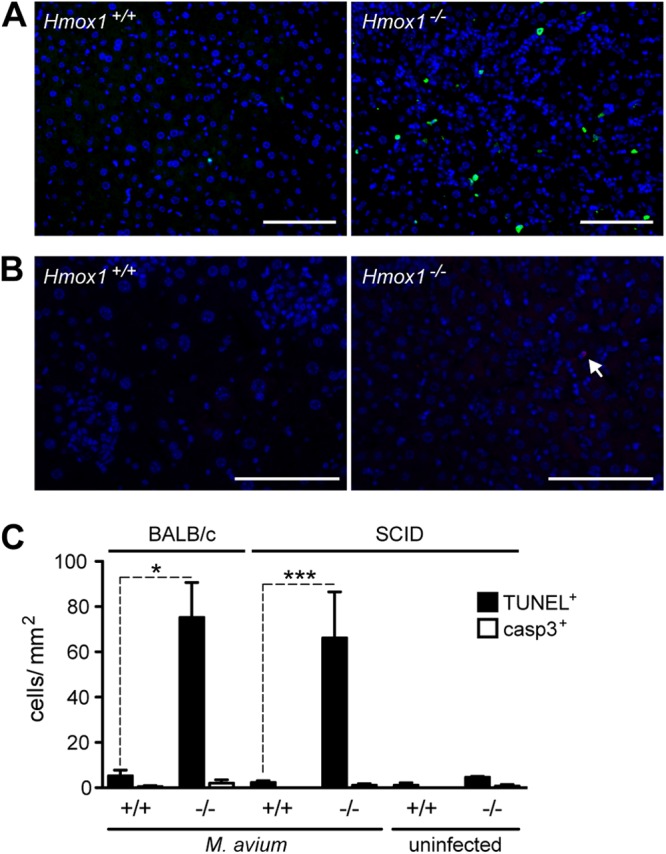
Hmox1-deficient mice have increased cell death. (A and B) Cell death in the livers of BALB/c Hmox1+/+ and Hmox1−/− mice was evaluated by TUNEL (green) (A) and by detecting cleaved caspase-3 (red) (B) at 60 days after M. avium infection. Nuclei were stained with DAPI (blue). Bar, 100 μm. (C) Quantification of the positive cells in panels A and B. Bars represent the average + standard deviation for each group. *, P < 0.05; ***, P < 0.001.
Heme induces infected Mϕ to undergo programmed cell death and impairs granuloma formation.
Given the results presented above, we reasoned that heme might sensitize mycobacterium-infected Mϕ to undergo programmed cell death (24), compromising granuloma formation and ultimately host resistance to infection. To test this hypothesis, we exposed bone marrow-derived Mϕ to heme before M. avium infection in vitro. Heme-treated Mϕ underwent programmed cell death, as assessed at 24 h after infection (Fig. 6A and B). This effect was dose dependent in that increasing the heme concentration progressively reduced Mϕ viability (Fig. 6B). Concomitantly with heme-induced cell death, Mϕ became more permissive to mycobacterial proliferation (Fig. 6C). Despite the increased growth of mycobacteria in heme-treated Mϕ, heme was equally efficient at inducing cell death in uninfected and infected Mϕ (Fig. 6B), suggesting that necroptosis is induced not by higher pathogen loads but by the pro-oxidant effect of heme. These results further suggest that heme impairs the bactericidal activity of Mϕ, compromising host resistance to mycobacterial infection.
Fig 6.
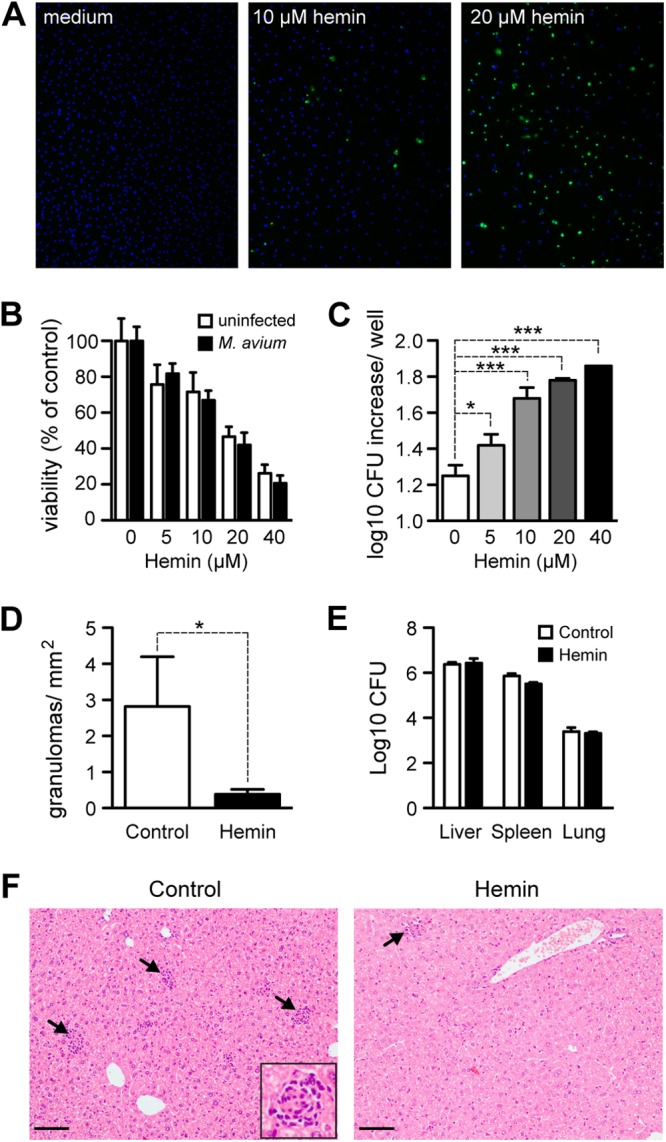
Heme administration triggers Mϕ cell death and hampers granuloma formation. Bone marrow-derived Mϕ (BMMϕ) from wild-type mice were exposed to heme 1 h prior to M. avium infection. (A) Cell death of BMMϕ visualized by TUNEL (green) at 24 h after M. avium infection. DAPI (blue) was used for total DNA staining. (B) BMMϕ viability was quantified by resazurin staining at 24 h after M. avium infection. Results are shown as mean + standard deviation for three culture wells per condition. (C) Bacterial load was quantified at 4 days after infection as CFU. Results are shown as mean + standard deviation of the log10 increase in CFU/well of three wells per condition. *, P < 0.05; ***, P < 0.001. (D) Number of granulomas in the livers of BALB/c mice treated with heme (40 mg/kg, every other day, i.v.) or vehicle and infected with M. avium for 16 days. Each bar represents the mean + one standard deviation (n = 3). *, P < 0.05. (E) Bacterial loads in the livers, spleens, and lungs of the mice described for panel D. Each bar represents the mean + one standard deviation of log10 CFU/organ. (F) Representative images of hematoxylin- and eosin-stained liver sections. Arrows indicate granulomas. The inset shows one typical granuloma. Bar, = 100 μm.
On the other hand, the in vivo administration of heme to M. avium-infected BALB/c mice inhibited granuloma formation (Fig. 6D and F). This was, however, not associated with the detection of cell death in the liver or the increase in bacterial load, as assessed at 15 days after infection (Fig. 6E). This suggests that heme suppresses granuloma formation at an early stage of infection, rather than disrupting preformed granulomas.
Host resistance to M. tuberculosis infection acts via a mechanism that requires the expression of HO-1.
Unlike M. avium, Mycobacterium tuberculosis actively manipulates the death pathway of its host cells (10). Therefore, we asked whether expression of HO-1 is also required to confer host resistance to M. tuberculosis infection. It was previously reported that intravenous infection with high doses (105 and 106 CFU) of M. tuberculosis induces the expression of HO-1 in the lung (27, 28). Here, we used a model of infection with a low dose of M. tuberculosis (30 CFU) via the aerosol route, which more closely replicates the natural route of infection. After 60 days of infection, we detected the expression of HO-1 by immunohistochemistry in the lung. Akin to the case for M. avium, HO-1 was induced in granulomas, while it is absent from healthy tissue (Fig. 7A). In the absence of HO-1, mice developed higher bacterial loads in the lung and spleen than wild-type mice at 60 days postinfection (Fig. 7B). Furthermore, all Hmox1−/− mice succumbed to M. tuberculosis infection, while wild-type (Hmox1+/+) and heterozygous (Hmox1+/−) mice all survived until day 240 postinfection, the last time point analyzed (Fig. 7C). The lung bacterial load at the time of death of Hmox1−/− mice ranged from 5.1 to 8.6 log10 CFU (Fig. 7C, values in brackets), compared to 3.4 to 4.3 log10 CFU in Hmox1+/+ mice at the same time range after infection. When infected with M. tuberculosis, Hmox1−/− mice also exhibited higher bacterial loads in other organs. At the time of death, the bacterial load of these mice ranged from 5.4 to 8.4 log10 CFU in the spleen and from 5.2 to 8.5 log10 CFU in the liver, while wild-type mice presented 4.1 to 4.3 log10 CFU in the spleen and below the level of detection (below 100 bacteria) in the liver at the same time of infection. Histological analysis of the lungs of Hmox1+/+ mice at 60 days after M. tuberculosis infection showed rare lesions, composed predominantly of lymphocytes, surrounding a central Mϕ core and corresponding to early-stage granulomas (Fig. 7D). Cell infiltrates were also observed in M. tuberculosis-infected Hmox1−/− mice, but these were composed predominantly of Mϕ and appeared to be less organized (Fig. 7D). Lung sections from Hmox1-deficient mice also showed high bacterial loads in this organ (Fig. 7E) and significantly increased levels of cell death, revealed by TUNEL staining (Fig. 7F). Overall, these observations reveal that expression of HO-1 also plays a critical role in controlling M. tuberculosis infection in the lung.
Fig 7.

Expression of HO-1 is required for host resistance to M. tuberculosis infection. (A) The expression of HO-1 was detected by immunohistochemistry in the lungs of BALB/c mice infected aerogenically with a low dose (30 CFU/mouse) of M. tuberculosis for 2 months. Note the expression of HO-1 (brown) within the granuloma. Bar, 200 μm. (B) Bacterial loads in the lungs and spleens of BALB/c mice infected aerogenically with a low dose (30 CFU/mouse) of M. tuberculosis for 2 months. *, P < 0.05; **, P < 0.01. Each symbol indicates an individual mouse analyzed, and horizontal bars indicate mean values. (C) Survival of M. tuberculosis-infected mice. Values in brackets indicate the log10 CFU in the lungs of moribund mice. Hmox1+/+, n = 5; Hmox1+/−, n = 4; and Hmox1−/−, n = 5. (D) Representative lung sections stained with hematoxylin and eosin from infected wild-type (left) and Hmox1−/− (right) mice at 60 days after infection. Notice the granuloma in formation in the wild-type mouse and the monocytic, less organized infiltrate in the Hmox1−/− mouse. (E) Representative Ziehl-Neelsen staining of the lung of a moribund Hmox1−/− mouse (bar, 50 μm). (F) Representative TUNEL staining of the lung of an Hmox1−/− mouse infected with M. tuberculosis for 2 months.
DISCUSSION
The present work shows that HO-1 plays a central role in the control of Mycobacterium infection. This heme-catabolizing enzyme is required to confer host resistance to Mycobacterium infection, acting via mechanisms that are independent of the classic IL-12–IFN-γ axis and the development of protective pathogen-specific T cells (Fig. 1D) (26, 29, 37). Instead, HO-1 is required to sustain Mϕ viability during infection (Fig. 5 and 6) and for the formation and/or maintenance of protective granulomas (Fig. 3).
Mechanisms regulating Mϕ viability can have a major impact on the outcome of Mycobacterium infections (10). Nonpathogenic mycobacterial species and attenuated strains of M. tuberculosis induce Mϕ to undergo apoptosis, which promotes host resistance to infection (38–40). In contrast to this, virulent M. tuberculosis strains suppress Mϕ apoptosis and instead trigger Mϕ to undergo necrosis (41, 42), allowing mycobacteria to evade and spread. The recent finding that heme causes Mϕ cell death with characteristics of programmed necrosis (24) is consistent with our observation that heme is cytotoxic to M. avium-infected Mϕ (Fig. 6A and B), which underlies the increase in mycobacterial growth (Fig. 6C). Furthermore, we show that this mechanism can occur in vivo, as observed during Mycobacterium infection in Hmox1-deficient mice (Fig. 3 and 7). Uninfected Hmox1−/− mice did not show increased programmed cell death (Fig. 5). It could be argued that the infection per se induces Mϕ to undergo programmed cell death. However, in the absence of infection, heme can trigger Mϕ programmed cell death (24) (Fig. 6).
The erythrocyte life span is reduced during anemia in chronic disease (43), a condition occurring during mycobacterial infections (44). Moreover, mice infected with Mycobacterium lepraemurium have increased erythrophagocytosis (45), implying a higher hemoglobin turnover and supporting the hypothesis that Mϕ are exposed to hemoglobin and therefore to heme during mycobacterial infection. In the case of HO-1 deficiency, heme is not adequately cleared, resulting in oxidative damage and programmed cell death. Heme-driven Mϕ programmed cell death should occur predominantly in Mϕ that recognize mycobacteria, based on the production of reactive oxygen species and TNF in response to mycobacteria (24). Presumably this explains why heme catabolism by HO-1 is required to mount a protective granulomatous response, a notion supported by two independent and complementary observations. First, granulomas are absent or significantly decreased in Mycobacterium-infected Hmox1-deficient mice, in contrast to wild-type controls, where these structures are observed (Fig. 3). Second, granuloma formation after M. avium infection in wild-type mice is drastically reduced by the administration of heme (Fig. 6). This suggests that the substrate of HO-1 activity, i.e., heme, impairs granuloma formation and/or maintenance, thus explaining why heme catabolism by HO-1 promotes granuloma formation and/or maintenance.
The molecular mechanisms regulating granuloma formation in response to Mycobacterium infection are not completely understood (46). It is clear, however, that expression of cytokines, e.g., TNF, IFN-γ, or IL-12, as well as adhesion molecules, e.g., ICAM-1, is required for the formation of protective granulomas (7, 9). The observation that the expression of these genes was not impaired in M. avium-infected Hmox1-deficient versus wild-type mice (Fig. 2) suggests that the mechanism via which HO-1 promotes the formation and/or maintenance of protective granulomas acts independently of a putative effect on the expression of these and probably other related inflammatory genes.
How the absence of granulomas in Hmox1−/− mice affects their resistance to the infection is not clear. The granuloma is thought to be protective to the host, by favoring a close contact between infected Mϕ and lymphocytes, and the lack of granulomas is usually associated with increased bacterial proliferation (7). Hence, the increased susceptibility of Hmox1−/− mice could be attributed to their inability to form these lesions. However, heme administration to BALB/c mice blocks the assembly of the granuloma, as assessed at day 16 after M. avium infection, without affecting bacterial growth (Fig. 6), whereas Hmox1−/− mice already have significantly higher bacterial loads at this time point (see Fig. S1 in the supplemental material). One possible interpretation is that the lack of granulomas is not entirely responsible for the increased susceptibility of Hmox1−/− mice, at least during the initial phase of infection. Similar observations have been made using Icam-1-deficient mice, which present a deficient recruitment of Mϕ to infected tissues. These mice do not form the granulomas after aerosol challenge with M. tuberculosis but are capable of mounting a Th1 cell-mediated response, with normal levels of IFN-γ and IL-12, which controls the infection until day 90 similarly to the case for wild-type mice (47). However, at 100 to 130 days after infection, ICAM-1-deficient mice undergo a surge in bacterial numbers, resulting in lung tissue damage and death (48), which suggests that although they are not necessary to control the infection initially, granulomas are necessary to control chronic infection. Our results show that Hmox1−/− mice have increased bacterial loads after 60 days of infection with M. tuberculosis, which are probably not attributable to a deficient granuloma formation. However, akin to Icam1−/− mice, Hmox1−/− mice infected with M. tuberculosis undergo a surge in bacterial numbers before the death of the host (Fig. 7), corroborating the notion that the faulty assembly of granulomas may impair the control of infection at late stages.
In addition to acting as a cytotoxic agonist, heme is a putative source of labile iron that can promote microbial growth, thus limiting host resistance to infection (49). Hmox1-deficient mice accumulate heme in plasma and develop tissue iron overload following M. avium infection (Fig. 4A), which could contribute to the higher pathogen load observed in Hmox1−/− than in Hmox1+/+ mice (Fig. 1D). However, iron overload per se is associated with at most a 10-fold increase in pathogen load (50–53), but Hmox1 deficiency and heme accumulation in plasma are associated with 100-fold increase in pathogen load, compared to that in wild-type mice (Fig. 1D). This suggests that HO-1 reduces the pathogen load by additional mechanisms, other than controlling iron overload, presumably through its cytoprotective effect exerted on infected Mϕ. It should be noted that while exogenously administered heme inhibits granuloma formation, this is not enough to cause a significant increase in M. avium growth in vivo at early infection time points (Fig. 6D and E). Only in the absence of the protective action of HO-1 does heme cause massive Mϕ death and concomitant uncontrolled bacterial growth.
Although the underlying mechanism via which HO-1 confers host protection against Mycobacterium infection is similar to the one described for other pathogens (16, 18, 19), i.e., cytoprotection against heme, HO-1 confers resistance rather than tolerance to Mycobacterium infection. The reason for this is most probably that HO-1 provides cytoprotection in a major cell compartment conferring resistance to Mycobacterium infection, i.e., Mϕ. These findings should have implications for our understanding of the pathogenesis of diseases triggered by intramacrophagic pathogens that have evolved mechanisms to subvert the Mϕ cell death for their advantage, such as Listeria monocytogenes, Salmonella spp., Shigella spp., and Yersinia pestis (54–56). Akin to the case for Mycobacterium, HO-1 may determine the pathological outcome of these infections by virtue of its cytoprotective effect exerted on Mϕ.
There are most probably other mechanisms via which HO-1 controls host microbe interaction in the context of mycobacterial infection. Carbon monoxide produced via heme catabolism by HO-1 induces the expression of the dormancy regulon during M. tuberculosis infection in vitro (27, 28), suggesting that HO-1 activity may contribute to latency. Therefore, reduced carbon monoxide should prevent the induction of the dormancy regulon, contributing to the increased proliferation of mycobacteria. While this hypothesis remains to be tested, M. tuberculosis deficient in the induction of the dormancy regulon establishes in vivo infections in mice that are indistinguishable from those caused by the parental strain (57) or even present a growth defect (58), suggesting that a putative effect of CO on the dormancy regulon should have a limited impact on bacterial growth in vivo.
While we were preparing this article, Regev et al. (32) reported that Hmox1−/− mice infected through the intratracheal route with M. avium fail to develop organized granulomas in the lung. Those authors attributed this effect to an increased expression of CCL2/CCR2 in HO-1 deficient mice, which while consistent with our findings (Fig. 2; see Fig. S2 in the supplemental material) may be attributed to a higher bacterial load in Hmox1−/− than in Hmox1+/+ mice, thus inducing CCL2 expression (59). However, it is not clear whether there is a causal effect of increased CCL2 and lack of granulomas. While our present data confirm that HO-1 regulates MCP-1/CCL2 expression in response to M. avium infection (Fig. 2; see Fig. S2 in the supplemental material) (32), this should not have a major impact on pathogen clearance since MCP-1/CCL2-deficient mice are indistinguishable from wild-type mice in their ability to clear Mycobacterium infection (60). Instead we propose that it is the oxidative stress induced by the accumulation of heme in Mϕ that recognize mycobacteria that inhibits granuloma formation. This argues for the existence of an inverse relationship between the levels of reactive oxygen and nitrogen species and granuloma development, a notion is strongly supported by our previous observation that inducible nitric oxide synthase-deficient mice form larger and denser granulomas in response to M. avium infection than wild-type mice (33, 61). Of note, when we treated mice with heme, although they were unable to form granulomas in response to M. avium infection, the level of expression of Ccl2 mRNA in the liver was not increased compared to that in control mice (data not shown).
In conclusion, we demonstrate that expression of HO-1 is strictly required to prevent Mϕ from undergoing programmed cell death induced by free heme, thus playing a key role in host resistance against mycobacterial infections. We propose that pharmacologic targeting of heme catabolism might be used therapeutically against Mycobacterium infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Fundação para a Ciência e Tecnologia (Portugal), FCT/FEDER grant POCI/SAUIMI/56578/2004 to M.S.G., and European Community 6th Framework grant LSH-2005-1.2.5-1 to M.P.S. S.S.-G. was supported by FCT grant SFRH/BD/29257/2006. Work by M.P.S. and R.L. was partially supported by European Research Council ERC-2011 Advanced Grant 294709 DAMAGECONTROL and Fundação para a Ciência e Tecnologia, PTDC/BIA-BCM/101311/2008, to M.P.S. and SFRH/BPD/25436/2005 to R.L.
We thank Silvia Cardoso and Sofia Rebelo (Instituto Gulbenkian de Ciência) for breeding and maintenance of the Hmox1-deficient mouse colonies, the collaboration of ICVS (Universidade do Minho, Braga, Portugal) and especially António Gil Castro for technical assistance with tuberculosis experiments, and the Laboratory of Veterinary Pathology at ICBAS for technical assistance with histopathological analysis.
Footnotes
Published ahead of print 29 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00251-13.
REFERENCES
- 1. Walzl G, Ronacher K, Hanekom W, Scriba TJ, Zumla A. 2011. Immunological biomarkers of tuberculosis. Nat. Rev. Immunol. 11:343–354 [DOI] [PubMed] [Google Scholar]
- 2. Johnson MM, Waller EA, Leventhal JP. 2008. Nontuberculous mycobacterial pulmonary disease. Curr. Opin. Pulm. Med. 14:203–210 [DOI] [PubMed] [Google Scholar]
- 3. Medzhitov R, Schneider DS, Soares MP. 2012. Disease tolerance as a defense strategy. Science 335:936–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Appelberg R. 2006. Pathogenesis of Mycobacterium avium infection: typical responses to an atypical mycobacterium? Immunol. Res. 35:179–190 [DOI] [PubMed] [Google Scholar]
- 5. Russell DG. 2011. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol. Rev. 240:252–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cooper AM, Mayer-Barber KD, Sher A. 2011. Role of innate cytokines in mycobacterial infection. Mucosal Immunol. 4:252–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saunders BM, Cooper AM. 2000. Restraining mycobacteria: role of granulomas in mycobacterial infections. Immunol. Cell Biol. 78:334–341 [DOI] [PubMed] [Google Scholar]
- 8. Ulrichs T, Kaufmann SH. 2006. New insights into the function of granulomas in human tuberculosis. J. Pathol. 208:261–269 [DOI] [PubMed] [Google Scholar]
- 9. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. 2009. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 10:943–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Behar SM, Divangahi M, Remold HG. 2010. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat. Rev. Microbiol. 8:668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paige C, Bishai WR. 2010. Penitentiary or penthouse condo: the tuberculous granuloma from the microbe's point of view. Cell. Microbiol. 12:301–309 [DOI] [PubMed] [Google Scholar]
- 12. Vile GF, Basu-Modak S, Waltner C, Tyrrell RM. 1994. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 91:2607–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, Grey ST, Colvin RB, Choi AM, Poss KD, Bach FH. 1998. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat. Med. 4:1073–1077 [DOI] [PubMed] [Google Scholar]
- 14. Tenhunen R, Marver HS, Schmid R. 1968. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. U. S. A. 61:748–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gozzelino R, Jeney V, Soares MP. 2010. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50:323–354 [DOI] [PubMed] [Google Scholar]
- 16. Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, Rebelo S, Henri A, Beuzard Y, Soares MP. 2011. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell 145:398–409 [DOI] [PubMed] [Google Scholar]
- 17. Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, Portugal S, Soares MP, Mota MM. 2007. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 13:703–710 [DOI] [PubMed] [Google Scholar]
- 18. Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, Soares MP. 2009. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. U. S. A. 106:15837–15842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, Marguti I, Cardoso S, Sepulveda N, Smith A, Soares MP. 2010. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2:51ra71. [DOI] [PubMed] [Google Scholar]
- 20. Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee ME. 1999. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J. Clin. Invest. 103:R23–R29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pfaffl MW, Horgan GW, Dempfle L. 2002. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Torrance JD, Bothwell TH. 1980. Tissue iron stores, p 104–109 In Cook JD. (ed), Methods in hematology. Churchill Livingstone Press, New York, NY [Google Scholar]
- 23. Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. 2010. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood 116:6054–6062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, Chan FK, Bozza MT. 2012. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 119:2368–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silva RA, Florido M, Appelberg R. 2001. Interleukin-12 primes CD4+ T cells for interferon-gamma production and protective immunity during Mycobacterium avium infection. Immunology 103:368–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Appelberg R, Castro AG, Pedrosa J, Silva RA, Orme IM, Minoprio P. 1994. Role of gamma interferon and tumor necrosis factor alpha during T-cell-independent and -dependent phases of Mycobacterium avium infection. Infect. Immun. 62:3962–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar A, Deshane JS, Crossman DK, Bolisetty S, Yan BS, Kramnik I, Agarwal A, Steyn AJ. 2008. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J. Biol. Chem. 283:18032–18039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shiloh MU, Manzanillo P, Cox JS. 2008. Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe 3:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castro AG, Silva RA, Appelberg R. 1995. Endogenously produced IL-12 is required for the induction of protective T cells during Mycobacterium avium infections in mice. J. Immunol. 155:2013–2019 [PubMed] [Google Scholar]
- 30. Roque S, Nobrega C, Appelberg R, Correia-Neves M. 2007. IL-10 underlies distinct susceptibility of BALB/c and C57BL/6 mice to Mycobacterium avium infection and influences efficacy of antibiotic therapy. J. Immunol. 178:8028–8035 [DOI] [PubMed] [Google Scholar]
- 31. Algood HM, Chan J, Flynn JL. 2003. Chemokines and tuberculosis. Cytokine Growth Factor Rev. 14:467–477 [DOI] [PubMed] [Google Scholar]
- 32. Regev D, Surolia R, Karki S, Zolak J, Montes-Worboys A, Oliva O, Guroji P, Saini V, Steyn AJ, Agarwal A, Antony VB. 2012. Heme oxygenase-1 promotes granuloma development and protects against dissemination of mycobacteria. Lab. Invest. 92:1541–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gomes MS, Florido M, Pais TF, Appelberg R. 1999. Improved clearance of Mycobacterium avium upon disruption of the inducible nitric oxide synthase gene. J. Immunol. 162:6734–6739 [PubMed] [Google Scholar]
- 34. Gozzelino R, Andrade BB, Larsen R, Luz NF, Vanoaica L, Seixas E, Coutinho A, Cardoso S, Rebelo S, Poli M, Barral-Netto M, Darshan D, Kuhn LC, Soares MP. 2012. Metabolic adaptation to tissue iron overload confers tolerance to malaria. Cell Host Microbe 12:693–704 [DOI] [PubMed] [Google Scholar]
- 35. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11:700–714 [DOI] [PubMed] [Google Scholar]
- 36. Vanlangenakker N, Vanden Berghe T, Vandenabeele P. 2012. Many stimuli pull the necrotic trigger, an overview. Cell death and differentiation. 19:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cooper AM. 2009. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 27:393–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duan L, Gan H, Arm J, Remold HG. 2001. Cytosolic phospholipase A2 participates with TNF-alpha in the induction of apoptosis of human macrophages infected with Mycobacterium tuberculosis H37Ra. J. Immunol. 166:7469–7476 [DOI] [PubMed] [Google Scholar]
- 39. Fairbairn IP, Stober CB, Kumararatne DS, Lammas DA. 2001. ATP-mediated killing of intracellular mycobacteria by macrophages is a P2X(7)-dependent process inducing bacterial death by phagosome-lysosome fusion. J. Immunol. 167:3300–3307 [DOI] [PubMed] [Google Scholar]
- 40. Pais TF, Appelberg R. 2000. Macrophage control of mycobacterial growth induced by picolinic acid is dependent on host cell apoptosis. J. Immunol. 164:389–397 [DOI] [PubMed] [Google Scholar]
- 41. Chen M, Gan H, Remold HG. 2006. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 176:3707–3716 [DOI] [PubMed] [Google Scholar]
- 42. Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, Fortune S, Behar SM, Remold HG. 2009. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 10:899–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mitlyng BL, Singh JA, Furne JK, Ruddy J, Levitt MD. 2006. Use of breath carbon monoxide measurements to assess erythrocyte survival in subjects with chronic diseases. Am. J. Hematol. 81:432–438 [DOI] [PubMed] [Google Scholar]
- 44. Rodrigues PN, Gomes SS, Neves JV, Gomes-Pereira S, Correia-Neves M, Nunes-Alves C, Stolte J, Sanchez M, Appelberg R, Muckenthaler MU, Gomes MS. 2011. Mycobacteria-induced anaemia revisited: a molecular approach reveals the involvement of NRAMP1 and lipocalin-2, but not of hepcidin. Immunobiology 216:1127–1134 [DOI] [PubMed] [Google Scholar]
- 45. Ha DK, Lawton JW, Gardner ID. 1986. Evaluation of phagocytic function in Mycobacterium lepraemurium infection. J. Comp. Pathol. 96:415–424 [DOI] [PubMed] [Google Scholar]
- 46. Saunders BM, Britton WJ. 2007. Life and death in the granuloma: immunopathology of tuberculosis. Immunol. Cell Biol. 85:103–111 [DOI] [PubMed] [Google Scholar]
- 47. Johnson CM, Cooper AM, Frank AA, Orme IM. 1998. Adequate expression of protective immunity in the absence of granuloma formation in Mycobacterium tuberculosis-infected mice with a disruption in the intracellular adhesion molecule 1 gene. Infect. Immun. 66:1666–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saunders BM, Frank AA, Orme IM. 1999. Granuloma formation is required to contain bacillus growth and delay mortality in mice chronically infected with Mycobacterium tuberculosis. Immunology 98:324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bullen J, Griffiths E, Rogers H, Ward G. 2000. Sepsis: the critical role of iron. Microbes Infect. 2:409–415 [DOI] [PubMed] [Google Scholar]
- 50. Gomes-Pereira S, Rodrigues PN, Appelberg R, Gomes MS. 2008. Increased susceptibility to Mycobacterium avium in hemochromatosis protein HFE-deficient mice. Infect. Immun. 76:4713–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gomes MS, Boelaert JR, Appelberg R. 2001. Role of iron in experimental Mycobacterium avium infection. J. Clin. Virol. 20:117–122 [DOI] [PubMed] [Google Scholar]
- 52. Lounis N, Truffot-Pernot C, Grosset J, Gordeuk VR, Boelaert 2001. Iron and Mycobacterium tuberculosis infection. J. Clin. Virol. 20:123–126 [DOI] [PubMed] [Google Scholar]
- 53. Schaible UE, Collins HL, Priem F, Kaufmann SH. 2002. Correction of the iron overload defect in beta-2-microglobulin knockout mice by lactoferrin abolishes their increased susceptibility to tuberculosis. J. Exp. Med. 196:1507–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao L, Abu Kwaik Y. 2000. Hijacking of apoptotic pathways by bacterial pathogens. Microbes Infect. 2:1705–1719 [DOI] [PubMed] [Google Scholar]
- 55. Labbe K, Saleh M. 2008. Cell death in the host response to infection. Cell Death Differ. 15:1339–1349 [DOI] [PubMed] [Google Scholar]
- 56. Haimovich B, Venkatesan MM. 2006. Shigella and Salmonella: death as a means of survival. Microbes Infect. 8:568–577 [DOI] [PubMed] [Google Scholar]
- 57. Rustad TR, Harrell MI, Liao R, Sherman DR. 2008. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS One 3:e1502. 10.1371/journal.pone.0001502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Converse PJ, Karakousis PC, Klinkenberg LG, Kesavan AK, Ly LH, Allen SS, Grosset JH, Jain SK, Lamichhane G, Manabe YC, McMurray DN, Nuermberger EL, Bishai WR. 2009. Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect. Immun. 77:1230–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rao SP, Hayashi T, Catanzaro A. 2000. Release of monocyte chemoattractant protein (MCP)-1 by a human alveolar epithelial cell line in response to mycobacterium avium. FEMS Immunol. Med. Microbiol. 29:1–7 [DOI] [PubMed] [Google Scholar]
- 60. Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. 1998. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 187:601–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lousada S, Florido M, Appelberg R. 2006. Regulation of granuloma fibrosis by nitric oxide during Mycobacterium avium experimental infection. Int. J. Exp. Pathol. 87:307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davis JM, Ramakrishnan L. 2009. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136:37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.