

Abstract
Hypoxia-inducible factor 1 (HIF-1) is the key transcription factor involved in the adaptation of mammals to hypoxia and plays a crucial role in cancer angiogenesis. Recent evidence suggests a leading role for HIF-1 in various inflammatory and infectious diseases. Here we describe the role of HIF-1 in Staphylococcus aureus infections by investigating the HIF-1-dependent host cell response. For this purpose, transcriptional profiling of HIF-1α-deficient HepG2 and control cells, both infected with Staphylococcus aureus, was performed. Four hours after infection, the expression of 190 genes, 24 of which were regulated via HIF-1, was influenced. LOX (encoding lysyl oxidase) was one of the upregulated genes with a potential impact on the course of S. aureus infection. LOX is an amine oxidase required for biosynthetic cross-linking of extracellular matrix components. LOX was upregulated in vitro in different cell cultures infected with S. aureus and also in vivo, in kidney abscesses of mice intravenously infected with S. aureus and in clinical skin samples from patients with S. aureus infections. Inhibition of LOX by β-aminopropionitrile (BAPN) did not affect the bacterial load in kidneys or blood but significantly influenced abscess morphology and collagenization. Our data provide evidence for a crucial role of HIF-1-regulated LOX in abscess formation.
INTRODUCTION
Hypoxia-inducible factor 1 (HIF-1) is the key transcription factor for the regulation of oxygen homoeostasis in all metazoan species (1). HIF-1 plays important roles not only in glycolysis, erythropoiesis, and angiogenesis but also in inflammation and immune response (2–4). HIF-1 is a heterodimer, consisting of constitutively expressed α and β subunits, is regulated mainly posttranscriptionally, and is rapidly degraded under normoxic conditions. Protein stability is regulated by oxygen-dependent hydroxylation of two proline residues (Pro402 and Pro564) in the oxygen-dependent degradation domain (ODD) of the α subunit, catalyzed by iron- and oxygen-dependent prolyl hydroxylases (PHDs). Hydroxylation events target the α subunit for ubiquitination by the von Hippel-Lindau protein and subsequent proteasomal degradation (5). Under hypoxia and iron deprivation, PHDs are less active, resulting in the stabilization of HIF-1α, heterodimer formation, and the subsequent activation of HIF-1-responsive genes by the binding of HIF-1 to hypoxia-responsive elements in their promoter regions (5, 6).
Recently, bacterial also infections have turned out to be potent stimuli for HIF-1 activation (7), and remarkably, HIF-1 activation seems to be a general phenomenon upon infection (8). HIF-1 activation has been shown in infections with bacteria such as Staphylococcus aureus, Escherichia coli, Streptococcus agalactiae, Pseudomonas aeruginosa, Bartonella henselae, Yersinia enterocolitica, and Chlamydophila pneumoniae. This HIF-1 activation seems to be induced by several overlapping mechanisms, including enhanced oxygen consumption at the site of infection, iron deprivation, and some bacterial components (e.g., outer membrane proteins, such as Bartonella adhesin A or lipopolysaccharides [LPS] from E. coli) (8–12). HIF-1 has a protective function in local infections; it is essential for the bactericidal capacity of phagocytes and controls the systemic spread of bacteria (13, 14). In contrast, HIF-1 activation seems to be disadvantageous in systemic infections, leading to higher mortality in, e.g., LPS sepsis and S. aureus peritonitis models (8, 15).
S. aureus is one of the most important human pathogens, causing life-threatening infections such as endocarditis or sepsis. A characteristic feature of S. aureus infections is the formation of abscesses (16). After entering the bloodstream (e.g., after a wound infection), S. aureus disseminates to organ tissues, where it elicits massive infiltration of polymorphonuclear leukocytes and other immune cells (17). During abscess maturation, a central accumulation of the pathogen is surrounded by a pseudocapsule of fibrin deposits, zones of necrotic and healthy polymorphonuclear neutrophils, and a rim of eosinophilic material consisting of collagens and fibroblasts (17–20). Finally, an abscess rupture can lead to novel dissemination of S. aureus via the bloodstream. Abscess formation is regarded as an important virulence strategy, and bacterial factors contributing to abscess formation have been shown to be crucial for S. aureus infection (17). Accordingly, therapeutic strategies interfering with abscess formation may become instrumental in combating staphylococcal diseases.
Lysyl oxidase (LOX) is a copper-dependent amine oxidase that catalyzes the final enzymatic step required for the cross-linking of collagen and elastin molecules in the extracellular matrix. Its expression has been shown to be regulated via HIF-1 (21). LOX is secreted from cells as an inactive proenzyme into the extracellular space, where it is cleaved to the mature, active LOX enzyme (22). LOX plays a leading role in metastatic niche formation in breast cancer (21) and enhances the motility of monocytes, vascular smooth muscle cells, and fibroblasts in vitro (23–25). Some evidence suggests that LOX may influence granulomatous and fibrotic infectious diseases: enhanced Lox expression has been detected in murine Schistosoma mansoni and Echinococcus multilocularis infections (26, 27), and increased LOX expression is also present in infected septic periprosthetic membranes (28). However, none of these studies were aimed at elucidating the biological function of LOX in the course of an infection.
Here we analyzed genes regulated by HIF-1 upon S. aureus infection. By the use of transcriptome analysis, we found 24 genes regulated by HIF-1 in S. aureus infections; of these, LOX was chosen to be analyzed in more detail. Our data revealed that HIF-1-dependent LOX induction is detectable in cell culture infection models and is also present in murine S. aureus-induced kidney abscesses and in human infections with S. aureus. Inhibition of LOX with the specific LOX inhibitor β-aminopropionitrile (BAPN) resulted in significantly reduced kidney abscess formation and less abscess collagenization.
MATERIALS AND METHODS
S. aureus growth conditions.
For in vitro experiments, S. aureus 8325-4 (29), a well-characterized, standard laboratory strain (8, 30, 31), was grown overnight in Luria-Bertani (LB) medium (Roth, Karlsruhe, Germany) with shaking at 37°C and was extensively washed in phosphate-buffered saline (PBS; Gibco, Karlsruhe, Germany), and bacterial numbers were determined by measuring the optical density at 600 nm (OD600) (an OD600 of 1.0 is equal to ∼3 × 108 bacteria/ml). For in vivo experiments, S. aureus 8325-4 was additionally subcultured by inoculating fresh BM medium (1% tryptone, 0.5% yeast extract, 0.5% NaCl, 0.1% K2HPO4, and 0.1% glucose) and allowing subsequent growth for 4 to 5 h so as to harvest bacteria in the exponential-growth phase. For control purposes, bacterial numbers were additionally determined by subcultivation of serial dilutions on BM agar plates (data not shown).
Cell culture and infection procedures for HepG2 cells and human keratinocytes.
HepG2 hepatocellular control cells (transfected with a control vector harboring a nonspecific nontarget sequence; referred to below as HepG2 cells) and HIF-1α−/− cells (32) were grown in minimal essential medium (Sigma-Aldrich, Taufkirchen, Germany) supplemented with 10% heat-inactivated fetal calf serum (FCS; Sigma-Aldrich), 2 mM l-glutamine (Gibco, Karlsruhe, Germany), 100 U/ml penicillin and 100 μg/ml streptomycin (Biochrom, Berlin, Germany), 0.1 mM nonessential amino acids, and 2 μg/ml puromycin dihydrochloride (Sigma-Aldrich). Normal human epidermal keratinocytes (NHEKs; PromoCell, Heidelberg, Germany) were grown in supplemented keratinocyte growth medium 2 (PromoCell) in the presence of 10 mg/ml streptomycin and 100 U penicillin. Cells were kept at 37°C under a humidified atmosphere with 5% CO2.
For infection experiments, cells were detached with 0.05% trypsin-EDTA (Gibco), which was neutralized by the addition of trypsin neutralizing solution (PromoCell) (for NHEKs) or by the addition of a medium containing FCS (for HepG2 cells). Cells were seeded the day before infection, and experiments were performed in cell culture medium without antibiotics (to allow bacterial growth) and without FCS (to avoid nonspecific HIF-1 activation). For protein extraction and RNA isolation, cells (1 × 106 HepG2 cells or 5.0 × 105 NHEKs; each cultivated in 6-well plates) were infected with S. aureus at a multiplicity of infection (MOI) of 20 for 4 h. Uninfected cells were used as negative controls, and desferrioxamine (DFO) (200 μM/liter; Sigma-Aldrich)-treated cells were used as positive controls. For the determination of secreted LOX levels, 2.0 × 105 cells were seeded in 24-well plates and were infected with S. aureus at MOI of 0.01 for 16 h.
Detection of HIF-1 activation.
For the detection of HIF-1 activation by immunoblotting, proteins from cell cultures were extracted as described (8), separated by 8% SDS-PAGE, and blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore, Schwalbach, Germany). Mouse anti-human HIF-1α antibodies (Becton Dickinson, Heidelberg, Germany) were used as primary antibodies, and horseradish peroxidase (HRP)-conjugated rabbit anti-mouse IgG antibodies (Dako, Hamburg, Germany) were used as secondary antibodies. Signals were visualized with the enhanced chemiluminescence (ECL) reagent (PJK, Kleinblittersdorf, Germany). As a loading control, mouse actin-specific antibodies (Sigma-Aldrich) were used (8).
Quantification of secreted LOX.
LOX concentrations from 100 μl of cell culture supernatants were measured by enzyme-linked immunosorbent assays (ELISA; USCN, Wuhan, China) according to the manufacturer's instructions. Plates were read on an ELISA reader (Tecan, Crailsheim, Germany) at 450 nm, and concentrations were calculated using Magellan software (version 6.0; Magellan Software GmbH, Dortmund, Germany).
RNA isolation, reverse transcription of mRNA, and PCR.
Total RNA was isolated from control and infected human cells or cell lines with the RNeasy or the miRNeasy minikit (Qiagen, Hilden, Germany). Contaminating DNA was removed from RNA preparations by using the Turbo DNA-free kit (Applied Biosystems, Darmstadt, Germany) or the RNase-free DNase kit (Qiagen). Reverse transcription was carried out using SuperScript III reverse transcriptase (Invitrogen, Karlsruhe, Germany), RNaseOUT recombinant RNase inhibitor (Invitrogen), oligo(dT) 18 mRNA primers (New England Biolabs, Frankfurt am Main, Germany) and nucleotides (10 mmol/liter deoxynucleoside triphosphate [dNTP] mixture; Fermentas, St. Leon-Rot, Germany). In cell culture experiments, the expression of the genes encoding HIF-1α, vascular endothelial growth factor A (VEGFA), LOX, β-2 microglobulin (B2M), prolyl hydroxylase 3 (PHD3), hexokinase 2 (HK2), 6-phosphofructo-2-kinase 3 (PFKFB3), and the Tet oncogene 1 protein (TET1) was analyzed using a TaqMan 7900HT Fast real-time PCR system (Applied Biosystems, Darmstadt, Germany) and the LightCycler 480 master mix (Roche, Mannheim, Germany). Primers for HIF-1α, VEGF, LOX, B2M, and PHD3 were obtained from Search LC (Heidelberg, Germany). The following primers were used for quantitative PCR of HK2, PFKFB3, and TET1: HK2f (5′-GTG GCT GTG GTG AAT GAC AC-3′) and HK2r (5′-ACA TCC CGC TGA TCA TCT TC-3′), PFKFB3f (5′-CTT GTC GCT GAT CAA GGT GA-3′) and PFKFB3r ((5′-TTC TGC TCC TCC ACG AAC TT-3′), and TET1f (5′-CTG CTT CAG CCA CAC CAG CTC-3′) and TET1r (5′-GGC TGA TGA GGC GTT AGC GGC-3′) (Metabion, Matrinsried, Germany), respectively. HIF-1α, VEGF, LOX, B2M, and PHD3 were amplified as follows: initial denaturation (10 min at 95°C) was followed by 35 cycles of denaturation (10 s at 95°C), primer annealing (10 s at 68°C), and elongation (16 s at 72°C), with subsequent melting-curve analysis. For the amplification of HK2, PFKFB3, and TET1, the annealing temperature was 64°C and the elongation time was 25 s.
For the isolation of total RNA from murine kidneys, PeqGold RNAPure buffer (PeqLab, Erlangen, Germany) was used, followed by phenol-chloroform extraction. Samples were further processed as described above. To analyze Lox gene expression in vivo, murine Lox and hypoxanthine phosphoribosyltransferase (Hprt) primers and probes (Applied Biosystems) and the TaqMan Universal master mix (Applied Biosystems) were used. Lox and Hprt were amplified as follows: initial incubation for 2 min at 50°C, initial denaturation for 10 min at 95°C, and 40 cycles of amplification (denaturation for 15 s at 95°C; annealing and amplification for 1 min at 60°C).
The relative gene expression was calculated using the 2−ΔΔCT method (33). B2M or Hprt was used as an internal control gene for cell culture or intravenous infection experiments, and relative gene expression was calculated in comparison to expression in uninfected control cells or organs of uninfected control animals.
Affymetrix Human Gene 1.0 ST array.
Whole-transcriptome analysis of HepG2 HIF-1α−/− and HepG2 cells was performed using an Affymetrix Human Gene 1.0 ST array. For this purpose, triplicates of infected and uninfected HepG HIF-1α−/− and HepG2 cells were analyzed, and total RNA was extracted as described above. RNA quality was evaluated on an Agilent 2100 Bioanalyzer with RNA integrity numbers (RIN) ranging from 7.1 to 10. Double-stranded cDNA was synthesized from 100 ng of total RNA and was subsequently linearly amplified and biotinylated by using the GeneChip WT cDNA synthesis and amplification kit (Affymetrix, Santa Clara, CA) according to the manufacturer's instructions. Fifteen micrograms of labeled and fragmented cDNA was hybridized to GeneChip Human Gene 1.0 ST arrays (Affymetrix). After hybridization, the arrays were washed and stained in a Fluidics Station 450 (Affymetrix) with the recommended washing procedure. Biotinylated cDNA bound to target molecules was detected with streptavidin-coupled phycoerythrin, followed by biotinylated anti-streptavidin IgG antibodies, and then streptavidin-coupled phycoerythrin again, according to the protocol described previously (34). Arrays were scanned using the GCS 3000 GeneChip scanner (Affymetrix) and AGCC 3.0 software. Scanned images were inspected visually to check for hybridization artifacts and proper grid alignment and were analyzed with Expression Console 1.0 (Affymetrix) to generate report files for quality control. Raw data were normalized with GeneSpring software, version 9.3, by applying an RMA (robust multiarray average) algorithm. Significance was calculated using a t test without multiple testing correction (GeneSpring), by selecting all transcripts with a minimum expression level change of 1.5-fold together with a P value of <0.05. In the evaluation of the total number of regulated genes, empty and double annotations were deleted for further analysis.
Intravenous infection of mice with S. aureus.
Mouse infection experiments were carried out in accordance with the animal experiment proposal (H2/06 and H1/12; Regierungspräsidium Tübingen, Tübingen, Germany). Female NMRI mice (weight, 33 to 35 g) were infected intravenously (i.v.) with S. aureus 8325-4 resuspended in 200 μl PBS (∼1.15 × 107 bacteria/mouse). The inoculum was verified by plating serial dilutions. Control mice were injected with the same volume of sterile PBS.
Five to 7 days postinfection (p.i.), mice were euthanized. Kidneys either were fixed in 5% buffered formalin (Roth) for histological staining or were frozen in PeqGold RNAPure buffer (PeqLab) in liquid nitrogen for RNA isolation. For analysis of bacterial loads, the weight of the kidneys was determined. Organs were homogenized in ice-cold PBS by using a mesh, and serial dilutions were plated in duplicate on BM agar plates. S. aureus numbers in circulating blood were determined by puncture of the right heart ventricle 5 days after infection. Coagulation of blood was inhibited by heparin, and bacterial loads were determined by counting CFU on BM agar in serial dilutions.
In some experiments, mice were infected with S. aureus and were simultaneously treated with the LOX inhibitor β-aminopropionitrile (BAPN; Sigma-Aldrich). BAPN was injected intraperitoneally (i.p.; 150 mg/kg of body weight in 200 μl PBS) daily, starting 1 day before infection, and the dosage was chosen according to previously published reports (21, 35, 36). Infected control mice were treated with sterile PBS (200 μl i.p.).
Selection of human abscess samples.
Formalin-fixed, paraffin-embedded diagnostic skin biopsy specimens were kindly provided by Amir Yazdi (Department of Dermatology, University of Tübingen, Tübingen, Germany). Samples were chosen according to the histological diagnosis “abscess.” To further ensure the presence of an S. aureus infection, deparaffinized tissue samples were subjected to S. aureus-specific real-time PCR as described previously (37). For this purpose, the following primers and probes for qualitative PCR of the S. aureus-specific Sa442 gene were used: primer Sa442 f (GTC GGG TAC ACG ATA TTC TTC ACG), primer Sa442 r (CTC TCG TAT GAC CAG CTT CGG TAC), probe Sa442 HP1 (TAC TGA AAT CTC ATT ACG TTG CAT CGG AA), and probe Sa442 HP2 (LC Red 705-ATT GTG TTC TGT ATG TAA AAG CCG TCT TG). PCR conditions were as follows: initial denaturation for 10 min at 95°C, followed by 50 cycles of amplification (denaturation for 10 s at 95°C; primer and probe annealing for 10 s at 50°C; elongation for 20 s at 72°C). This analysis was done in the diagnostic laboratories of the Institute of Medical Microbiology and Infection Control (Frankfurt am Main, Germany) under strict quality control conditions (DIN ISO 15189:2007; laboratory certificate, 13102-01-00). Only samples with the histological diagnosis “abscess” and the molecular detection of S. aureus were included in histological studies.
Histology and LOX IHC of patient and murine samples.
For murine kidney and human skin samples, serial sections were processed by LOX immunohistochemistry (IHC) (see below) and hematoxylin-eosin (H&E) or Masson's trichrome staining according to standard protocols. Tissue sections (2 μm) were deparaffinized and dehydrated by two successive 5-min treatments with xylene, followed by one 5-min treatment each with 100%, 95%, 80%, and 75% ethanol, deionized water, and PBS. Antigen was retrieved by microwave heat treatment of slides in 10 mM citric acid buffer (pH 6.0). Slides were allowed to cool and were washed with PBS for 5 min. Sections were treated with 3% hydrogen peroxide in methanol for 30 min, followed by two 5-min washing steps (PBS) and blocking in 10% normal goat serum for 30 min at room temperature. Slides were again washed twice with PBS (5 min each time). Sections were stained either with nonimmune rabbit IgG (10 μg/ml) as a negative control or with an affinity-purified rabbit anti-rat lysyl oxidase antibody raised against amino acid residues 78 to 115 contained in the unique lysyl oxidase proregion (38) and were visualized with immunoperoxidase-based technology by using the anti-rabbit Elite VectaStain kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Corresponding sections were counterstained with hematoxylin-eosin by using standard laboratory procedures.
Microscopy.
LOX and H&E staining patterns were evaluated using an AxioImager M2 microscope (Zeiss, Jena, Germany). Pictures were taken with a Spot camera system (Visitron, Puchheim, Germany) using Spot Advanced software, version 4.7. To avoid observer-specific bias, abscess morphology was classified by sorting abscesses into three different morphologies: (i) well-defined (abscess formation with clear abscess borders), (ii) diffuse (massive infiltration of granulocytes in the surrounding tissue without a clear abscess border), and (iii) intermediate. Microscopic evaluation was carried out by two independent researchers.
Statistical analysis.
All experiments were repeated (except for triplicate transcriptome analysis) and revealed comparable results. For statistical analysis, the unpaired, two-tailed Student t test was used. A P value of <0.05 was considered statistically significant.
Microarray data accession number.
The results of the transcriptome analysis have been deposited in Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE44943.
RESULTS
S. aureus infection activates HIF-1 and causes HIF-1-dependent gene programming.
To detect the host cell genes that are regulated via HIF-1 in the course of an S. aureus infection, we first analyzed the transcriptome profile of epithelial cells upon infection. For this purpose, HepG2 HIF-1α−/− cells and HepG2 cells, which were generated previously (32), were infected for 4 h, and HIF-1 activation was determined using HIF-1α-specific immunoblotting. The data revealed strong activation of HIF-1 in HepG2 but not in HepG2 HIF-1α−/− cells at this time point of infection (Fig. 1), indicating that HepG2 cells are suitable for further analysis of HIF-1-regulated gene programming.
Fig 1.
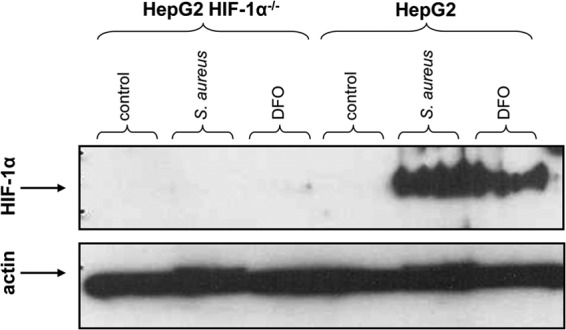
HIF-1 activation in HepG2 cells upon S. aureus infection. Four hours after infection of HepG2 HIF-1α−/− and HepG2 cells with S. aureus 8325-4 (MOI, 20), proteins were extracted, and HIF-1 expression was detected by immunoblotting. Actin was used as a loading control. Uninfected cells were used as the negative control, and DFO (200 μM) was used as the positive control.
To screen for HIF-1-regulated genes, the transcriptomes of S. aureus-infected HepG2 HIF-1α−/− cells and HepG2 control cells were determined using an Affymetrix microarray-based approach. For this purpose, total RNAs from three independent samples each of uninfected or infected HepG2 or HIF-1α−/− cells were prepared, transcribed to cDNA, cRNA, and biotinylated cDNA using random hexamer primers, and hybridized to Affymetrix ST 1.0 microarrays. Signals were analyzed with Affymetrix Expression Console, version 1.0, and differentially expressed transcripts were defined by t tests based on GeneSpring, version 9.3.
This transcriptome analysis revealed that in HepG2 cells, a total of 190 genes were regulated 4 h after infection (Fig. 2), and 24 of these genes turned out to be regulated via HIF-1 (Table 1). The expression of 21 of these genes was increased in infected HepG2 control cells over that in HIF-1α−/− cells, whereas 3 genes appeared to be downregulated. For the majority (n = 20) of these genes, HIF-1-dependent gene regulation has been described previously (the exceptions are TET1, FUT11, TUBB1, and KRT18P49) (Table 1). Most of the functions of these upregulated genes, such as maintaining glucose metabolism and cell energy homoeostasis (e.g., HK2 and PHD3), have been described. Furthermore, the tumor growth-relevant genes VEGF and LOX were also expressed in a HIF-1-dependent manner.
Fig 2.
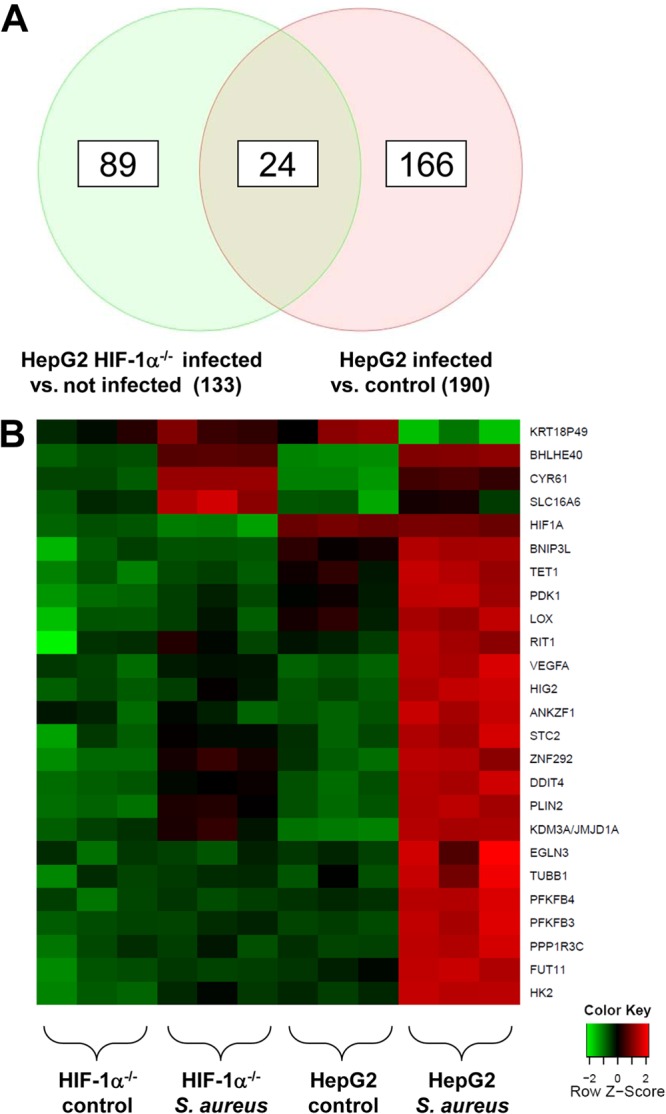
HIF-1-dependent gene expression in HepG2 cells upon S. aureus infection. HepG2 and HepG2 HIF-1α−/− cells were infected with S. aureus (MOI, 20). Four hours after infection, total RNA was isolated, transcribed, and hybridized to Affymetrix Human Gene 1.0 ST arrays (see Materials and Methods). Analysis was performed in triplicate. (A) Venn diagram. After infection, 190 transcripts were regulated in total in HepG2 cells, 133 transcripts were differentially regulated in infected HepG2 HIF-1α−/− cells and infected control cells, and 24 transcripts were regulated by HIF-1. (B) Heat map of HIF-1-regulated genes after S. aureus infection (for details, see Table 1). Cluster analysis for selected probe sets was performed using R2.15.1 software. Signal intensities were scaled and centered, and the distance between two expression profiles was calculated using the Manhattan distance measure. Hierarchical cluster analysis was performed with average linkages. Heat maps were generated with geneplotter in the Bioconductor package.
Table 1.
HIF-1-regulated genes in HepG2 cells 4 h after S. aureus infection
| No. | Protein | Genea | Differential inductionb | Biological functionc (reference) | Reference sequence | Known HIF-1 target (reference) |
|---|---|---|---|---|---|---|
| 1 | DNA damage-inducible transcript 4 | DDIT4 | 5.18 | Stress responsec | NM_019058 | Yes (50) |
| 2 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 | PFKFB3 | 3.97 | Energy metabolismc | NM_004566 | Yes (51) |
| 3 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 | PFKFB4 | 3.46 | Energy metabolismc | NM_004567 | Yes (51) |
| 4 | Pyruvate dehydrogenase kinase, isozyme 1 | PDK1 | 3.02 | Energy metabolismc | NM_002610 | Yes (52) |
| 5 | Fucosyltransferase 11 [α(1,3) fucosyltransferase] | FUT11 | 2.70 | Fucosylation (53) | NM_173540 | No |
| 6 | Protein phosphatase 1, regulatory (inhibitor) subunit 3C | PPP1R3C | 2.51 | Glycogen synthesisc | NM_005398 | Yes (54) |
| 7 | BCL2/adenovirus E1B 19-kDa interacting protein 3-like | similar to p40 | BNIP3L | LOC440258 | 2.38 | Autophagy (55) | NM_004331 | Yes (56) |
| 8 | Hexokinase 2 | HK2 | 2.31 | Glucose metabolismc | NM_000189 | Yes (52) |
| 9 | Egl nine homolog 3 (Caenorhabditis elegans) | EGLN3 | 2.05 | Prolyl hydroxylase 3, hypoxic regulationc | NM_022073 | Yes (57) |
| 10 | Lysyl oxidase | LOX | 2.03 | Modulator of extracellular matrix, progression and metastasis of breast cancer (21, 44, 58) | NM_002317 | Yes (21) |
| 11 | Tet oncogene 1 protein | TET1 | 1.98 | Potential epigenetic regulatorc | NM_030625 | No |
| 12 | Vascular endothelial growth factor A | VEGFA | 1.89 | Angiogenesis (42) | NM_001025366 | NM_003376 | NM_001025367 | NM_001025368 | NM_001025369 | NM_001025370 | NM_001033756 | Yes (59) |
| 13 | Ankyrin repeat and zinc finger domain containing 1 | ANKZF1 | 1.83 | Unknown | NM_018089 | NM_001042410 | Yes (60) |
| 14 | Jumonji domain containing 1A | JMJD1A | 1.71 | Demethylasec | NM_018433 | Yes (61) |
| 15 | Basic helix-loop-helix domain containing, class B, 2 | BHLHE40 | 1.71 | Differentiationc | NM_003670 | Yes (62) |
| 16 | Tubulin beta-1 chain | TUBB1 | 1.65 | Platelet assemblyc | NM_030773 | No |
| 17 | Hypoxia-inducible protein 2 | HIG2 | 1.69 | Cell growth, lipid droplet protein (63, 64) | NM_013332 | NM_001098786 | Yes (64) |
| 18 | Adipose differentiation-related protein | PLIN2/ADFP | 1.66 | Lipid droplet protein (65) | NM_001122 | Yes (62) |
| 19 | Stanniocalcin 2 | STC2 | 1.62 | Potential role for glucose homeostasis and cell proliferation (66, 67) | NM_003714 | Yes (67) |
| 20 | Ras-like without CAAX 1 | RIT1 | 1.59 | GTPasec | NM_006912 | Yes (68) |
| 21 | Zinc finger protein 292 | ZNF292 | 1.53 | Zinc finger protein, unknown function (69) | NM_015021 | Yes (62) |
| 22 | Solute carrier family 16, member 6 (monocarboxylic acid transporter 7) | SLC16A6 | −2.05 | Potential monocarboxylic acid transporterc | NM_004694 | Yes (70) |
| 23 | Cysteine-rich, angiogenic inducer, 61 | CYR61 | −1.65 | Vasculo- and angiogenesisc | NM_001554 | Yes (70) |
| 24 | Keratin 18 pseudogene 49 protein | KRT18P49 | −1.63 | Unknown function | XR_016590 | XR_018216 | XM_001714362 | No |
Three independent data sets were analyzed on Affymetrix Human Gene ST 1.0 arrays by using GeneSpring, version 9.3, as described in Materials and Methods.
Differential expression in HepG2 and HepG2 HIF-1α−/− cells after S. aureus infection.
According to OMIM.
To confirm the data of the microarray analysis, HIF-1-dependent regulation of various selected genes (VEGF, LOX, TET1, PHD3, HK2, PFKFB3) was analyzed by using conventional real-time PCRs. According to the gene array data, the expression of all genes analyzed was upregulated upon infection in HepG2 control cells but not in HIF-1α−/− cells (2.7-fold for LOX, 2.7-fold for VEGF, 3.4-fold for HK2, 2.1-fold for PFKFB3, 2.0-fold for TET1, and 11.0-fold for PHD3). As expected, the transcription of the HIF-1α gene itself was not regulated upon infection (since HIF-1 is known to be predominantly posttranslationally regulated [1]), but HIF-1α gene transcription was highly suppressed (10.0-fold) in HepG2 HIF-1α−/− cells relative to that in control HepG2 cells (Fig. 3).
Fig 3.

HIF-1-dependent gene expression in HepG2 HIF-1α−/− and HepG2 cells. Four hours after infection with S. aureus (MOI, 20), total RNA was isolated from HepG2 HIF-1α−/− and HepG2 cells and was transcribed into cDNA, and the relative expression of the LOX, VEGF, HK2, PFKFB3, TET1, PHD3, and HIF-1α genes was determined by real-time PCR analysis in triplicate (filled bars). Expression was calculated in relation to that in noninfected HepG2 cells (open bars). The β-2-microglobulin gene (B2M) was used for normalization. Asterisks indicate significant differences from values for control cells (P < 0.01); ns, not significant.
LOX expression is strongly upregulated in S. aureus infection models in vitro.
LOX turned out to represent one of the HIF-1-regulated genes in S. aureus infections (see above). LOX encodes the enzyme lysyl oxidase, a copper-dependent amine oxidase that catalyzes the final enzymatic step required for the cross-linking of collagen and elastin molecules in the extracellular matrix (22). Currently, nothing is known about the function of LOX in bacterial infections. Since tissue remodelling occurs during S. aureus infections (e.g., abscess formation), and LOX might possibly be involved in this process, the role of LOX in S. aureus infections was analyzed in greater detail.
First, to exclude cell line-specific and artificial results, we analyzed whether the increased LOX gene transcription in S. aureus infections occurs in cells other than HepG2 cells. For this purpose, primary human keratinocytes were infected with S. aureus, and LOX expression was determined by real-time PCR. The data revealed that LOX expression was 5.6-fold increased 4 h after infection (Fig. 4A), thus confirming our results in HepG2 cells (see above). Moreover, to verify that increased LOX gene expression is also followed by increased LOX protein synthesis, LOX secreted from supernatants of S. aureus-infected and uninfected HepG2 cells was quantified by a LOX-specific ELISA. Here, a 12.7-fold increase in the concentration of secreted LOX was detectable (Fig. 4B). Both results clearly demonstrate that LOX transcription is a general phenomenon upon infection of epithelial cells with S. aureus and that mature LOX protein is present in the supernatants of infected cells.
Fig 4.
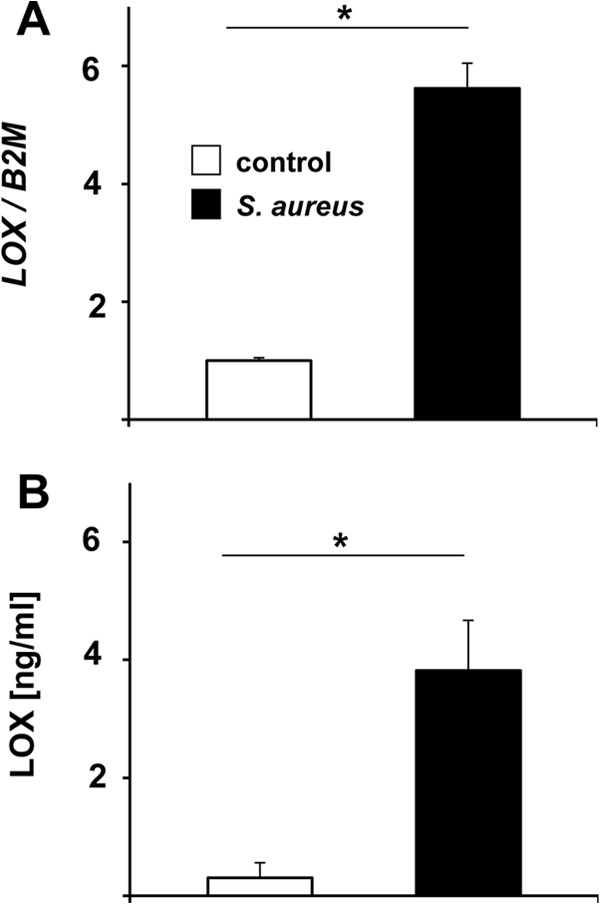
Induction of LOX upon S. aureus infection. (A) LOX expression in NHEKs upon S. aureus infection. NHEKs were infected with S. aureus (MOI, 20) for 4 h; total RNA was isolated and was transcribed to cDNA; and the relative expression of the LOX gene was determined using real-time PCR analysis. Expression was calculated relative to that in uninfected control cells (triplicates). The β-2-microglobulin gene (B2M) was used for normalization. Asterisks indicate significant differences from control cells (P < 0.01). (B) LOX secretion from S. aureus-infected HepG2 cells (MOI, 0.01). Supernatants were taken 16 h after infection and were analyzed by LOX-ELISA (triplicates). Asterisks indicate significant differences from control cells (P < 0.01).
LOX expression is strongly upregulated in S. aureus infections in vivo.
Next, we analyzed whether LOX expression is increased not only in vitro in cell culture infection models but also in vivo. For this purpose, Lox gene transcription and LOX expression in kidneys containing abscesses due to S. aureus were analyzed using a murine S. aureus infection model. In this model, mice are infected intravenously with a sublethal number of bacteria and develop kidney abscesses 4 to 6 days after infection (8). Such abscesses were also detectable in our experiments (data not shown).
Lox transcripts in kidneys positive for S. aureus abscesses were quantified using Lox real-time PCR analysis (Fig. 5A). Lox expression was strongly enhanced in total-kidney tissue samples of infected mice (∼16.8-fold) over that for uninfected control mice. Moreover, a significant quantity of murine kidney abscesses due to S. aureus stained positive for LOX by use of immunohistochemistry (IHC). In these experiments, sequential sets of hematoxylin-eosin-stained slides (used in the first step to detect abscesses) and LOX-IHC-stained slides were evaluated in parallel. S. aureus-infected mice reliably developed abscesses 5 to 7 days after infection, which were of typical morphology in hematoxylin-eosin staining (Fig. 5B). LOX-positive cells were detected in kidney abscesses, especially at the abscess borders (Fig. 5C and D). Internal IgG subtype staining controls remained negative (data not shown), underlining the specificity of this IHC staining.
Fig 5.

Detection of LOX in murine kidney abscesses due to S. aureus. NMRI mice were intravenously infected with S. aureus 8325-4 (1.0 × 107 bacteria; 200 μl) for 5 to 7 days. Uninfected control mice were treated i.v. with sterile PBS (200 μl). (A) Lox expression in kidneys. Total RNA was isolated from kidneys, transcribed to cDNA, and analyzed by real-time PCR. Lox expression in infected mice (n = 12) was calculated relative to that in the uninfected, PBS-treated control group (n = 13). The hypoxanthine phosphoribosyltransferase gene (Hprt) was used for normalization. The asterisk indicates a significant difference from the control group (P < 0.01). (B through D) Hematoxylin-eosin (H/E) staining (B) and LOX immunohistochemistry (C and D) of kidney abscesses due to S. aureus. Images represent the results of two independent experiments, each with three control mice and three infected mice. Kidneys were formalin fixed and paraffin embedded, and sections were stained with H&E and with a LOX-IHC stain. LOX-positive cells appear abundant at abscess borders. Arrows indicate abscesses. Bars, 100 μm.
To transfer these results from murine infection models to human infections, we applied LOX IHC to samples of human abscesses due to S. aureus. Histopathologically diagnosed abscesses in skin samples were selected and subjected to S. aureus-specific real-time PCR analysis. Strong LOX staining was detected in biopsy specimens of human abscesses due to S. aureus (n = 3) in the area of the abscess, whereas healthy, unaffected tissues from the same patient or tissues from healthy individuals (n = 3) remained negative throughout LOX staining (Fig. 6).
Fig 6.
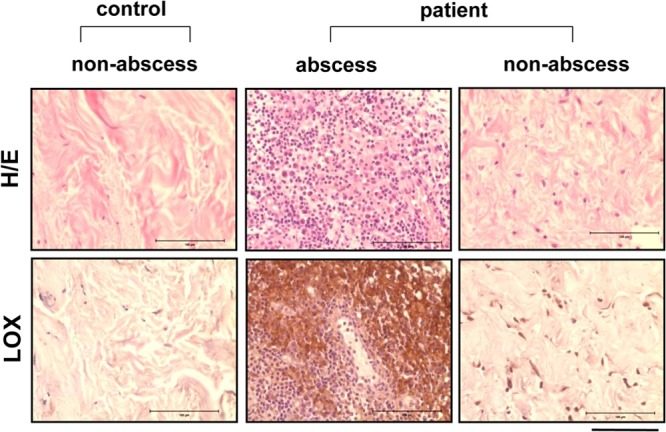
Detection of LOX in skin biopsy specimens from patients with cutaneous S. aureus infections. Human skin biopsy specimens of healthy tissue and of abscesses due to S. aureus were formalin fixed and paraffin embedded. Sections were analyzed by H&E staining and by LOX-IHC. Strong LOX signals were detectable in all S. aureus-infected human tissue in the area of abscess formation, whereas no signals appeared in healthy tissue from the same patients or in healthy control samples (n, 3 per group). Bars, 100 μm.
Taken together, mRNA transcript and protein levels were increased in murine kidney abscesses due to S. aureus and in human tissue samples from infected skin, demonstrating conclusively that LOX is expressed both in vitro (human cell culture infection models) and in vivo (murine S. aureus abscess-forming infection models; human skin infections).
Inhibition of LOX by BAPN affects the formation of abscesses due to S. aureus.
Since LOX expression appeared to be increased during the formation of abscesses due to S. aureus, we examined the biological role of LOX during this process. For this purpose, BAPN was used to modify the course of an intravenous S. aureus infection. BAPN is a well-recognized inhibitor of LOX activity (35); the oxidized intermediate of BAPN is covalently bound to the active site of LOX and inhibits its enzymatic activity (39).
First, antimicrobial side effects of BAPN were excluded by in vitro testing of the compound. Clearly, even at concentrations higher than those used in the subsequent in vivo infection models, BAPN did not affect the growth of S. aureus (data not shown). Intravenous infection experiments were performed as described above in the presence of the LOX inhibitor BAPN, which was administered daily by intraperitoneal injection starting the day before infection. Infected control mice were injected with PBS. Data on the survival of mice, bacterial clearance in the kidneys, and abscess morphology were collected. First, the experiments demonstrated that the LOX inhibitor BAPN did not influence the course of S. aureus infection, since treatment with BAPN had no effect on the survival rate (100% survival after 5 days of infection for both PBS- and BAPN-treated mice). Second, bacterial loads were analyzed by determining bacterial CFU in the kidneys and blood of BAPN-treated and control mice. The CFU counts for PBS- and BAPN-treated mice did not differ (5.77 versus 5.58 log10 CFU in the kidneys and 0.14 versus 0.64 log10 CFU in the blood); thus, no effect on bacterial clearance was observed within the first 5 days after infection (data not shown).
Finally, to analyze the influence of BAPN on abscess morphology, hematoxylin-eosin-stained kidney sections were analyzed microscopically, and the percentages of abscesses with the following three morphologies were determined by microscopy: (i) well-defined, round, with clear borders, (ii) intermediate, and (iii) diffuse, without clear borders. By this classification, well-defined abscesses were characterized by a local accumulation of immune cells and a clearly defined abscess border, diffuse abscesses by massive infiltration of immune cells into the surrounding tissue without a clear abscess border, and intermediate abscesses by a phenotype in between. Representative examples of these abscess phenotypes are depicted in Fig. 7A. The data revealed that all three abscess morphologies were present in both PBS- and BAPN-treated mice. However, PBS-treated mice more frequently showed well-defined abscesses (55.1% ± 30.5%) and only a minor proportion of diffuse abscesses (1.7% ± 3.9%). In contrast, the abscesses of BAPN-treated mice appeared more diffuse than those of PBS-treated mice. In BAPN-treated mice, the incidence of diffuse abscesses was significantly greater (12.7% ± 11.4%) and the percentage of well-defined abscesses was smaller (33.4% ± 20.4%). When the kidneys of control mice were analyzed, abscess borders appeared to be stained blue in Masson's trichrome staining, indicating collagen deposition, whereas this phenomenon was much less detectable in mice treated with BAPN (Fig. 8). These data demonstrate that administration of BAPN affects the formation of murine kidney abscesses and the collagenization of abscess borders in an intravenous S. aureus infection model.
Fig 7.

Influence of the LOX inhibitor BAPN on kidney abscess morphology. NMRI mice were infected i.v. with S. aureus (∼1 × 107 bacteria; 200 μl) for 5 days and were treated i.p. daily with the LOX inhibitor BAPN (150 mg/kg; 200 μl) starting 1 day before infection. Control mice (also infected with S. aureus) were treated with sterile PBS (200 μl). Five days after infection, kidneys were excised, formalin fixed, and paraffin embedded, and sections were stained with H&E. (A) Sections were analyzed microscopically and were classified as follows: (i) defined (clear abscess formation), (ii) intermediate, and (iii) diffuse (massive infiltration of granulocytes in the surrounding tissue without a clear abscess border). Bar, 100 μm. (B) Percentages of different abscess phenotypes from two independent experiments. Abscesses (100 abscesses from the kidneys of 12 mice treated with PBS and 123 abscesses from the kidneys of 10 mice treated with BAPN) were quantified microscopically, and the percentage of each phenotype was determined microscopically (*, P < 0.05).
Fig 8.
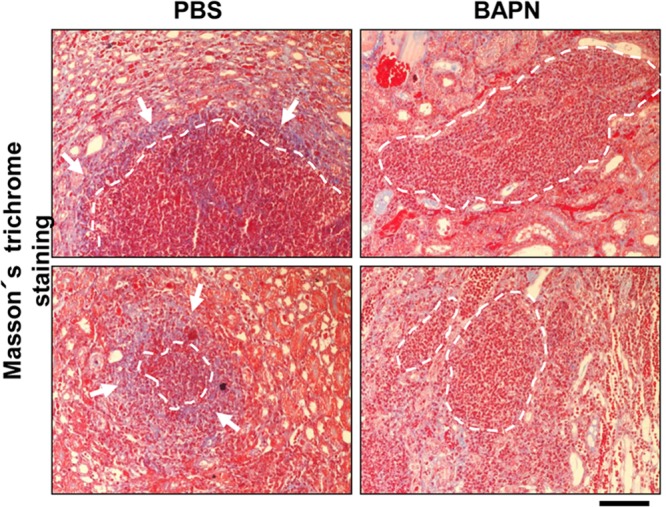
Influence of the LOX inhibitor BAPN on the collagenization of abscesses. NMRI mice were treated as described in the legend to Fig. 7. Five days after infection, kidneys were excised, formalin fixed, and paraffin embedded, and sections were stained with Masson's trichrome stain. Note the clear blue lining (arrows) of abscess borders (dashed white lines) in the control group, indicating collagen, which is much less evident in BAPN-treated mice. Bar, 100 μm.
DISCUSSION
In this work, we demonstrate a HIF-1-dependent gene expression pattern upon S. aureus infection by using HIF-1 knockdown cells, showing that 24 genes (including LOX) were regulated in a HIF-1-dependent manner. Furthermore, we demonstrate that LOX transcription and expression are strongly induced during abscess formation due to S. aureus and upon S. aureus infection by using murine infection models and by analyzing samples of infected skin from human patients. Moreover, chemical inhibition of bacterially induced LOX by BAPN resulted in the formation of a more diffuse abscess phenotype and less collagen deposition at abscess borders, suggesting that LOX inhibition might influence the course of abscess formation in S. aureus infections.
The finding that cellular reactions to infections are similar to those known from hypoxia is a recent finding. In particular, the activation of HIF-1, which is known to be regulated in infections by hypoxia or hypoxia-mimicking conditions (8, 10), is an exciting phenomenon, linking the scientific topic “infection and inflammation” to the area of “cancer research,” where HIF-1-driven pathways have been objects of major interest for years (40).
The role of HIF-1 in bacterial infections has been elucidated only partly so far. The activation of HIF-1 does not argue for a clear beneficial or detrimental effect on the course of infections. On the one hand, HIF-1 mediates phagocytosis and the elimination of bacteria in skin infection models, thereby preventing the spread of infection (13, 14), and similarly, an intestinal HIF-1α deletion led to higher susceptibility of mice to intestinal yersiniosis (10). On the other hand, HIF-1 promotes overwhelming immune responses in systemic infections, leading to higher survival rates for HIF-1α knockout mice than for wild-type mice in lipopolysaccharide (LPS)-induced sepsis (15) or for mice treated with a HIF-1 inhibitor in an S. aureus peritonitis model (8).
In a previous study, we showed that S. aureus infection leads to HIF-1 activation, most likely mediated via oxygen consumption, suggesting inactivation of PHD activity (8). Here, using HepG2 cells with a stable HIF-1α knockdown, we identified 24 genes regulated by HIF-1 upon S. aureus infection, including not only genes important for the maintenance of cell homeostasis and energy metabolism (e.g., HK2, PFKFB3, PFKFB4, PDK1) and oxygen supply (e.g., VEGF, PHD3) but also other genes with less clear functions (e.g., TET1, JMJD1A, BHLHB2, HIG2). This HIF-1-dependent reprogramming of infected host cells seems to be a robust process; VEGF expression in S. aureus infections, for example, has been described previously (8, 41). Interestingly, two HIF-1 target genes found to be induced in S. aureus infections are VEGF and LOX. VEGF has long been known as a HIF-1 target gene crucial for promoting tumor angiogenesis (42). Recently, LOX was also found to be regulated by HIF-1 and to contribute greatly to the progress of cancer by facilitating the metastasis of cancer cells in the organism (21).
LOX is a key component in extracellular matrix remodelling and is also involved in the development of fibrotic diseases. LOX is required for the biosynthetic cross-linking of collagen and elastin molecules, a reaction leading to the formation of H2O2 as a by-product (43). In mice, Lox knockout is lethal, since the development of the cardiovascular and respiratory system is severely affected (44). Only very limited information is available regarding the role of LOX in infections. Enhanced Lox mRNA levels were detected in a murine schistosomiasis model (Schistosoma mansoni) during liver granuloma formation (26), and increased LOX expression was also detected in the livers of mice infected with Echinococcus multilocularis (27). Elevated LOX levels were also present in human septic periprosthetic membranes, which were found to be infected with organisms such as S. aureus (28). Our data here are principally in line with these observations: we were able to detect an increase in LOX transcription and LOX expression in vitro using human cell lines, in vivo using murine infection models, and ex vivo using human infected-skin biopsy samples. From the studies cited above and our own results, it is apparent that LOX expression occurs in a variety of infections with human-pathogenic microorganisms. Moreover, LOX has been demonstrated to be a HIF-1 target gene (21), and our own work has revealed that HIF-1 activation upon infection with S. aureus and other human pathogens appears to be a general phenomenon (8). All these facts suggest that HIF-1-driven LOX activation might represent a conserved reaction of host cells in the course of various infections, although the biological function of LOX is not clear yet.
Our data represent the first results demonstrating a potential role of LOX in the course of an infection and were generated in a murine S. aureus infection model in the presence of BAPN, a well-known covalent-binding inhibitor of LOX (35, 39). Treatment of infected mice with BAPN influenced neither survival nor bacterial clearance from the kidneys or circulating blood relative to those for corresponding PBS-treated control groups. Interestingly, we observed two effects of BAPN treatment on abscess morphology on day 5 p.i.: first, BAPN-treated mice showed a more diffuse and less well defined abscess morphology than untreated control groups, and second, BAPN prevented dense collagen deposition at the abscess borders, which was detectable in kidneys from infected control mice.
Abscess formation is the most characteristic clinical manifestation of S. aureus infections. Abscesses are defined by a purulent liquefaction process whereby S. aureus is surrounded by a pseudocapsule, zones of healthy and necrotic neutrophils, and finally, an abscess membrane consisting of eosinophilic material, macrophages, and fibroblasts (17–19). On the one hand, abscess formation is a hallmark of the attempt of the host to protect itself from S. aureus infections; an abscess is a local demarcation of infected and healthy tissue. On the other hand, such an abscess represents a niche for the pathogen, in which S. aureus can replicate shielded from the immune response of the host and antibiotic treatment (17).
The exact role of LOX in abscess formation awaits further analysis. It can be assumed that LOX activation leads to enhanced demarcation and encapsulation of the infected tissue through a collagen-cross-linking process. In fact, collagen has been demonstrated previously to represent an essential compound of the abscess membrane (18), and enhanced collagen deposition at the borders of abscesses was detectable in our infection model (analyzed by Masson's trichrome staining [Fig. 8]). Therefore, in addition to other mechanisms, enhanced LOX expression might be crucial for abscess formation. Furthermore, one could speculate that by the production of H2O2, which is generated during the oxidative deamination of substrates, LOX might enhance the migration and motility of fibroblasts and monocytes to the abscess border, since this has been demonstrated previously to occur in vitro (23, 24). This process could contribute to the formation of a fully developed abscess membrane and to a well-defined abscess phenotype. Moreover, the modulation of abscess formation by LOX inhibition might be of therapeutic relevance, since the number of well-defined kidney abscesses upon BAPN treatment is lower than that in untreated controls. Under such conditions, S. aureus should not be shielded as efficiently against antibiotics, which do not reach therapeutic levels in the centers of abscesses.
Possibly, overwhelming LOX expression might complicate the course of the healing process, even when an infection has been cleared from the organism. It is known from a multitude of different fibrosis models that LOX plays a crucial role in the process of organ fibrosis characterized by high levels of collagen accumulation and subsequent replacement of tissue with extracellular matrix components (36, 43, 45). Remarkably, a frequent complication of infections is “postinfectious organ fibrosis,” which is best known from chronic infectious diseases (e.g., from viral hepatitis [46] and lung infections [47]) and also from the Schistosoma and Echinococcus infection models published previously, in which LOX activation has been described (26, 27, 48). Moreover, a linkage between the activation of HIF-1, LOX, and fibrosis has been described already: mice with HIF-1 knockout showed less kidney fibrosis in a ureter ligation model, and fibrosis was prevented when was BAPN administered (36). Other evidence for this hypothesis comes from transgenic mice overexpressing HIF-1 in adipose tissue: these mice showed enhanced LOX levels and suffered from tissue fibrosis, the severity of which was reduced by BAPN treatment (49).
Taken together, our data demonstrate that LOX is expressed in S. aureus infections. Our in vitro, ex vivo, and in vivo data point toward an important and formerly unrecognized role of LOX in the formation of abscesses due to S. aureus. LOX expression appears to be a HIF-1-regulated process, and HIF-1 and LOX might turn out to be attractive molecules potentially representing new therapeutic targets in bacterial infections and subsequent fibrotic complications.
ACKNOWLEDGMENTS
We thank Amir Yazdi (Universitäts-Hautklinik Tübingen, Germany) for providing human skin biopsy specimens and Nathalie Dehne (Institut für Biochemie I, Goethe-Universität, Frankfurt am Main, Germany) and Erwin Bohn (Institut für Medizinische Mikrobiologie und Hygiene, Universitätsklinikum Tübingen, Germany) for expert discussions. We also thank Wibke Ballhorn (Institut für Medizinische Mikrobiologie und Krankenhaushygiene, Klinikum der Goethe-Universität, Frankfurt am Main, Germany) and Ralf Lieberz (Senkenbergisches Institut für Pathologie, Klinikum der Goethe-Universität, Frankfurt am Main, Germany) for excellent technical assistance.
This work was funded by the DFG (SFB 815 [to C.B., J.A.E., B.B., and V.A.J.K.] and TRR34 [to A.P.]), by a grant from the King Adulazi University, Jeddah, Saudi Arabia (to M.A.), and by the NIH (R01 DE11004 and R01 DE14066 [to P.C.T.]).
Footnotes
Published ahead of print 6 May 2013
REFERENCES
- 1. Semenza GL. 2011. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harbor Symp. Quant. Biol. 76:347–353 [DOI] [PubMed] [Google Scholar]
- 2. Weidemann A, Johnson RS. 2008. Biology of HIF-1α. Cell Death Differ. 15:621–627 [DOI] [PubMed] [Google Scholar]
- 3. Imtiyaz HZ, Simon MC. 2010. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 345:105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nizet V, Johnson RS. 2009. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 9:609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pugh CW, Ratcliffe PJ. 2003. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9:677–684 [DOI] [PubMed] [Google Scholar]
- 6. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr 2001. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468 [DOI] [PubMed] [Google Scholar]
- 7. Kempf VA, Lebiedziejewski M, Alitalo K, Wälzlein JH, Ehehalt U, Fiebig J, Huber S, Schütt B, Sander CA, Müller S, Grassl G, Yazdi AS, Brehm B, Autenrieth IB. 2005. Activation of hypoxia-inducible factor-1 in bacillary angiomatosis: evidence for a role of hypoxia-inducible factor-1 in bacterial infections. Circulation 111:1054–1062 [DOI] [PubMed] [Google Scholar]
- 8. Werth N, Beerlage C, Rosenberger C, Yazdi AS, Edelmann M, Amr A, Bernhardt W, von Eiff C, Becker K, Schäfer A, Peschel A, Kempf VA. 2010. Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS One 5:e11576. 10.1371/journal.pone.0011576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Riess T, Andersson SG, Lupas A, Schaller M, Schäfer A, Kyme P, Martin J, Wälzlein JH, Ehehalt U, Lindroos H, Schirle M, Nordheim A, Autenrieth IB, Kempf VA. 2004. Bartonella adhesin A mediates a proangiogenic host cell response. J. Exp. Med. 200:1267–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hartmann H, Eltzschig HK, Wurz H, Hantke K, Rakin A, Yazdi AS, Matteoli G, Bohn E, Autenrieth IB, Karhausen J, Neumann D, Colgan SP, Kempf VA. 2008. Hypoxia-independent activation of HIF-1 by Enterobacteriaceae and their siderophores. Gastroenterology 134:756–767 [DOI] [PubMed] [Google Scholar]
- 11. Frede S, Stockmann C, Freitag P, Fandrey J. 2006. Bacterial lipopolysaccharides induce HIF-1 activation in human monocytes via p44/42 MAPK and NF-κB. Biochem. J. 396:517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rupp J, Gieffers J, Klinger M, van Zandbergen G, Wrase R, Maass M, Solbach W, Deiwick J, Hellwig-Bürgel T. 2007. Chlamydia pneumoniae directly interferes with HIF-1α stabilization in human host cells. Cell. Microbiol. 9:2181–2191 [DOI] [PubMed] [Google Scholar]
- 13. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. 2005. HIF-1α expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest. 115:1806–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. 2003. HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112:645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. 2007. Essential role of hypoxia inducible factor-1α in development of lipopolysaccharide-induced sepsis. J. Immunol. 178:7516–7519 [DOI] [PubMed] [Google Scholar]
- 16. Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532 [DOI] [PubMed] [Google Scholar]
- 17. Cheng AG, Dent AC, Schneewind O, Missiakas D. 2011. A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol. 19:225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flaris NA, Hickey WF. 1992. Development and characterization of an experimental model of brain abscess in the rat. Am. J. Pathol. 141:1299–1307 [PMC free article] [PubMed] [Google Scholar]
- 19. Lam GT, Sweeney FJ, Jr, Witmer CM, Wise RI. 1963. Abscess-forming factor(s) produced by Staphylococcus aureus. I. Collodion bag implantation technique. J. Bacteriol. 86:611–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Freedlander SO, Toomey JA. 1928. The role of clasmatocytes and connective tissue cells in non-specific local cutaneous immunity to Staphylococcus. J. Exp. Med. 47:663–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ. 2006. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440:1222–1226 [DOI] [PubMed] [Google Scholar]
- 22. Kagan HM, Li W. 2003. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J. Cell. Biochem. 88:660–672 [DOI] [PubMed] [Google Scholar]
- 23. Lazarus HM, Cruikshank WW, Narasimhan N, Kagan HM, Center DM. 1995. Induction of human monocyte motility by lysyl oxidase. Matrix Biol. 14:727–731 [DOI] [PubMed] [Google Scholar]
- 24. Nelson JM, Diegelmann RF, Cohen IK. 1988. Effect of beta-aminopropionitrile and ascorbate on fibroblast migration. Proc. Soc. Exp. Biol. Med. 188:346–352 [DOI] [PubMed] [Google Scholar]
- 25. Li W, Liu G, Chou IN, Kagan HM. 2000. Hydrogen peroxide-mediated, lysyl oxidase-dependent chemotaxis of vascular smooth muscle cells. J. Cell. Biochem. 78:550–557 [PubMed] [Google Scholar]
- 26. Sommer P, Gleyzal C, Raccurt M, Delbourg M, Serrar M, Joazeiro P, Peyrol S, Kagan H, Trackman PC, Grimaud JA. 1993. Transient expression of lysyl oxidase by liver myofibroblasts in murine schistosomiasis. Lab. Invest. 69:460–470 [PubMed] [Google Scholar]
- 27. Guerret S, Vuitton DA, Liance M, Pater C, Carbillet JP. 1998. Echinococcus multilocularis: relationship between susceptibility/resistance and liver fibrogenesis in experimental mice. Parasitol. Res. 84:657–667 [DOI] [PubMed] [Google Scholar]
- 28. Morawietz L, Weimann A, Schroeder JH, Kuban RJ, Ungethuem U, Kaps C, Slevogt H, Gehrke T, Krukemeyer MG, Krenn V. 2008. Gene expression in endoprosthesis loosening: chitinase activity for early diagnosis? J. Orthop. Res. 26:394–403 [DOI] [PubMed] [Google Scholar]
- 29. Novick R. 1967. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33:155–166 [DOI] [PubMed] [Google Scholar]
- 30. Berscheid A, Sass P, Weber-Lassalle K, Cheung AL, Bierbaum G. 2012. Revisiting the genomes of the Staphylococcus aureus strains NCTC 8325 and RN4220. Int. J. Med. Microbiol. 302:84–87 [DOI] [PubMed] [Google Scholar]
- 31. von Eiff C, McNamara P, Becker K, Bates D, Lei XH, Ziman M, Bochner BR, Peters G, Proctor RA. 2006. Phenotype microarray profiling of Staphylococcus aureus menD and hemB mutants with the small-colony-variant phenotype. J. Bacteriol. 188:687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Menrad H, Werno C, Schmid T, Copanaki E, Deller T, Dehne N, Brüne B. 2010. Roles of hypoxia-inducible factor-1α (HIF-1α) versus HIF-2α in the survival of hepatocellular tumor spheroids. Hepatology 51:2183–2192 [DOI] [PubMed] [Google Scholar]
- 33. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 34. Kurz A, Rabbani N, Walter M, Bonin M, Thornalley P, Auburger G, Gispert S. 2011. Alpha-synuclein deficiency leads to increased glyoxalase I expression and glycation stress. Cell. Mol. Life Sci. 68:721–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eliades A, Papadantonakis N, Bhupatiraju A, Burridge KA, Johnston-Cox HA, Migliaccio AR, Crispino JD, Lucero HA, Trackman PC, Ravid K. 2011. Control of megakaryocyte expansion and bone marrow fibrosis by lysyl oxidase. J. Biol. Chem. 286:27630–27638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. 2007. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 117:3810–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reischl U, Linde HJ, Metz M, Leppmeier B, Lehn N. 2000. Rapid identification of methicillin-resistant Staphylococcus aureus and simultaneous species confirmation using real-time fluorescence PCR. J. Clin. Microbiol. 38:2429–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hurtado PA, Vora S, Sume SS, Yang D, St Hilaire C, Guo Y, Palamakumbura AH, Schreiber BM, Ravid K, Trackman PC. 2008. Lysyl oxidase propeptide inhibits smooth muscle cell signaling and proliferation. Biochem. Biophys. Res. Commun. 366:156–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tang SS, Trackman PC, Kagan HM. 1983. Reaction of aortic lysyl oxidase with beta-aminopropionitrile. J. Biol. Chem. 258:4331–4338 [PubMed] [Google Scholar]
- 40. Eltzschig HK, Carmeliet P. 2011. Hypoxia and inflammation. N. Engl. J. Med. 364:656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jonkam CC, Bansal K, Traber DL, Hamahata A, Maybauer MO, Maybauer DM, Cox RA, Lange M, Connelly RL, Traber LD, Djukom CD, Salsbury JR, Herndon DN, Enkhbaatar P. 2009. Pulmonary vascular permeability changes in an ovine model of methicillin-resistant Staphylococcus aureus sepsis. Crit. Care 13:R19. 10.1186/cc7720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carmeliet P. 2005. VEGF as a key mediator of angiogenesis in cancer. Oncology 69(Suppl. 3):4–10 [DOI] [PubMed] [Google Scholar]
- 43. Kagan HM. 2000. Intra- and extracellular enzymes of collagen biosynthesis as biological and chemical targets in the control of fibrosis. Acta Trop. 77:147–152 [DOI] [PubMed] [Google Scholar]
- 44. Mäki JM. 2009. Lysyl oxidases in mammalian development and certain pathological conditions. Histol. Histopathol. 24:651–660 [DOI] [PubMed] [Google Scholar]
- 45. Kagan HM. 1994. Lysyl oxidase: mechanism, regulation and relationship to liver fibrosis. Pathol. Res. Pract. 190:910–919 [DOI] [PubMed] [Google Scholar]
- 46. Mormone E, George J, Nieto N. 2011. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem. Biol. Interact. 193:225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vanfleteren LE, Linssen CF. 2010. Role of microorganisms in interstitial lung disease. Curr. Opin. Pulm. Med. 16:489–495 [DOI] [PubMed] [Google Scholar]
- 48. Andrade ZA. 2009. Schistosomiasis and liver fibrosis. Parasite Immunol. 31:656–663 [DOI] [PubMed] [Google Scholar]
- 49. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE. 2009. Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 29:4467–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ellisen LW. 2005. Growth control under stress: mTOR regulation through the REDD1-TSC pathway. Cell Cycle 4:1500–1502 [DOI] [PubMed] [Google Scholar]
- 51. Bartrons R, Caro J. 2007. Hypoxia, glucose metabolism and the Warburg's effect. J. Bioenerg. Biomembr. 39:223–229 [DOI] [PubMed] [Google Scholar]
- 52. Semenza GL. 2010. HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20:51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mollicone R, Moore SE, Bovin N, Garcia-Rosasco M, Candelier JJ, Martinez-Duncker I, Oriol R. 2009. Activity, splice variants, conserved peptide motifs, and phylogeny of two new α1,3-fucosyltransferase families (FUT10 and FUT11). J. Biol. Chem. 284:4723–4738 [DOI] [PubMed] [Google Scholar]
- 54. Shen GM, Zhang FL, Liu XL, Zhang JW. 2010. Hypoxia-inducible factor 1-mediated regulation of PPP1R3C promotes glycogen accumulation in human MCF-7 cells under hypoxia. FEBS Lett. 584:4366–4372 [DOI] [PubMed] [Google Scholar]
- 55. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. 2009. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29:2570–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Aminova LR, Siddiq A, Ratan RR. 2008. Antioxidants, HIF prolyl hydroxylase inhibitors or short interfering RNAs to BNIP3 or PUMA, can prevent prodeath effects of the transcriptional activator, HIF-1α, in a mouse hippocampal neuronal line. Antioxid. Redox. Signal. 10:1989–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. del Peso L, Castellanos MC, Temes E, Martin-Puig S, Cuevas Y, Olmos G, Landazuri MO. 2003. The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. J. Biol. Chem. 278:48690–48695 [DOI] [PubMed] [Google Scholar]
- 58. Rodriguez C, Martinez-Gonzalez J, Raposo B, Alcudia JF, Guadall A, Badimon L. 2008. Regulation of lysyl oxidase in vascular cells: lysyl oxidase as a new player in cardiovascular diseases. Cardiovasc. Res. 79:7–13 [DOI] [PubMed] [Google Scholar]
- 59. Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E. 1998. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394:485–490 [DOI] [PubMed] [Google Scholar]
- 60. Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. 2011. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117:e207–e217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. 2009. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. U. S. A. 106:4260–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. 2006. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 66:5641–5647 [DOI] [PubMed] [Google Scholar]
- 63. Togashi A, Katagiri T, Ashida S, Fujioka T, Maruyama O, Wakumoto Y, Sakamoto Y, Fujime M, Kawachi Y, Shuin T, Nakamura Y. 2005. Hypoxia-inducible protein 2 (HIG2), a novel diagnostic marker for renal cell carcinoma and potential target for molecular therapy. Cancer Res. 65:4817–4826 [DOI] [PubMed] [Google Scholar]
- 64. Gimm T, Wiese M, Teschemacher B, Deggerich A, Schödel J, Knaup KX, Hackenbeck T, Hellerbrand C, Amann K, Wiesener MS, Höning S, Eckardt KU, Warnecke C. 2010. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 24:4443–4458 [DOI] [PubMed] [Google Scholar]
- 65. Chang BH, Chan L. 2007. Regulation of triglyceride metabolism. III. Emerging role of lipid droplet protein ADFP in health and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 292:G1465–G1468 [DOI] [PubMed] [Google Scholar]
- 66. Chang AC, Jellinek DA, Reddel RR. 2003. Mammalian stanniocalcins and cancer. Endocr. Relat. Cancer 10:359–373 [DOI] [PubMed] [Google Scholar]
- 67. Law AY, Wong CK. 2010. Stanniocalcin-2 is a HIF-1 target gene that promotes cell proliferation in hypoxia. Exp. Cell Res. 316:466–476 [DOI] [PubMed] [Google Scholar]
- 68. Warnecke C, Weidemann A, Volke M, Schietke R, Wu X, Knaup KX, Hackenbeck T, Bernhardt W, Willam C, Eckardt KU, Wiesener MS. 2008. The specific contribution of hypoxia-inducible factor-2α to hypoxic gene expression in vitro is limited and modulated by cell type-specific and exogenous factors. Exp. Cell Res. 314:2016–2027 [DOI] [PubMed] [Google Scholar]
- 69. Furney SJ, Simmons A, Breen G, Pedroso I, Lunnon K, Proitsi P, Hodges A, Powell J, Wahlund LO, Kloszewska I, Mecocci P, Soininen H, Tsolaki M, Vellas B, Spenger C, Lathrop M, Shen L, Kim S, Saykin AJ, Weiner MW, Lovestone S. 2011. Genome-wide association with MRI atrophy measures as a quantitative trait locus for Alzheimer's disease. Mol. Psychiatry 16:1130–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wan J, Ma J, Mei J, Shan G. 2009. The effects of HIF-1α on gene expression profiles of NCI-H446 human small cell lung cancer cells. J. Exp. Clin. Cancer Res. 28:150. 10.1186/1756-9966-28-150 [DOI] [PMC free article] [PubMed] [Google Scholar]