

Abstract
Cardiomyopathy is a serious complication of Chagas' disease, caused by the protozoan parasite Trypanosoma cruzi. The parasite often infects cardiac myocytes, causing the release of inflammatory mediators, including eicosanoids. A recent study from our laboratory demonstrated that calcium-independent phospholipase A2γ (iPLA2γ) accounts for the majority of PLA2 activity in rabbit ventricular myocytes and is responsible for arachidonic acid (AA) and prostaglandin E2 (PGE2) release. Thus, we hypothesized that cardiac iPLA2γ contributes to eicosanoid production in T. cruzi infection. Inhibition of the isoform iPLA2γ or iPLA2β, with the R or S enantiomer of bromoenol lactone (BEL), respectively, demonstrated that iPLA2γ is the predominant isoform in immortalized mouse cardiac myocytes (HL-1 cells). Stimulation of HL-1 cells with thrombin, a serine protease associated with microthrombus formation in Chagas' disease and a known activator of iPLA2, increased AA and PGE2 release, accompanied by platelet-activating factor (PAF) production. Similarly, T. cruzi infection resulted in increased AA and PGE2 release over time that was inhibited by pretreatment with (R)-BEL. Further, T. cruzi-infected iPLA2γ-knockout (KO) mice had lower survival rates and increased tissue parasitism compared to wild-type (WT) mice, suggesting that iPLA2γ-KO mice were more susceptible to infection than WT mice. A significant increase in iPLA2 activity was observed in WT mice following infection, whereas iPLA2γ-KO mice showed no alteration in cardiac iPLA2 activity and produced less PGE2. In summary, these studies demonstrate that T. cruzi infection activates cardiac myocyte iPLA2γ, resulting in increased AA and PGE2 release, mediators that may be essential for host survival during acute infection. Thus, these studies suggest that iPLA2γ plays a cardioprotective role during the acute stage of Chagas' disease.
INTRODUCTION
Worldwide, almost 10 million people are infected with Trypanosoma cruzi, the causative agent of Chagas' disease (1). Initially confined mostly to South America, global migration, blood products, organ transplantation, and congenital transmission have contributed to the spread of the disease, particularly in areas of nonendemicity. Millions of people are now at risk for contracting the infection, and in the United States alone, it is estimated that more than 300,000 people are infected with T. cruzi (2).
The organ primarily affected by Chagas' disease is the heart. The disease progresses from an acute stage, followed by the indeterminate stage, to the chronic stages. The acute stage of the disease may or may not be symptomatic, and if present, symptoms are usually nonspecific, such as fever, chills, and nausea. Cardiac involvement at the acute stage is rare but can occur in a small number of infected people, leading to myocarditis and sudden death. The indeterminate phase is devoid of any clinical presentation and may last for several decades. Among infected patients, approximately 30% develop chronic disease with cardiac involvement (3, 4). Chronic Chagas' heart disease includes arrhythmias, thromboembolic events, and congestive heart disease, leading to sudden death (5). A key pathological feature of Chagas' disease is inflammation, which is regulated by several mediators, including adhesion molecules, cytokines, eicosanoids, and nitric oxide (6–8). In the acute stage, these molecules are seen to be important for the development of host resistance, as well as regulating progression to the chronic stages of the disease (9). However, much remains unknown about the host-parasite interactions that contribute to Chagas' disease via these mediators. A better understanding of the complex relationship between the parasite, its target organ, and the cascade of events following infection will give us valuable information regarding a major human disease that we still have very limited means to treat.
Following infection, the parasites move across the endothelium and vascular tissue to gain access to the myocardium. After penetrating cardiac myocytes, the parasites divide intracellularly before causing the cell to rupture, releasing more parasites that can then spread and infect other cells. This cycle of infection and multiplication of the parasites, resulting in host cell lysis, directly causes tissue injury in the myocardium. Myocytes also contribute indirectly to disease progression via cytokine, chemokine, and reactive oxygen species (ROS) production, accompanied by changes in myocyte morphology (8, 10, 11).
The release of arachidonic acid from membrane phospholipids, catalyzed by phospholipase A2 (PLA2), is the rate-limiting step in eicosanoid formation. Arachidonic acid is converted to prostaglandins via the activity of cyclooxygenase enzymes. The accompanying lysophospholipid can be converted to platelet-activating factor (PAF) following a transacetylation reaction. PAF is a potent molecule involved in the recruitment and activation of inflammatory cells under a variety of pathophysiological circumstances. Hence, using a single substrate, PLA2 enzymes can contribute to the generation of a variety of bioactive lipids with a well-established role in inflammation.
Several studies have described the presence of platelet aggregation and thrombus formation with the progress of Chagas' disease (5, 12). Thrombin is an important serine protease involved in thrombus formation that is known to activate calcium-independent PLA2 (iPLA2) in ventricular myocytes (13) and thus could exacerbate the production of bioactive lipid metabolites following T. cruzi infection. Previous studies from our laboratory have shown that the majority of the PLA2 activity in rabbit ventricular myocytes is membrane associated, calcium independent, and mediated by the gamma isoform (iPLA2γ) of the enzyme (14, 15). Additionally, iPLA2γ mediates arachidonic acid and prostaglandin E2 (PGE2) release from rabbit ventricular myocytes following stimulation with tryptase, another important protease that is released from activated mast cells (16).
The role of iPLA2 in Chagas' disease remains unexplored, despite the fact that it is an important enzyme that produces several membrane phospholipid-derived inflammatory mediators in the myocardium, including eicosanoids and PAF. Since previous studies have demonstrated an increase in eicosanoids in T. cruzi infection, it is valuable to know the signal transduction pathways involved in eicosanoid production. It is not known whether the parasite activates iPLA2 directly during infection. However, T. cruzi infection is associated with increased thrombin activation, and we have demonstrated increased cardiomyocyte iPLA2 activity in response to thrombin. The aim of this study was to determine the contribution of iPLA2 in arachidonic acid release and eicosanoid production in response to T. cruzi infection and to compare this outcome to the effect of thrombin stimulation. In vitro studies using immortalized mouse cardiac myocytes (HL-1 cells) were performed in the presence of the pharmacological inhibitors (R)- and (S)-bromoenol lactone (BEL) to discriminate between the two major isoforms, iPLA2γ and iPLA2β, respectively. To understand the contribution of this enzyme in a more physiologically relevant setting, in vivo studies were carried out using T. cruzi-infected iPLA2γ-knockout (KO) and wild-type (WT) mice.
MATERIALS AND METHODS
Cell culture.
The HL-1 cardiomyocyte cell line, derived from the AT-1 mouse atrial cardiac myocyte cell line, was obtained from W. Claycomb (Louisiana State University Medical Center, New Orleans, LA, USA) (17). Cells were grown in flasks/culture dishes coated overnight, at 37°C, with 0.5% fibronectin in 0.02% gelatin. Cells were propagated in Claycomb medium (SAFC Biosciences, Lenexa, KS) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 0.1 mM norepinephrine, 100 units penicillin, and 100 μg streptomycin/ml. Cells were infected with T. cruzi when they reached confluence and attained a beating phenotype.
Parasites and infection of HL-1 cells.
Tissue culture trypomastigotes (TCT) from the Brazil strain of T. cruzi were propagated in 3T3 mouse embryonic fibroblasts grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2% neonatal calf serum. The 3T3 cells were infected with T. cruzi on reaching 60% confluence. Infected cells ruptured following parasite multiplication, releasing a large number of parasites. The supernatant containing the parasites was collected, and parasite numbers were determined using a Neubauer hemocytometer. HL-1 cells, grown to confluence in the appropriate culture dish, were counted, and a multiplicity of infection (MOI) of 0.2 was used to infect the cells. Parasites and HL-1 cells were fed Claycomb medium supplemented with 2% FBS, and the appropriate number of parasites were then added to each culture dish. To prepare Tulahuén strain blood form trypomastigotes (BFT), BALB/c or C.B-17 SCID mice were infected subcutaneously with 5,000 to 20,000 BFT. After 12 to 16 days, the animals were sacrificed and bled by decapitation into 50-ml conical tubes containing 5 μl of heparin (1,000 U/ml)/animal. The concentration of BFT/ml was determined by hemocytometer counts with heparinized blood samples diluted 1:10 with phosphate-buffered saline (PBS).
Mice and infections.
Animal protocols were in strict accordance with the National Institutes of Health guidelines for humane treatment of animals and were reviewed and approved by the Animal Care and Use Committee of Saint Louis University. C57BL6 WT or iPLA2γ-KO mice were used for in vivo studies. iPLA2γ-KO mice were generated in the laboratory of Richard Gross and have been described previously (18). Mice were infected with the Tulahuén strain blood form trypomastigotes (5,000 BFT; subcutaneous infection) and sacrificed 14 days postinfection. Hearts were collected and frozen in liquid nitrogen before processing for biochemical analysis.
Phospholipase A2 activity.
HL-1 cells were grown to confluence and exposed to the different experimental conditions. At the end of the experimental intervention, the medium was rapidly removed; the cells were washed with ice-cold PBS; 1 ml of ice-cold PLA2 activity buffer containing (per liter) 250 mmol sucrose, 10 mmol KCl, 10 mmol imidazole, 5 mmol EDTA, and 2 mmol dithiothreitol (DTT) with 10% glycerol, pH 7.8, was added to each well; and the cells were removed from the plate using a cell scraper. Similarly, mouse hearts were flash frozen and homogenized in PLA2 buffer following collection at day 14 postinfection.
The suspension was sonicated on ice six times for 10 s each time (using a microtip probe at 20% power output; 500 Sonic Dismembrator; Fisher Scientific), and the sonicate was centrifuged at 20,000 × g for 20 min to remove cellular debris and nuclei. The pellet was resuspended in PLA2 activity buffer, and activity was determined by incubating the enzyme (50 μg protein) with 100 μM (16:0, [3H]18:1) plasmenylcholine substrate in assay buffer containing (per liter) 10 mmol Tris, 4 mmol EGTA, 10% glycerol, pH 7.0, at 37°C for 5 min in a total volume of 200 μl. The radiolabeled phospholipid substrate was introduced into the incubation mixture by injection in 5 μl ethanol to initiate the assay. Reactions were terminated by the addition of 100 μl butanol, and the released radiolabeled fatty acid was isolated by the application of 25 μl of the butanol phase to channeled Silica Gel G plates, development in petroleum ether-diethyl ether-acetic acid (70/30/1 [vol/vol/vol]), and subsequent quantification by liquid scintillation spectrometry. The protein content of each sample was determined by the Lowry method utilizing freeze-dried bovine serum albumin as the protein standard.
Measurement of total arachidonic acid release.
HL-1 cells grown in 35-mm culture dishes were incubated at 37°C with 3 μCi [3H]arachidonic acid for 18 h. This incubation resulted in >70% incorporation of radioactivity into the myocytes. After incubation, the myocytes were washed three times with Tyrode solution containing 0.36% bovine serum albumin to remove unincorporated [3H]arachidonic acid. The cells were incubated at 37°C for 15 min before being subjected to the experimental conditions. At the end of the stimulation period, the supernatant was removed and centrifuged at 2,000 × g to remove any detached myocytes. Myocytes attached to the tissue culture well were solubilized in 10% sodium dodecyl sulfate, and radioactivity in both supernatant and cells was quantified by liquid scintillation spectrometry.
Measurement of PGE2 release from HL-1 cells.
Myocytes were grown to confluence in 16-mm tissue culture dishes. The cells were washed twice with Hanks balanced salt solution (HBSS) containing (per liter) 135 mmol NaCl, 0.8 mmol MgSO4, 10 mmol HEPES (pH 7.6), 1.2 mmol CaCl2, 5.4 mmol KCl, 0.4 mmol KH2PO4, and 6.6 mmol glucose. After washing, 0.5 ml of HBSS with 0.36% bovine serum albumin (BSA) was added to each culture well. The myocytes were then subjected to the experimental conditions, and the surrounding buffer was removed from the cells after selected time intervals. Detached myocytes were removed by centrifugation at 2,000 × g, and PGE2 release was measured immediately using an immunoassay kit (R&D Systems, Minneapolis, MN).
Measurement of PGE2 content by MS.
PGE2 was quantified by liquid chromatography-tandem mass spectrometry (LC–MS-MS) using a method adapted from Degousee and coworkers (19). In brief, 25 mg of freeze-clamped and pulverized ventricular tissue was homogenized on ice in 125 μl of phosphate-buffered saline and 40 μl of methanol (containing deuterated PGE2 internal standard). Eicosanoids were then extracted twice into a volume of 600 μl, and ethyl acetate was subsequently added to the tissue homogenate and thoroughly mixed for 30 s. The pooled ethyl acetate extracts were then dried under nitrogen and resuspended in 50 μl acetonitrile-water-formic acid (20:80:0.0025 [vol/vol/vol]) prior to LC–MS-MS. Thirty microliters was injected onto a Supelco Discovery HS C18 column (150 mm by 2.1-mm inside diameter [i.d.]; 5-μm particle size) as the stationary phase, with binary discontinuous linear gradient elution using mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in methanol) at a flow rate of 0.4 ml/min. The initial mobile phase was comprised of 80% A with sequential linear gradients to 62% A (over 5 min), 50% A (over 10 min), 40% A (over 13 min), and 10% A (over 2 min). Following the linear gradients, the mobile phase was maintained for 3 min at 10% A, followed by a return to the initial conditions. A Thermo Fisher Quantum Ultra triple-quadrupole tandem mass spectrometer was operated in negative ion mode. Argon was used as the collision gas at 1 atm with collision energy at 17 eV. Electrospray ionization conditions included voltage set at 4,000 eV, sheath pressure at 40 arbitrary units, auxiliary pressure at 10 arbitrary units, and temperature at 310°C. Selected reaction monitoring of m/z 351.23 to m/z 270.95 for [3,3,4,4-d4]-PGE2 and m/z 355.25 to m/z 275.03 for PGE2 was used to detect PGE2 (Rt = 17 min). Authentic PGE2 (14010) and the internal standard, [3,3,4,4-d4]-PGE2 (10007273), were purchased from Cayman Chemicals and were used to confirm calibration factors for quantification of PGE2 from peak areas.
PAF assay.
Cardiac myocytes were grown in 16-mm tissue culture dishes and washed twice with Hanks balanced salt solution containing 135 mM NaCl, 0.8 mM MgSO4, 10 mM HEPES (pH 7.4), 1.2 mM CaCl2, 5.4 mM KCl, 0.4 mM KH2PO4, 0.3 mM Na2HPO4, and 6.6 mM glucose and incubated with 50 μCi [3H]acetic acid for 20 min. After the selected time interval for incubation with the appropriate agents, lipids were extracted from the cells by the method of Bligh and Dyer (20). The chloroform layer was concentrated by evaporation under N2, applied to a silica gel 60 (Merck) thin-layer chromatography (TLC) plate, and developed in chloroform-methanol-acetic acid-water (50/25/8/4 [vol/vol/vol/vol]). The region corresponding to PAF was scraped, and radioactivity was quantified using liquid scintillation spectrometry. Loss of PAF during extraction and chromatography was corrected for by adding a known amount of [14C]PAF as an internal standard. [14C]PAF was synthesized by acetylating the sn-2 position of lyso-PAF with [14C]acetic anhydride using 0.33 M dimethylaminopyridine as a catalyst. The synthesized [14C]PAF was purified by high-performance liquid chromatography (HPLC).
RAW cell adherence.
RAW 264.7 cells were grown to confluence in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. Cell suspensions (10 × 106/ml) were labeled with 4 μg/ml calcein-AM for 45 min at 37°C. The cells were washed three times with HEPES buffer and resuspended at a concentration of 4 × 106/ml, and 0.5 ml was added to the confluent mouse cardiac myocyte cell layer. At the end of the incubation time, nonadherent cells were removed and adherent cells and cardiac myocytes were lysed in 1 ml of 0.2% Triton X-100. Calcein fluorescence in each sample was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. The percent cell adherence in each sample was calculated based upon the fluorescence measured in 0.5 ml of RAW 264.7 cell suspension.
Measurement of parasitemia.
Circulating parasites from samples of fresh blood collected from each mouse at day 14 postinfection were counted. Blood was obtained by cardiac puncture, 1.5 μl was placed under a 12-mm round coverslip, and 25 high-power (×40) fields were counted. This method of parasitemia estimation has a lower-sensitivity detection limit of 16,740 BFT/ml.
Real-time PCR detection of T. cruzi in mouse tissue.
The hearts of WT and iPLA2γ-KO mice were harvested 14 days postinfection with T. cruzi. DNA samples were purified using a DNeasy kit (Qiagen, Valencia, CA), and the total DNA concentration was adjusted to 40 ng/μl. Primers (5′-AACCACCACGACAACCACAA-3′ and 5′-TGCAGGACATCTGCACAAAGTA-3′) were used to specifically amplify a 65-bp fragment of cruzipain, as described previously (21). However, instead of SYBR green detection, as described in the previous method, the amplicon generation was detected using a 6-carboxyfluorescein–6-carboxytetramethylrhodamine (FAM-TAMRA) probe (5′-TGCCCCAGGACCGTCCCCA-3′; Synthegen, Houston, TX) and TaqMan PCR master mix (Applied Biosystems, Carlsbad, CA) (a 900-nm concentration of each primer and 200 ng of sample DNA). A standard curve was generated using positive-control DNA harvested from a known concentration of T. cruzi epimastigotes grown in pure culture. These reactions were run in an Applied Biosystems 7500 Fast Instrument under the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Analysis of T. cruzi molecular equivalents (mEq) was performed by using Applied Biosystems 7500 software v2.0.1.
Statistical analysis.
Statistical comparison of values was performed by Student's t test or one-way analysis of variance, with post hoc analysis performed using Dunnet's test. All results are expressed as mean ± standard error of the mean (SEM). Statistical significance was considered to be a P value of <0.05.
RESULTS
Measurement of PLA2 activity in cardiac myocytes.
HL-1 cells were incubated with increasing concentrations of either (R)- or (S)-BEL for 10 min, and iPLA2 activity was measured. The R and S enantiomers of BEL are 10-fold more selective for iPLA2γ and iPLA2β, respectively (22). PLA2 activity was measured in the absence of Ca2+ (4 mM EGTA) and using (16:0, [3H] 18:1) plasmenylcholine as the substrate. As shown in Fig. 1, pretreatment with (R)-BEL to inhibit iPLA2γ resulted in significantly greater inhibition of iPLA2 activity than when cells were pretreated with (S)-BEL at the corresponding concentration. Thus, we conclude that the majority of iPLA2 activity in HL-1 cells is iPLA2γ.
Fig 1.
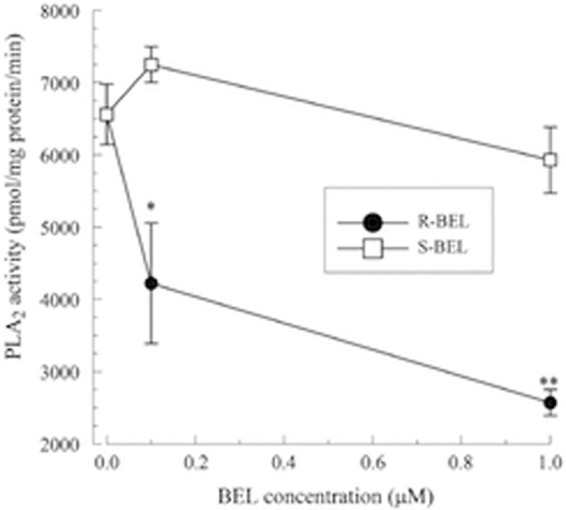
Inhibition of iPLA2 activity in mouse cardiac myocytes treated with either (R)- or (S)-BEL (10-min pretreatment). Cardiac myocytes were incubated with BEL (10 min). PLA2 activity was measured using 100 μM (16:0, [3H]18:1) plasmenylcholine in the presence of 4 mM EGTA. *, P < 0.05, and **, P < 0.01, compared to activity measured in the absence of BEL. The values shown are means ± SEM for 4 separate cell cultures.
Arachidonic acid release.
HL-1 cells were pretreated with either (R)- or (S)-BEL (2 μM; 10 min) prior to the addition of T. cruzi (MOI, 0.2), after which arachidonic acid release was measured for up to 24 h. T. cruzi infection resulted in a significant increase in arachidonic acid release that was evident after 12 h (Fig. 2A). Increased arachidonic acid release in response to T. cruzi infection was not affected by (S)-BEL pretreatment, but a significant inhibition of T. cruzi-induced arachidonic acid release was observed with (R)-BEL pretreatment. In similar experiments, thrombin (0.1 IU/ml) was added to HL-1 cells and arachidonic acid release was measured for up to 30 min, with or without pretreatment with either (R)- or (S)-BEL. Arachidonic acid release from cardiac myocytes was increased in response to thrombin (Fig. 2B). Pretreatment with (R)-BEL inhibited thrombin-induced arachidonic acid release, whereas pretreatment with (S)-BEL did not affect arachidonic acid release significantly. Thus, iPLA2γ is predominantly responsible for arachidonic acid release from mouse atrial myocytes following either T. cruzi infection or thrombin stimulation.
Fig 2.
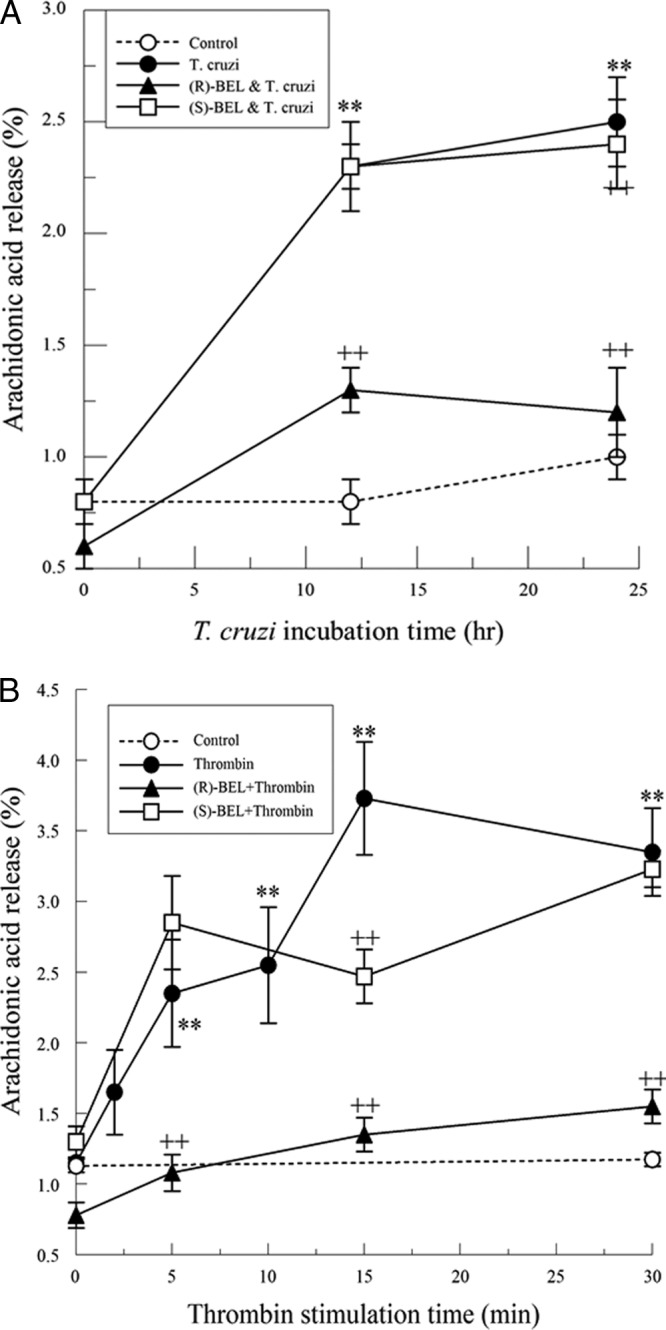
(A) Increase in arachidonic acid release from T. cruzi (MOI, 0.2)-infected cardiac myocytes in the presence or absence of pretreatment with (R)- or (S)-BEL (2 μM; 10 min). **, P < 0.01 compared to unstimulated release, and ++, P < 0.01 when comparing results for T. cruzi-infected myocytes in the presence or absence of BEL. The values shown are means ± SEM for 4 separate cell cultures. (B) Increase in arachidonic acid release from thrombin-stimulated (0.1 IU/ml) mouse cardiac myocytes with or without pretreatment (10 min) with (R)- or (S)-BEL (2 μM). **, P < 0.01 compared to unstimulated release, and ++, P < 0.01 when comparing results for thrombin-stimulated myocytes in the presence or absence of (R)-BEL. The values shown are means ± SEM for 4 separate cell cultures.
PGE2 release.
Cardiac myocytes infected with T. cruzi show a significant increase in PGE2 release 24 to 48 h postinfection (Fig. 3A). Similar to what was seen for arachidonic acid release, (S)-BEL pretreatment did not affect PGE2 release; however, pretreatment with (R)-BEL resulted in significantly reduced PGE2 release. Likewise, thrombin (0.1 IU/ml)-treated cells had a significant increase in PGE2 release, observed over time (Fig. 3B). As with T. cruzi infection, (S)-BEL pretreatment did not affect thrombin-stimulated PGE2 release; however, (R)-BEL inhibits thrombin-stimulated PGE2 release (Fig. 3B). These data suggest that iPLA2γ is critical in regulating release of PGE2 from cardiac myocytes following T. cruzi infection.
Fig 3.
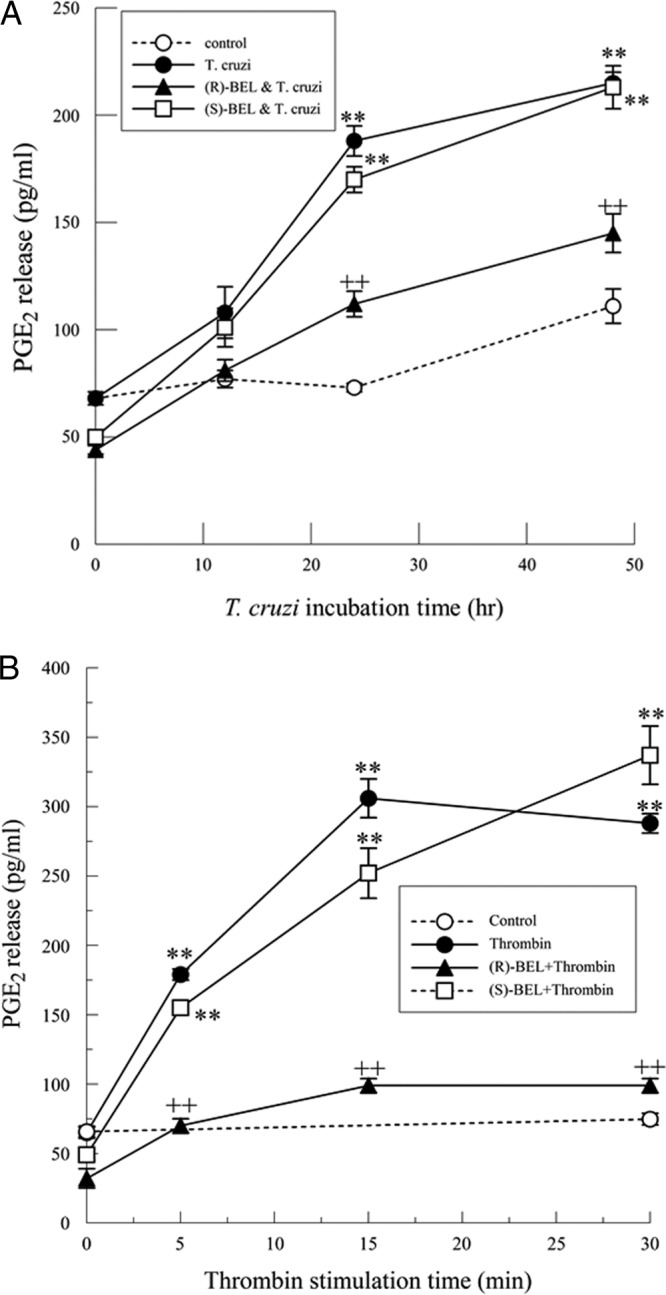
(A) Increase in PGE2 release from T. cruzi-infected (MOI, 0.2) mouse cardiac myocytes with or without pretreatment with (R)- or (S)-BEL (2 μM; 10 min). **, P < 0.01 compared to unstimulated release. and ++, P < 0.01 when comparing results for T. cruzi-infected myocytes in the presence or absence of (R)-BEL. The values shown are means ± SEM for 6 separate cell cultures. (B) Increase in PGE2 release from thrombin-stimulated (0.1 IU/ml) mouse cardiac myocytes with or without pretreatment with (R)- or (S)-BEL (2 μM; 10 min). **, P < 0.01 compared to unstimulated release, and ++, P < 0.01 when comparing results for thrombin-stimulated myocytes in the presence or absence of (R)-BEL. The values shown are means ± SEM for 6 separate cell cultures.
Arachidonic acid and PGE2 release were measured under similar experimental conditions using H9C2 cells, a rat cardiomyoblast cell line. H9C2 cells infected with T. cruzi also showed a significant increase in arachidonic acid and PGE2 release that was inhibited by pretreatment with (R)-BEL, but not by pretreatment with (S)-BEL (data not shown).
PAF production.
In response to T. cruzi infection, there was a slight increase in PAF production that did not reach significance and was unaffected by pretreatment with either (R)- or (S)-BEL (Fig. 4A). However, incubating myocytes with thrombin (1 IU/ml) resulted in a significant increase in PAF production over time (Fig. 4B). In contrast to arachidonic acid and PGE2 release, (R)-BEL does not affect thrombin-mediated PAF production, whereas (S)-BEL significantly inhibits it. Thus, iPLA2γ does not contribute significantly to PAF production in cardiac myocytes, and the mediator is produced by iPLA2β activation in response to thrombin stimulation (Fig. 4B). Hence, in response to thrombin stimulation, PAF production is regulated by iPLA2β and not by iPLA2γ.
Fig 4.
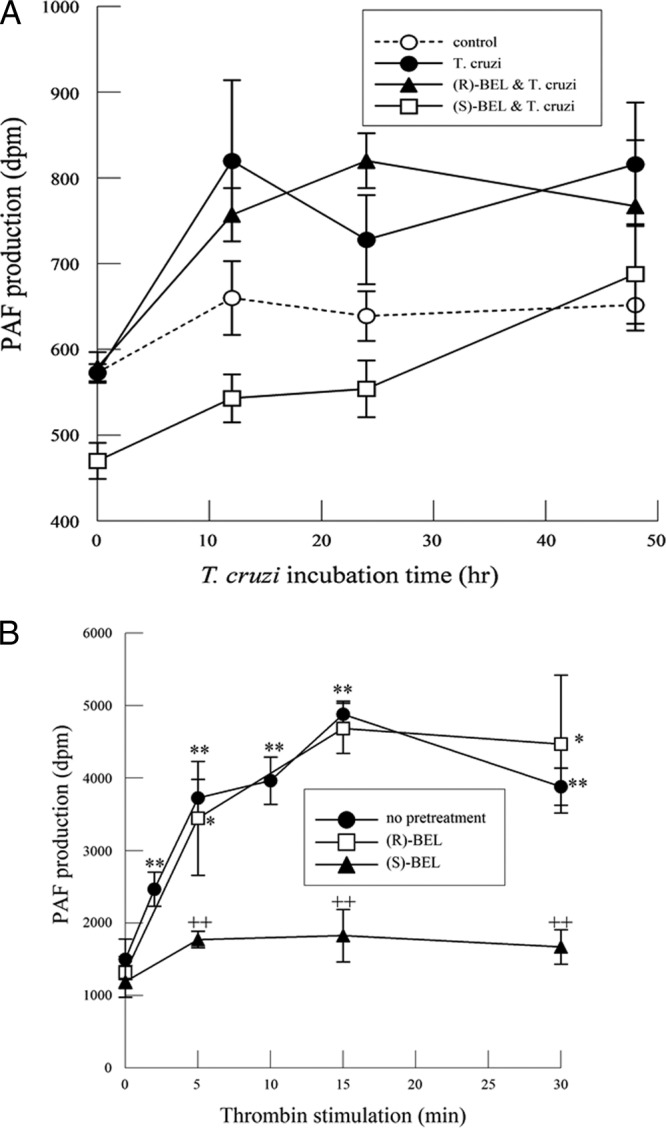
(A) PAF production in mouse cardiac myocytes infected with T. cruzi (MOI, 0.2) with or without (R)- or (S)-BEL pretreatment (2 μM; 10 min). The values shown are means ± SEM for 6 separate cell cultures. (B) PAF production in mouse cardiac myocytes stimulated with thrombin (1 IU/ml) with or without pretreatment with (R)- or (S)-BEL (2 μM; 10 min). *, P < 0.05, and **, P < 0.01 compared with values for unstimulated myocytes, and ++, P < 0.01 when comparing values in the presence or absence of BEL. The values shown are means ± SEM for 8 separate cell cultures.
RAW cell adherence.
To evaluate the functional impact of PAF production on recruitment of inflammatory cells, we measured the adherence of calcein-labeled mouse monocytes/macrophages, RAW 264.7 cells, to cardiac myocytes following T. cruzi infection or thrombin stimulation. Infection of cardiac myocytes with T. cruzi caused no significant changes in RAW 264.7 cell adherence (Fig. 5A). This is in agreement with the absence of PAF production in T. cruzi-infected myocytes (Fig. 4A). Pretreating RAW 264.7 cells with CV3988, a PAF receptor antagonist, completely inhibits macrophage/monocte adherence to T. cruzi-infected HL-1 cells. Following thrombin stimulation, a significant increase in RAW 264.7 cell adherence was observed (Fig. 5B) with a time course similar to that observed for increased PAF production (Fig. 4B). RAW 264.7 cell adherence to thrombin-stimulated HL-1 cells was inhibited by (S)-BEL pretreatment, but not by pretreatment with (R)-BEL (Fig. 5B).
Fig 5.
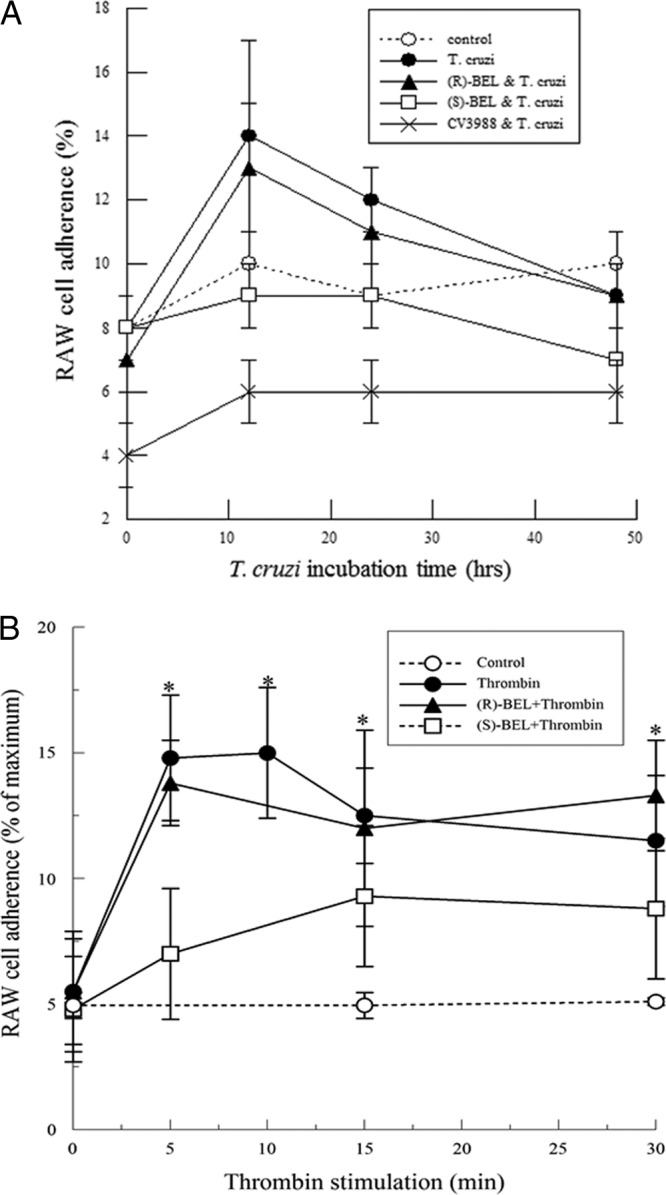
(A) RAW 264.7 cell adherence to mouse cardiac myocytes infected with T. cruzi (MOI, 0.2) with or without pretreatment with (R)- or (S)-BEL (2 μM; 10 min). Where indicated, RAW 264.7 cells were incubated with the PAF receptor antagonist CV3988 (10 μM; 10 min). (B) RAW 264.7 cell adherence to mouse cardiac myocytes stimulated with thrombin (1 IU/ml) with or without pretreatment with (R)- or (S)-BEL (2 μM; 10 min). Where indicated, RAW 264.7 cells were incubated with the PAF receptor antagonist CV3988 (10 μM; 10 min). *, P < 0.05 compared to unstimulated control; *, P < 0.05 compared to unstimulated RAW 264.7 cell adherence. The values shown are means ± SEM for 4 separate cell cultures.
Studies in iPLA2γ-KO mice.
In vitro studies indicate that iPLA2γ is crucial for the production of arachidonic acid and PGE2. To understand its physiological relevance in Chagas' disease, WT and iPLA2γ-KO mice were infected with T. cruzi and observed over a period of 42 days (6 weeks). By day 21, all the iPLA2γ-KO mice died, but over 50% of the WT mice were still alive (Fig. 6). Thus, iPLA2γ-KO mice are more susceptible to infection than WT mice. Given this time course, differences in pathology and biochemical changes may be pronounced just prior to the onset of mortality, i.e., day 14. Hence, in the subsequent experiments, mice were sacrificed 14 days postinfection; fresh blood samples were collected to measure parasitemia; and hearts were isolated to measure tissue parasitism, enzyme activity, and eicosanoid production.
Fig 6.
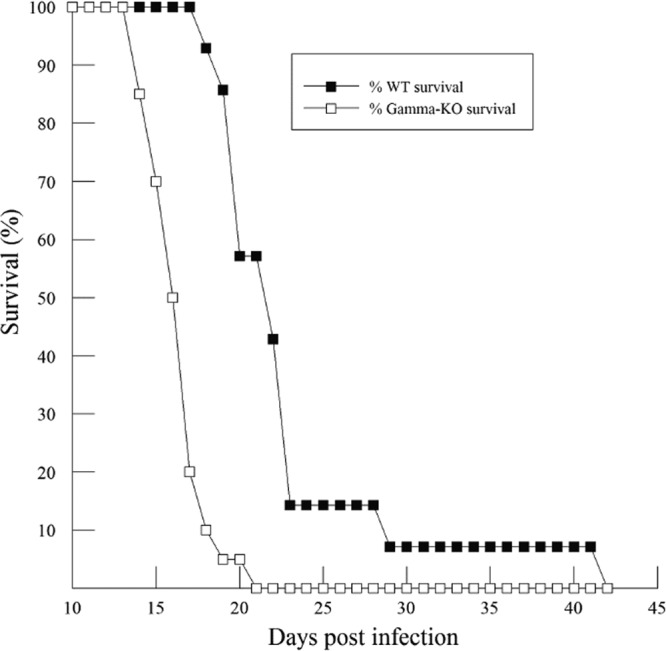
Survival curve (%) for WT (n = 13) and iPLA2γ-KO (n = 19) mice following infection with the Tulahuén strain of T. cruzi (5,000 BFT; subcutaneous injection).
Parasitemia and tissue parasitism.
The number of circulating parasites was significantly higher in iPLA2γ-KO mice than in WT mice after 14 days of infection (Fig. 7A). Since microscopic examination of tissues has limited sensitivity, a PCR-based amplification technique was used to accurately identify the presence of parasites in tissues. Corresponding to the higher levels of circulating parasites, an increase in the number of parasites in the hearts of iPLA2γ-KO mice compared to WT mice was also observed (Fig. 7B).
Fig 7.
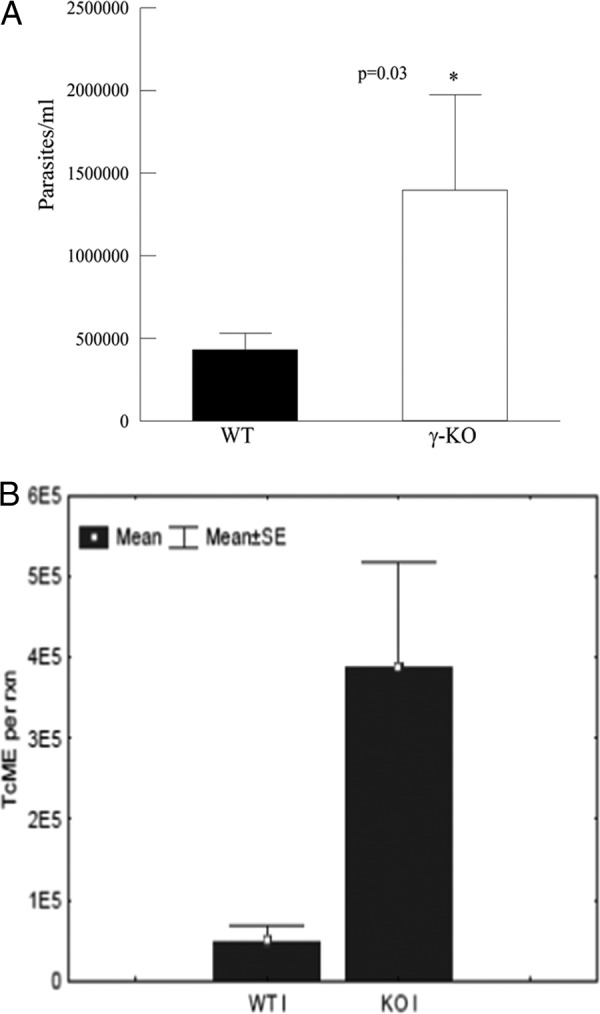
(A) Parasitemia at day 14 in WT and iPLA2γ-KO mice following infection with the Tulahuén strain of T. cruzi (5,000 BFT; subcutaneous injection). *, P < 0.05 compared to the WT. The values shown are means and SEM; n = 6. (B) T. cruzi molecular equivalents (TcME), quantified using T. cruzi-specific quantitative PCR (qPCR), from DNA isolated from the hearts of WT and iPLA2γ-KO mice 14 days postinfection. *, P < 0.05 compared to the WT. The values shown are means and SEM; n = 6.
Biochemical changes in the heart.
A significant increase was observed in cardiac iPLA2 activity in infected WT mice compared to uninfected mice. Further, the iPLA2γ-KO mice showed no change in cardiac iPLA2 activity following infection (Fig. 8). Thus, T. cruzi infection leads to activation of iPLA2γ in the heart, the absence of which is detrimental to survival. Reduced iPLA2 activity in the KO hearts suggests that these mice could also have impaired eicosanoid production. On measuring the PGE2 content in the heart, the iPLA2γ-KO mice were found to have significantly less of the eicosanoid than WT mice (Fig. 9). These data are in agreement with our in vitro observations and demonstrate the importance of myocardial iPLA2γ and eicosanoid production following T. cruzi infection.
Fig 8.
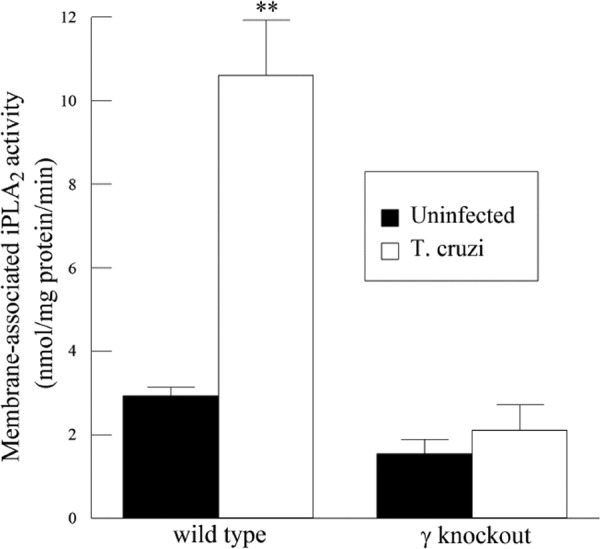
iPLA2 activity in the hearts of WT and iPLA2γ-KO mice 14 days postinfection with T. cruzi. Shown is PLA2 activity, measured using 100 μM (16:0, [3H]18:1) plasmenylcholine in the presence of 4 mM EGTA. **, P < 0.01 compared to activity measured in the uninfected heart. The values shown are means and SEM; n = 6.
Fig 9.
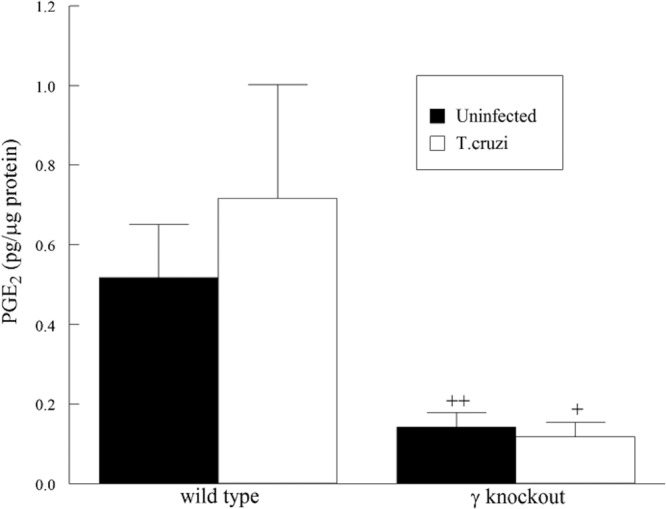
PGE2 content in the hearts of WT and iPLA2γ-KO mice 14 days postinfection with T. cruzi. +, P < 0.05, and ++, P < 0.01, compared to activity measured in the WT heart. The values shown are means and SEM; n = 6.
DISCUSSION
Despite knowing the etiology of Chagas' disease for over a century, successful treatment strategies are still elusive. So far, only two drugs, benznidazole and nifurtimox, are widely used for the treatment of the disease, and both are known to cause severe side effects. Adverse effects include digestive problems, hematological changes, dermatological alterations and neurological alterations (23, 24). Though both drugs are recommended, their efficacies are variable, and more information is needed to verify their ability to clear the parasites. The fact that only two drugs exist to treat a disease that has been prevalent for so long highlights the “neglected” status of Chagas' disease. Several challenges have impaired the development of therapeutic strategies, and much remains unknown about vector-host interactions and the mechanisms behind the various pathological symptoms associated with the disease. Only recently has it been determined that the chronic stages of the disease are associated with parasite persistence and that autoimmune mechanisms may not be responsible for organ damage, as hypothesized for several decades previously (25). Since the presence of parasites is critical for the disease, it is necessary to decipher the cascade of events that occur following infection, increasing our understanding and the potential to create drugs that can effectively treat the disease.
Several downstream metabolites of PLA2 have been implicated in Chagas' disease, but little is known about the direct contribution of the enzyme to the pathogenesis of the disease. Bioactive lipids, such as eicosanoids and PAF, are potent inflammatory molecules produced by the action of PLA2 on membrane phospholipids. Previous studies have outlined the varying roles of eicosanoids in Chagas' disease. Michelin et al. have suggested an immunosuppressive role for PGE2, upregulated via concanavalin A stimulation of COX-2 in T. cruzi-infected spleen cells (26). Abdalla et al. demonstrated that inhibition of COX-2-mediated PGE2 production resulted in reduced inflammation, parasite pseudocysts, and fibrosis in the hearts of infected mice, suggesting that PGE2 contributes to cardiac damage in the acute phase of infection (27). Cardoni et al. concluded from their studies that COX-2-derived mediators are protective during the acute stages of T. cruzi infection but could be responsible for tissue damage in the chronic stages. More recent studies by Ashton et al. have suggested that TXA2 is an important parasite-derived eicosanoid that contributes to parasite proliferation and hence the inflammatory response (28). Several studies also suggest that eicosanoids, in conjunction with cytokines, chemokines, and other inflammatory mediators, are essential for the progression and perpetuation of the chronic phase of the disease (9, 29). Since PLA2 is critical for arachidonic acid release and eicosanoid production, it could contribute to the pathogenesis of Chagas' disease via the formation of these mediators.
The predominant phospholipase A2 in the heart, iPLA2, is known to have two major isoforms in mammalian cells, iPLA2β and iPLA2γ. While both isoforms are present in cardiac tissue, it has been determined in these studies and in previous work from our laboratory that most of the cardiac iPLA2 activity is attributable to the γ isoform of the enzyme (15). In order to differentiate between the functions of the two isoforms, pharmacological inhibitors, (R)- and (S)-BEL, are commonly used (22). BEL has been demonstrated to have 100-fold selectivity for iPLA2 versus cytosolic PLA2 (cPLA2) and secretory PLA2 (sPLA2) isoforms, and its enantiomers, (R)- and (S)-BEL, are 10-fold more selective for iPLA2γ and iPLA2β, respectively (30). Previously, we determined that iPLA2 has a preference for the arachidonylated plasmalogen phospholipid species, which are abundant in the sarcolemma of cardiac myocytes (14). Serine proteases, such as thrombin, were shown to activate iPLA2 via the protease-activated receptors (PARs) present on the myocyte surface, and based on pharmacological inhibition, iPLA2γ was established as the predominant isoform in cardiac myocytes (13, 16). Since arachidonic acid release, the rate-limiting step in eicosanoid production, is generated by the action of phospholipases, it is likely that iPLA2γ contributes to the production of this critical mediator. However, until the present study, the effect of T. cruzi on cardiac iPLA2 has not been investigated.
Other members of the PLA2 family also exist in the myocardium and are grouped based on their structures, substrate preferences, and requirements for calcium. cPLA2 enzymes are selective for arachydonylated choline phospholipids but do not demonstrate any preference for sn-1 vinyl ether linkages (plasmalogens) and contribute to the formation of eicosanoids in response to extracellular signals (31). In the heart, cPLA2 is known to regulate cardiac hypertrophy and plays a protective role in ischemia reperfusion injury (32, 33). sPLA2s do not exhibit any preference for the sn-2 fatty acid but are proposed to play a role in eicosanoid formation in the heart (31). It has also been suggested that sPLA2 may be involved in regulating cardiac hypertrophy (34). iPLA2 enzymes are selective for plasmalogen phospholipids, which are selectively enriched in the sarcolemma of myocardial cells. Also, the majority of the myocardial arachidonic acid is esterified to plasmalogen species, making plasmalogens a rich source of the arachidonic acid required for eicosanoid production following hydrolysis by iPLA2. The contribution of any of the PLA2 isoforms to T. cruzi infection remains largely unknown. It is possible that both cPLA2 and sPLA2 contribute to the progression of Chagas' heart disease; however, since the majority of the myocardial PLA2 activity is attributable to iPLA2, we have explored the role of this isoform in acute Chagas' disease.
We used the mouse atrial cardiac myocyte cell line (HL-1 cells), as well as a knockout mouse model, to demonstrate the role of iPLA2 in acute T. cruzi infection. In mouse cardiac myocytes, the inhibition of iPLA2 activity is much more sensitive to (R)-BEL than to (S)-BEL, suggesting that iPLA2γ is the predominant isoform in these cells. An iPLA2γ-mediated increase in arachidonic acid and PGE2 release was also observed following treatment with thrombin or T. cruzi infection of cardiac myocytes. Hence, this isoform may be critical for controlling the rate-limiting step in eicosanoid production. The mouse model of infection revealed that iPLA2γ is important in acute infection, since the iPLA2γ-KO mice exhibited early mortality compared to WT mice. Prior to the onset of mortality in either group (day 14 postinfection), WT mice showed increased cardiac iPLA2 activity and less parasitemia and tissue parasitism, unlike that observed in the iPLA2γ-KO mice. Hence, the absence of iPLA2γ results in a higher parasite load and mortality, suggesting a protective role for the isoform in acute T. cruzi infection.
Previous studies by Mukherjee et al. suggest that COX-1 inhibition by aspirin leads to increased parasitemia and mortality in T. cruzi-infected mice. These findings were not mimicked entirely in COX-1 knockout mice, which show increased parasitemia, but not enhanced mortality. Based on these findings, it was hypothesized that COX-1-mediated inhibition of eicosanoids contributed to increased parasitemia but did not affect mortality and that aspirin-mediated increase in mortality is likely due to “off-target” effects of the drug. Our findings, in the absence of any pharmacological inhibitors, show that the reduced eicosanoids and increased parasite load associated with mortality are due to the absence of iPLA2γ.
Another important PLA2-derived mediator, PAF, is responsible for the activation and transmigration of inflammatory cells to relevant sites (35). The role of PAF in cardiac inflammation following T. cruzi infection is largely unknown. It has been suggested that PAF induces the production of nitric oxide in T. cruzi-infected macrophages and hence is protective (36). PAFR−/− mice infected with T. cruzi were shown to have an increased number of parasite pseudocysts and inflammation in the heart and hence are more susceptible to infection than WT mice (37). In this study, cardiac myocytes infected with T. cruzi did not show a significant increase in PAF production. Thrombin treatment however, did result in a considerable increase in PAF production, which is mediated by iPLA2β and not iPLA2γ.
The presence of microthrombi and platelet aggregation have been observed in Chagasic hearts, suggesting an increase in thrombin in the myocardium (12, 38). Pinazo et al. have also demonstrated that a prothrombotic state exists in chronic Chagas' disease patients, as they have significantly increased endogenous thrombin potential and prothrombin fragment 1 + 2, which serves as a marker for thrombin generation (39). Thus, while the parasite itself may not directly affect PAF production in cardiac myocytes, other associated factors present in the vicinity of infection could enhance the process. These findings suggest a protective role for iPLA2β in acute infection via the production of PAF and its effects on macrophages, but this needs further investigation. The disparity in PAF production with T. cruzi infection versus thrombin pretreatment is not known but may be related to differences in intracellular events initiated by activation of the thrombin receptor and those initiated by infection with T. cruzi. In previous studies, we have demonstrated that there is an immediate increase in PAF production in response to thrombin stimulation of endothelial cells (35). Our data here show a similar increase in PAF production in cardiac myocytes stimulated with thrombin. We propose that this is due to tight coupling between the thrombin receptor and iPLA2β activation. Our data here suggest that thrombin activates HL-1 cell iPLA2γ and iPLA2β, whereas T. cruzi infection results in iPLA2γ activation only and may be related to more delayed intracellular processes. Regardless of changes in PAF production, RAW cell adherence remained unchanged, suggesting that increased PAF production does not play a direct role in inflammatory-cell adherence to cardiac myocytes. In conclusion, these studies suggest that, following T. cruzi infection, the subsequent increase in arachidonic acid and eicosanoid levels is mediated by iPLA2γ, the predominant PLA2 isoform in these cells. Thrombin stimulation has a similar effect on iPLA2γ but, additionally, causes increased PAF production, mediated by iPLA2β.
Several studies have suggested that eicosanoids produced during the acute stage of the disease are important immunomodulators, enabling the disease to progress into its chronic stages. Thus, iPLA2γ contributes to this process via eicosanoid production, which in turn affects parasite persistence and disease progression, forming a critical link between the parasite and the host. Our studies indicate that iPLA2γ activation is beneficial during acute infection, elucidated by the early mortality observed in iPLA2γ-KO mice following infection. While many questions remain, clearly, iPLA2γ is critical in the setting of T. cruzi infection. Understanding its role during the acute stages of the disease may thus help us recognize important events during infection that may impact the chronic stages and present us with more points of intervention and treatment of Chagas' disease.
Footnotes
Published ahead of print 19 February 2013
REFERENCES
- 1. WHO 2012. WHO factsheet for Chagas' disease. WHO, Geneva, Switzerland [Google Scholar]
- 2. Bern C, Montgomery SP. 2009. An estimate of the burden of Chagas disease in the United States. Clin. Infect. Dis. 49:e52–e54. 10.1086/605091 [DOI] [PubMed] [Google Scholar]
- 3. Tanowitz HB, Machado FS, Jelicks LA, Shirani J, de Carvalho AC, Spray DC, Factor SM, Kirchhoff LV, Weiss LM. 2009. Perspectives on Trypanosoma cruzi-induced heart disease (Chagas disease). Prog. Cardiovasc. Dis. 51:524–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rassi A, Jr, Rassi A, Marin-Neto JA. 2010. Chagas disease. Lancet 375:1388–1402 [DOI] [PubMed] [Google Scholar]
- 5. Rossi MA, Tanowitz HB, Malvestio LM, Celes MR, Campos EC, Blefari V, Prado CM. 2010. Coronary microvascular disease in chronic Chagas cardiomyopathy including an overview on history, pathology, and other proposed pathogenic mechanisms. PLoS Negl. Trop. Dis. 4:e674. 10.1371/journal.pntd.0000674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang H, Calderon TM, Berman JW, Braunstein VL, Weiss LM, Wittner M, Tanowitz HB. 1999. Infection of endothelial cells with Trypanosoma cruzi activates NF-kappaB and induces vascular adhesion molecule expression. Infect. Immun. 67:5434–5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Talvani ARC, Aliberti JC, Michailowsky V, Santos PV, Murta SM, Romanha AJ, Almeida IC, Farber J, Lannes-Vieira J, Silva JS, Gazzinelli RT. 2000. Kinetics of cytokine gene expression in experimental chagasic cardiomyopathy: tissue parasitism and endogenous IFN-gamma as important determinants of chemokine mRNA expression during infection with Trypanosoma cruzi. Microbes Infect. 2:851–866 [DOI] [PubMed] [Google Scholar]
- 8. Machado FS, Souto JT, Rossi MA, Esper L, Tanowitz HB, Aliberti J, Silva JS. 2008. Nitric oxide synthase-2 modulates chemokine production by Trypanosoma cruzi-infected cardiac myocytes. Microbes Infect. 10:1558–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mukherjee S, Machado FS, Huang H, Oz HS, Jelicks LA, Prado CM, Koba W, Fine EJ, Zhao D, Factor SM, Collado JE, Weiss LM, Tanowitz HB, Ashton AW. 2011. Aspirin treatment of mice infected with Trypanosoma cruzi and implications for the pathogenesis of Chagas disease. PLoS One 6:e16959. 10.1371/journal.pone.0016959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Novaes RD, Penitente AR, Goncalves RV, Talvani A, Neves CA, Maldonado IR, Natali AJ. 2011. Effects of Trypanosoma cruzi infection on myocardial morphology, single cardiomyocyte contractile function and exercise tolerance in rats. Int. J. Exp. Pathol. 92:299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Machado FS, Martins GA, Aliberti JC, Mestriner FL, Cunha FQ, Silva JS. 2000. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation 102:3003–3008 [DOI] [PubMed] [Google Scholar]
- 12. Tanowitz HB, Burns ER, Sinha AK, Kahn NN, Morris SA, Factor SM, Hatcher VB, Bilezikian JP, Baum SG, Wittner M. 1990. Enhanced platelet adherence and aggregation in Chagas' disease: a potential pathogenic mechanism for cardiomyopathy. Am. J. Trop. Med. Hyg. 43:274–281 [DOI] [PubMed] [Google Scholar]
- 13. McHowat J, Creer MH. 1998. Thrombin activates a membrane-associated calcium-independent PLA2 in ventricular myocytes. Am. J. Physiol. 274:C447–C454 [DOI] [PubMed] [Google Scholar]
- 14. McHowat J, Creer MH. 1998. Calcium-independent phospholipase A2 in isolated rabbit ventricular myocytes. Lipids 33:1203–1212 [DOI] [PubMed] [Google Scholar]
- 15. Beckett CS, McHowat J. 2008. Calcium-independent phospholipase A2 in rabbit ventricular myocytes. Lipids 43:775–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharma J, McHowat J. 2011. PGE2 release from tryptase-stimulated rabbit ventricular myocytes is mediated by calcium-independent phospholipase A2γ. Lipids 46:391–397 [DOI] [PubMed] [Google Scholar]
- 17. Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr 1998. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U. S. A. 95:2979–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mancuso DJ, Kotzbauer P, Wozniak DF, Sims HF, Jenkins CM, Guan S, Han X, Yang K, Sun G, Malik I, Conyers S, Green KG, Schmidt RE, Gross RW. 2009. Genetic ablation of calcium-independent phospholipase A2{gamma} leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J. Biol. Chem. 284:35632–35644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Degousee N, Fazel S, Angoulvant D, Stefanski E, Pawelzik SC, Korotkova M, Arab S, Liu P, Lindsay TF, Zhuo S, Butany J, Li RK, Audoly L, Schmidt R, Angioni C, Geisslinger G, Jakobsson PJ, Rubin BB. 2008. Microsomal prostaglandin E2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation 117:1701–1710 [DOI] [PubMed] [Google Scholar]
- 20. Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917 [DOI] [PubMed] [Google Scholar]
- 21. Schnapp AR, Eickhoff CS, Sizemore D, Curtiss R, III, Hoft DF. 2002. Cruzipain induces both mucosal and systemic protection against Trypanosoma cruzi in mice. Infect. Immun. 70:5065–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jenkins CM, Han X, Mancuso DJ, Gross RW. 2002. Identification of calcium-independent phospholipase A2 (iPLA2) beta, and not iPLA2gamma, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J. Biol. Chem. 277:32807–32814 [DOI] [PubMed] [Google Scholar]
- 23. Le Loup G, Pialoux G, Lescure FX. 2011. Update in treatment of Chagas disease. Curr. Opin. Infect. Dis. 24:428–434 [DOI] [PubMed] [Google Scholar]
- 24. Apt W. 2010. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Des. Devel. Ther. 4:243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Machado FS, Tyler KM, Brant F, Esper L, Teixeira MM, Tanowitz HB. 2012. Pathogenesis of Chagas disease: time to move on. Front. Biosci. 4:1743–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michelin MA, Silva JS, Cunha FQ. 2005. Inducible cyclooxygenase released prostaglandin mediates immunosuppression in acute phase of experimental Trypanosoma cruzi infection. Exp. Parasitol. 111:71–79 [DOI] [PubMed] [Google Scholar]
- 27. Abdalla GK, Faria GE, Silva KT, Castro EC, Reis MA, Michelin MA. 2008. Trypanosoma cruzi: the role of PGE2 in immune response during the acute phase of experimental infection. Exp. Parasitol. 118:514–521 [DOI] [PubMed] [Google Scholar]
- 28. Ashton AW, Mukherjee S, Nagajyothi FN, Huang H, Braunstein VL, Desruisseaux MS, Factor SM, Lopez L, Berman JW, Wittner M, Scherer PE, Capra V, Coffman TM, Serhan CN, Gotlinger K, Wu KK, Weiss LM, Tanowitz HB. 2007. Thromboxane A2 is a key regulator of pathogenesis during Trypanosoma cruzi infection. J. Exp. Med. 204:929–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sterin-Borda L, Gorelik G, Goren N, Cappa SG, Celentano AM, Borda E. 1996. Lymphocyte muscarinic cholinergic activity and PGE2 involvement in experimental Trypanosoma cruzi infection. Clin. Immunol. Immunopathol. 81:122–128 [DOI] [PubMed] [Google Scholar]
- 30. Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. 1991. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J. Biol. Chem. 266:7227–7232 [PubMed] [Google Scholar]
- 31. McHowat J, Creer MH. 2001. Comparative roles of phospholipase A2 isoforms in cardiovascular pathophysiology. Cardiovasc. Toxicol. 1:253–265 [DOI] [PubMed] [Google Scholar]
- 32. Kerkela R, Boucher M, Zaka R, Gao E, Harris D, Piuhola J, Song J, Serpi R, Woulfe KC, Cheung JY, O'Leary E, Bonventre JV, Force T. 2011. Cytosolic phospholipase A(2)alpha protects against ischemia/reperfusion injury in the heart. Clin. Transl Sci. 4:236–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haq S, Kilter H, Michael A, Tao J, O'Leary E, Sun XM, Walters B, Bhattacharya K, Chen X, Cui L, Andreucci M, Rosenzweig A, Guerrero JL, Patten R, Liao R, Molkentin J, Picard M, Bonventre JV, Force T. 2003. Deletion of cytosolic phospholipase A2 promotes striated muscle growth. Nat. Med. 9:944–951 [DOI] [PubMed] [Google Scholar]
- 34. Reddy ST, Herschman HR. 1994. Ligand-induced prostaglandin synthesis requires expression of the TIS10/PGS-2 prostaglandin synthase gene in murine fibroblasts and macrophages. J. Biol. Chem. 269:15473–15480 [PubMed] [Google Scholar]
- 35. McHowat J, Kell PJ, O'Neill HB, Creer MH. 2001. Endothelial cell PAF synthesis following thrombin stimulation utilizes Ca(2+)-independent phospholipase A(2). Biochemistry 40:14921–14931 [DOI] [PubMed] [Google Scholar]
- 36. Aliberti JC, Machado FS, Gazzinelli RT, Teixeira MM, Silva JS. 1999. Platelet-activating factor induces nitric oxide synthesis in Trypanosoma cruzi-infected macrophages and mediates resistance to parasite infection in mice. Infect. Immun. 67:2810–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Talvani A, Santana G, Barcelos LS, Ishii S, Shimizu T, Romanha AJ, Silva JS, Soares MB, Teixeira MM. 2003. Experimental Trypanosoma cruzi infection in platelet-activating factor receptor-deficient mice. Microbes Infect. 5:789–796 [DOI] [PubMed] [Google Scholar]
- 38. Tanowitz HB, Kaul DK, Chen B, Morris SA, Factor SM, Weiss LM, Wittner M. 1996. Compromised microcirculation in acute murine Trypanosoma cruzi infection. J. Parasitol. 82:124–130 [PubMed] [Google Scholar]
- 39. Pinazo MJ, Tassies D, Munoz J, Fisa R, Posada Ede J, Monteagudo J, Ayala E, Gallego M, Reverter JC, Gascon J. 2011. Hypercoagulability biomarkers in Trypanosoma cruzi-infected patients. Thromb. Haemost. 106:617–623 [DOI] [PubMed] [Google Scholar]