

Abstract
Brucella abortus is an intracellular pathogen that uses a crafty strategy to invade and proliferate within host cells, but the distinct signaling pathways associated with phagocytic mechanisms of B. abortus remain unclear. The present study was performed to test the hypothesis that Toll-like receptor 4 (TLR4)-linked signaling interacting with Janus kinase 2 (JAK2) plays an essential role in B. abortus phagocytosis by macrophages. The effects of TLR4-JAK2 signaling on B. abortus phagocytosis in murine macrophage RAW 264.7 cells were observed through an infection assay and confocal microscopy. We determined that the uptake of B. abortus was negatively affected by the dysfunction of TLR4 and JAK2. F-actin polymerization detected by flow cytometry and F-actin assay was amplified for B. abortus entry, whereas that event was attenuated by the disruption of TLR4 and JAK2. Importantly, JAK2 phosphorylation and actin skeleton reorganization were suppressed immediately after B. abortus infection in bone marrow-derived macrophages (BMDMs) from TLR4−/− mice, showing the cooperation of JAK2 with TLR4. Furthermore, small GTPase Cdc42 participated in the intermediate pathway of TLR4-JAK2 signaling on B. abortus phagocytosis. Consequently, TLR4-associated JAK2 activation in the early cellular signaling events plays a pivotal role in B. abortus-induced phagocytic processes in macrophages, implying the pathogenic significance of JAK2-mediated entry. Here, we elucidate that this specific phagocytic mechanism of B. abortus might provide achievable strategies for inhibiting B. abortus invasion.
INTRODUCTION
Brucella species are Gram-negative, facultative intracellular bacteria that cause abortion and infertility in many domestic and wild mammals and a disease known as undulant fever in humans (1). These bacteria invade and replicate within professional and nonprofessional phagocytes, suggesting that Brucella undergoes many interactions with host cells (2–4). The virulence associated with bacterial invasion and chronic infections by Brucella are assumed to be due to their ability to evade the killing mechanisms within macrophages (2, 5), but the molecular mechanisms by which Brucella survives within phagocytes are incompletely understood.
Phagocytosis is a critical step for a successful immune reaction against microbial pathogens, because it provokes both degradation of pathogens and the subsequent presentation of pathogen peptide antigens. Ligation of various phagocytic receptors, such as Fc gamma receptors or complement receptor 3, activates a series of intracellular signal transductions that induce dynamic and rapid rearrangement of the actin cytoskeleton, which is essential for phagocytic uptake (6). An early study established that M cells, macrophages, and neutrophils ingest Brucella by zipper-like phagocytosis (7). It has also been determined that Brucella invades macrophages through lipid raft microdomains (8). F-actin polymerization is implicated in the phagocytosis of Brucella in both epithelial cells and macrophages (9–11).
Toll-like receptors (TLRs) are part of a skillful system that detects invasion by microbial pathogens. Recognition of microbial components by TLRs triggers signaling pathways that promote the expression of genes and regulate innate immune responses (12). It has been reported that B. abortus signals through TLR2 and TLR4, but TLR2 plays no role in controlling the infection (3, 13, 14). Ligation of TLR4 promotes the activation of complex signaling pathways, including mitogen-activated protein kinases (MAPKs) (p38α, JNK, and ERK), phosphoinositide 3-kinase (PI3K), and GTPases, all of which play important roles in phagocytosis (15, 16).
The Janus kinase (JAK) family consists of four members: JAK1, JAK2, JAK3, and TYK2. All four members of the JAK family are ubiquitously expressed in most cells, although macrophages predominantly express JAK2 (17). The JAK pathway is initiated by various ligands, including cytokines, and it promotes proliferation, migration, inflammatory and immune responses, and other cellular events (18, 19). JAK2 is an essential mediator of cytokine-dependent signal transduction and a modulator of immune responses (20). It has been confirmed that JAK2 function is required for the activation of Src-kinase, MAPKs, PI3K-AKT, and STAT signaling following the interaction of cytokine/interferon receptors with their ligands (19) and infection by pathogens (21, 22).
The small GTPases of the Rho subfamily, such as Rho, Rac, and Cdc42, were characterized for their roles in regulating the actin cytoskeleton (16). Activated GTP-bound proteins of the Rho subfamily interact with effector proteins to process biological responses comprising actin restructure. Some of these responses are associated with membrane reorganizations implicated in diverse functions, including phagocytosis (23). Nevertheless, the downstream signaling cascades subsequent to the entry of B. abortus into epithelial cells (3, 9) and murine macrophages (24) have not been fully elucidated.
The stimulation of TLRs activates multiple signaling pathways, but information on how distinct signaling pathways are associated with the TLR-mediated phagocytosis of Brucella is still limited. The objective of this study was to address the novel phagocytic mechanism of B. abortus involved in intracellular signal pathways. In the current study, we suggest that the activation of JAK2 via TLR4 is required for the internalization of viable B. abortus and is accompanied by enhanced actin polymerization. Notably, our findings underscore the importance of a complex interplay between TLR4 and JAK2 as signaling platforms for B. abortus in a way that favors its invasion. Collectively, we clarified the phagocytic mechanism of B. abortus, implying that the pathogenic significance of receptor-mediated entry is linked with the specific cellular signaling events.
MATERIALS AND METHODS
Mice.
TLR4 mutants (TLR4−/−; HeJ) and wild-type mice (WT; HeSlc) on a C3H background were obtained from Japan SLC, Inc. (Makoto Yanabe). All experimental protocols in this study were reviewed and approved by the Institutional Animal Care and Use Committee and performed according to the National Institutes of Health guidelines (permit number GNU-120423-M0013). Bone marrow-derived macrophages (BMDMs) from female C3H mice were prepared as described previously (25).
Cells and culture conditions.
Murine macrophage RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and grown at 37°C in a 5% CO2 atmosphere in RPMI 1640 (Gibco, Carlsbad, CA) containing 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (all provided by Gibco). BMDMs and RAW 264.7 cells were seeded (1 × 105 cells/well) in cell culture plates and incubated for 24 h before infection for all assays.
Transfection of siRNA specific for JAK2 and TLR4.
For silencing of mouse JAK2 and TLR4 expression, RAW 264.7 cells were transfected with a pool of short interfering RNA (siRNA)-specific JAK2 and TLR4 (Santa Cruz, Santa Cruz, CA), wherein siRNA products generally consist of pools of three to five target-specific, 19- to 25-nucleotide (nt) siRNAs designed to knock down gene expression, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. As a control, control-siRNA duplexes were transfected in parallel into cells. The optimal concentration (50 nM) of siRNA for JAK2 and TLR4 silencing was confirmed by reverse transcription-PCR (RT-PCR) of mouse JAK2 and TLR4 mRNA. For the RT-PCR analysis, 1 μg of mRNA isolated from cultured murine macrophages was reverse transcribed into cDNA by using 0.5 μg of oligo(dT) primers (Promega) and 200 U of Superscript II reverse transcriptase (Gibco-BRL, Life Technologies) at 45°C for 50 min and then at 95°C for 4 min. The cDNA was amplified by PCR using the specific murine primers for the following: mouse JAK2 (GenBank accession no. L16956 and L16956.1), 5′-TTTCAGAGCTGTCATCCGTG-3′ (forward) and 5′-CTTTCCCCTGGCTCCTTTAC (reverse); mouse TLR4 (GenBank accession no. AF110133 and AF110133.1), 5′-AGCAGAGGAGAAAGCATCTAT-3′ (forward) and 5′-GGTTTAGGCCCCAGAGTTTTG-3′ (reverse). The PCR was conducted in 30 incubation cycles of 1 min at 94°C, 1 min at 60°C, and 1 min at 72°C. The amplified DNA products were electrophoresed on a 2% agarose gel, and the band densities were quantified using an image analyzer. The housekeeping gene encoding mouse β-actin served as an internal control for sample loading and mRNA integrity.
Bacterial strains and culture conditions.
The Brucella abortus strain was derived from 544 (ATCC 23448), a smooth, virulent B. abortus biovar 1 strain. The B. abortus organisms were maintained as frozen glycerol stocks (80% [vol/vol] glycerol) at −70°C. In all experiments, the contents of freshly thawed vials were cultured in Brucella broth (Becton, Dickinson, Franklin Lakes, NJ) or Brucella broth containing 1.5% agar without antibiotics for 3 days at 37°C with aeration. Bacteria were grown at 37°C with vigorous shaking until they reached the stationary phase, and bacteria were suspended in phosphate-buffered saline (PBS). Viable counting was measured by plating serial dilutions on Brucella agar.
Lipopolysaccharide (LPS) from Escherichia coli (0111:B4), purified by phenol extraction, was obtained from Sigma (St. Louis, MO) and reconstituted by adding sterile culture medium to 1 mg/ml of stock concentration. The reconstituted product was further diluted to desired working concentrations using the same medium.
Bacterial internalization and intracellular replication assay.
To determine internalization of bacteria, macrophage cells were infected with B. abortus as described previously (8). Prior to infection, cells were washed three times and replaced with new media without antibiotics, and then bacteria were deposited onto cells at a multiplicity of infection (MOI) of 10, centrifuged at 150 × g at 22°C for 10 min, and incubated at 37°C in 5% CO2 for 0, 15, and 30 min. Cells were washed once with medium and then incubated with RPMI 1640 containing 10% fetal bovine serum (FBS) with gentamicin (30 μg/ml) for 30 min to kill any remaining extracellular and adherent bacteria. For evaluation of viable bacteria at different periods of time, infected cells were washed three times with PBS and then lysed with distilled water so that resultant cell lysates contained only internalized bacteria. The serial dilutions of cell lysates were carried out for accurate colony counts, and the last dilution was spread on Brucella agar plates. The number of viable internalized bacteria was evaluated by CFU, which were determined by serial dilutions on plates. To determine intracellular replication of bacteria, the infected cells were incubated at 37°C for 1 h, and the media were replaced with RPMI 1640 containing 10% FBS and gentamicin (30 μg/ml) and then incubated for 2, 24, and 48 h. The cell washing, lysis, and plating procedures were the same as those for the detection of bacterial uptake efficiency. All assays were conducted in triplicate and repeated at least three times on different days.
Immunofluorescence microscopy.
Macrophages were cultured in 12-well plates with 18-mm-diameter glass coverslips (105 cells/well) for 24 h before the infection. Cells were infected with unconjugated or Alexa Fluor 405 (Molecular Probes, Eugene, OR)-conjugated B. abortus. To monitor bacterial infection within 5 min, samples were fixed and then permeabilized with 0.1% Triton X-100 for 10 min at 22°C. After 30 min of incubation with a blocking buffer (2% goat serum in PBS), the preparations were stained with different antibodies in blocking buffer for 1 h at 37°C. For staining of F-actin, cells were incubated with 0.1 μM rhodamine-phalloidin (Cytoskeleton, Inc., Denver, CO) for 30 min at 22°C. For staining of internalized bacteria, fixed cells on coverslips were stained as previously described (8). For detection of intracellular proteins, localized and internalized B. abortus cells were incubated with phospho-JAK2 (Tyr1007/1008; Abcam, Cambridge, United Kingdom) and then fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (Sigma-Aldrich, St. Louis, MO). Finally, preparations were washed and mounted with fluorescent mounting medium (DakoCytomation, Glostrup, Denmark). Fluorescence images were collected with an Olympus FV1000 laser-scanning confocal microscope. Images were processed with Adobe Photoshop and NIH ImageJ software.
Immunoblot analysis.
The cultured cells in 6-well plates were infected with B. abortus for the indicated times. Cells were then washed twice with ice-cold PBS and lysed in ice-cold lysis buffer for 30 min at 4°C. Protein concentration was determined by Bradford protein assay (Bio-Rad, Richmond, CA). Samples were separated by SDS-PAGE and electrically transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). Blots were blocked for 1 h with 5% (wt/vol) bovine serum albumin in TBS-T (20 mM Tris-HCl, 150 mM NaCl, Tween 0.1%, pH 7.4) and proved by phosphospecific antibodies against JAK2, ERK1/2, p38α (Thr180/Tyr182; Cell Signaling), JNK (Thr183/Tyr185; Cell Signaling), PI3K, and AKT (Ser473; Cell Signaling). Pan-specific antibodies and β-actin antibody were obtained by Cell Signaling and applied on stripped blots to verify that equivalent amounts of proteins were loaded per lane. The binding of primary antibody was visualized using horseradish peroxidase (HRP)-conjugated anti-rabbit IgG and anti-mouse IgG secondary antibodies (Sigma-Aldrich) and then was detected by ECL (Amersham, Little Chalfont, United Kingdom). The immunoblot ECL signals were quantified using NIH Image J software.
Determination of Cdc42 activation by pull-down GST assay.
The recombinant glutathione S-transferase (GST)-tagged PBD fusion protein was expressed from a derivative pGEX-2T plasmid (obtained from Gary M. Bokoch, Scripps Research Institute) (26). Escherichia coli BL21 cells transformed with the GST-tagged PBD construct were grown at 37°C. Expression of recombinant protein was induced by addition of 0.1 mM isopropylthiogalactoside (IPTG) for 2 h and later was purified. The ProFound pull-down GST assay (Pierce, Rockford) was conducted according to the manufacturer's instructions. Samples were resolved on SDS-PAGE, and pulled-down GTP-bound forms of Cdc42 were detected by Western blot analysis using anti-Cdc42 antibodies and visualized using ECL.
FACS assay for F-actin.
To evaluate the relative content of F-actin in B. abortus-infected cells, we performed fluorescence-activated cell sorter (FACS) assay for F-actin as previously described (27). In brief, cells (1.5 × 106 cells/ml) were harvested and fixed with 4% (wt/vol) paraformaldehyde at room temperature for 30 min. They were then permeabilized and stained with 20 μg/ml lysophosphatidylcholine (Sigma-Aldrich) containing 1 μM tetramethyl rhodamine isothiocyanate (TRITC)-phalloidin (Sigma-Aldrich). After centrifugation at 300 × g and 4°C for 5 min, cells were washed with PBS and their F-actin content quantified by FACS analysis using a FACSCalibur flow cytometer (Becton, Dickinson, Mountain View, CA). Data were collected as log-scaled fluorescence histograms from 10,000 cells, and the average F-actin content of a population was expressed as the mean of the fluorescence intensity. Experiments were performed in duplicate and repeated at least three times.
Statistical analysis.
The data are expressed as the means ± standard deviations (SD) for the replicate experiments. Statistical analysis was carried on using GraphPad-Prism software, version 4.00 (Graphpad Software, Inc., San Diego, CA). Student's t test or one-way analysis of variance (ANOVA), followed by the Newman-Keuls test, were used to make a statistical comparison between the groups. Results of P < 0.05 were considered statistically significant differences. Asterisks in figures (*, **, and ***) denote P < 0.05, P < 0.01, and P < 0.001, respectively.
RESULTS
TLR4 signaling promotes internalization of B. abortus into macrophages.
It has been reported previously that TLR4 mediates cellular responses to Gram-negative bacteria, especially the LPS of Gram-negative bacteria (28–30). We investigated whether TLR4 affects the phagocytosis of B. abortus. The internalization of B. abortus into RAW 264.7 cells was significantly inhibited by the neutralization of TLR4 with blocking antibody (MTS510) compared to that for the control (Fig. 1A). The knockdown of TLR4 using 50 nM TLR4-siRNA in RAW 264.7 cells (Fig. 1B) also decreased the uptake of B. abortus (Fig. 1C). In addition, BMDMs from TLR4−/− mice led to a remarkably diminished uptake of B. abortus compared to that of the WT (Fig. 1D).
Fig 1.

TLR4-involved signaling promotes the internalization of B. abortus into macrophages. (A to D) Bacterial internalization was assessed in anti-mouse TLR4-neutralized RAW 264.7 cells (A), TLR4-siRNA-transfected RAW 264.7 cells under optimal conditions of transfection (B and C), and WT or TLR4−/− BMDMs (D). (E and F) Stimulation of TLR4 signaling was conducted by treatment of E. coli LPS in RAW 264.7 cells (E) and in WT or TLR4−/− BMDMs (F). Bacterial internalization efficiency was determined by evaluating the log10 CFU. Data represent the means ± SD of triplicate trials from three independent experiments. Statistically significant differences from the untreated samples are indicated by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
TLR4 is an essential sensor and signal transducer for LPS, which is one of the strongest immunostimulatory components of Gram-negative bacteria (12, 31, 32). Thus, we hypothesized that stimulation of TLR4 with LPS alters the uptake of B. abortus. RAW 264.7 cells were stimulated with various concentrations (50 to 200 ng/ml) of E. coli LPS for 30 min prior to B. abortus infection. LPS preincubation elicited a small (1.3-fold) but statistically significant increase in uptake of B. abortus (Fig. 1E). Additionally, we tested whether the reduction of B. abortus uptake by the inhibition of TLR4 was changed by stimulation with LPS. In contrast to the significant increase of B. abortus uptake by LPS stimulation in WT cells, the reduced uptake of B. abortus in TLR4-deficient cells was rarely altered by LPS stimulation (Fig. 1F).
Moreover, we investigated whether the attenuation of B. abortus uptake by TLR4 blocking is involved in the intracellular replication of bacteria. As a result, the numbers of intracellular bacteria in TLR4-neutralized cells and TLR4 knockdown cells were lower than those of the control, but the patterns of intracellular replication in both cell types were similar to those of the control (see Fig. S1 in supplemental material).
JAK2-associated signaling pathways are required for B. abortus internalization into macrophages.
Previous studies have shown that JAK2 signaling pathways are associated with the triggering receptor expressed on myeloid cells 1 (TREM-1), an activating receptor that interacts with the TLR4/LPS-receptor complex through its engagement upon LPS stimulation in eliciting various host defenses, including phagocytosis (33, 34). Nevertheless, JAK2 signaling events that contribute to bacterial engulfment and the engagement of their phagocytic receptor are less explored. The activation of JAK2 was reduced by 35.42% after cells pretreated with a specific JAK2 inhibitor, AG490 (75 μM), were infected with B. abortus for 5 min (Fig. 2A and B).
Fig 2.

Roles of JAK2-associated signaling pathways in B. abortus infection. (A) Immunoblot analysis of total cell lysates from RAW 264.7 cells pretreated with AG490 (75 μM) combined with E. coli LPS (100 ng/ml), at the indicated times postinfection (p.i.) with B. abortus, was assessed with phosphospecific and pan-specific antibodies against JAK2. (B) The immunoblot ECL signals were quantified using NIH Image J software and the densitometry ratios of phosphor signal to pan-specific signal. (C to E) Bacterial internalization was assessed in AG490-pretreated RAW 264.7 cells (1 to 75 μM) (C) or JAK2-siRNA-transfected RAW 264.7 cells under optimal conditions of transfection for the indicated times (D and E). Data represent the means ± SD of triplicate trials from three independent experiments. Statistically significant differences from the untreated samples are indicated by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Because LPS stimulation induces the JAK2 signaling pathways (33–35), we investigated whether LPS stimulation could supplement JAK2 activation during B. abortus phagocytosis. As shown in Fig. 2A and B, in contrast to the increased intensity of JAK2 activation by LPS stimulation in control cells (activation increased by 31.22% at 5 min), the diminished JAK2 phosphorylation in JAK2-inactivated cells was not reversed by LPS stimulation (recovery values of 3.20%).
To assess the effect of JAK2 on the phagocytosis of B. abortus, RAW 264.7 cells were pretreated with different concentrations (1 to 75 μM) of AG490 and then infected with B. abortus. The inhibition of JAK2 activation notably decreased the uptake of B. abortus in a dose-dependent manner compared to untreated cells (Fig. 2C). Additionally, when the role of JAK2 in the uptake of B. abortus was assessed using JAK2-siRNA (Fig. 2D), JAK2 knockdown also significantly reduced (2.2-, 1.4-, and 1.5-fold changes at 0, 15, and 30 min, respectively) the uptake of B. abortus (Fig. 2E).
In addition, LPS (100 ng/ml) stimulation increased (1.4-, 1.3-, and 1.3-fold changes at 0, 15, and 30 min, respectively) B. abortus uptake in control cells but did not change the B. abortus uptake in JAK2 (75 μM)-inactivated cells (1.1-, 1.1-, and 1.0-fold changes at 0, 15, and 30 min, respectively), indicating that the decline of B. abortus uptake by JAK2 inhibition is seldom recovered by LPS stimulation.
Activation of Cdc42 in B. abortus internalization into macrophage is TLR4-dependent.
An earlier report had shown that Cdc42 is directly activated by virulent B. abortus (9). In the present study, we evaluated whether the activation of Cdc42 accounts for TLR4-associated phagocytosis of B. abortus. The knockdown of TLR4 in RAW 264.7 cells showed a prominent reduction in the activation of Cdc42, which was substantially enhanced by B. abortus infection (Fig. 3). These findings demonstrated that the activation of small GTPase, especially Cdc42, is TLR4 dependent for the uptake of B. abortus into macrophage cells.
Fig 3.
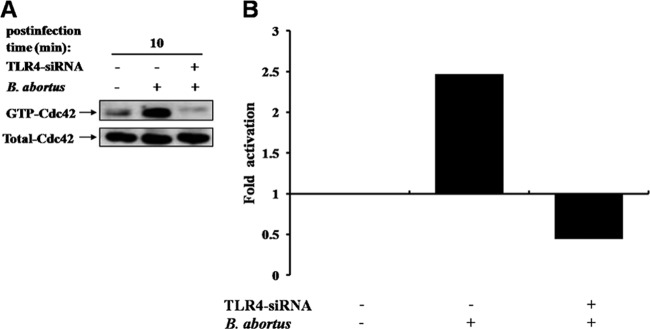
Engagement of TLR4 with Cdc42 of the Rho subfamily contributes to internalization of B. abortus into macrophage. (A) TLR4-siRNA- or control siRNA (50 nM)-transfected RAW 264.7 cells were infected with B. abortus for the indicated times. The GTP-bound, active molecules of endogenous Cdc42 were precipitated by GST-tagged PBD fusion protein and detected by Western blot analysis using anti-Cdc42 antibody. Cell lysates probed for total Cdc42 are shown as a loading control. (B) GTP-Cdc42 levels, quantified by fold activation, were detected upon uptake with B. abortus in cells transfected with control siRNA or TLR4-siRNA over basal levels present in resting cells. Data shown are the means ± SD of triplicate trials from three independent experiments.
The activation of intracellular JAK2-associated signaling pathways plays an essential role in the internalization of B. abortus by macrophages.
MAPKs (ERK1/2, p38α, and JNK) cooperate with the TLR and JAK2 signaling pathways, which are triggered by various extracellular stimuli (36, 37). In addition, pathogen-associated PI3K activation has been demonstrated to be necessary for induction of phagocytosis (38). To address the notion that the activations of JAK2 and downstream proteins are involved in the uptake of B. abortus by macrophages, we analyzed the induction of the phosphorylation of JAK2 and downstream signaling proteins, including MAPKs and PI3K-AKT, following B. abortus infection. B. abortus infection resulted in the enhanced phosphorylation of JAK2 and downstream target proteins (Fig. 4A). B. abortus-induced activation of JAK2 was reduced by 34.11 and 39.50% at 5 and 15 min postinfection (p.i.), respectively, in JAK2 inhibitor-pretreated cells compared to the control (see Fig. S2A in supplemental material), and those of downstream proteins were correspondingly decreased by JAK2 inhibition.
Fig 4.

TLR4-mediated downstream signaling pathways after B. abortus infection. (A to D) Immunoblot analysis of total cell lysates was assessed with phosphospecific and pan-specific antibodies against JAK2, ERK1/2, p38α, JNK, PI3K, and AKT in RAW 264.7 cells pretreated with AG490 (75 μM) (A) or E. coli LPS (100 ng/ml) (B) and WT or TLR4−/− BMDMs pretreated with AG490 (C) or E. coli LPS (D) at the indicated times p.i. Images shown are representative of three independent experiments.
Furthermore, the abrogation of TLR4 suppressed the phosphorylation of JAK2 (by 27.17% at 5 min and 9.19% at 15 min) and downstream proteins compared to the normal condition of TLR4 (Fig. 4C; also see Fig. S2C in supplemental material). Additionally, LPS stimulation induced the enhancement of the phosphorylation of JAK2 and downstream proteins in RAW 264.7 cells (Fig. 4B; also see Fig. S2B). However, in contrast to the increased activation of JAK2 by LPS stimulation in control cells, diminished JAK2 phosphorylation in TLR4-deficient cells (a recovery of 0.69%) was not reversed by LPS stimulation (Fig. 4D; also see Fig. S2D).
AK2 signaling pathways cooperate with TLR4 in phagocytosis of B. abortus by macrophages through enhancing actin polymerization.
Because previous studies have shown that TLR4 participates in the phagocytosis of bacteria (28, 39, 40), we determined whether the phagocytosis of B. abortus by macrophages through F-actin polymerization is mediated by TLR4. B. abortus-induced F-actin polymerization was diminished in TLR4−/− BMDMs compared to WT levels (Fig. 5A). We also evaluated whether stimulating TLR4 with E. coli LPS affects F-actin polymerization during B. abortus uptake. Both the reorganization of F-actin and the colocalization of B. abortus with F-actin were remarkably amplified by E. coli LPS. As an additional approach to confirm the role of JAK2 activation in B. abortus phagocytosis, we further examined the redistribution of cytosolic JAK2 and its colocalization with F-actin in B. abortus-infected macrophages. The results showed that B. abortus-induced F-actin polymerization and redistribution of cytosolic JAK2 were weakened in TLR4−/− BMDMs compared to those of the WT (Fig. 5B). On the basis of the result that the activation of JAK2 promoted the uptake of B. abortus by macrophages, we hypothesized that the same event affects the reorganization of the actin cytoskeleton. The observation revealed that B. abortus-augmented F-actin polymerization during uptake was weakened by the disruption of JAK2 (Fig. 5C). Furthermore, the redistribution of activated JAK2 and the colocalization of JAK2 with F-actin were reduced in cells treated with AG490 compared to those of control cells.
Fig 5.

JAK2 signaling pathways cooperate with TLR4 for phagocytosis of B. abortus through enhancing actin polymerization. (A to C) F-actin polymerization, bacterial colocalization, and redistribution of activated JAK2 were observed in WT or TLR4−/− BMDMs (A and B) and RAW 264.7 cells (C) pretreated with AG490 (75 μM) at 5 min p.i. Cells were fixed and stained with both rhodamine-conjugated phalloidin for F-actin (red) and FITC-labeled B. abortus (green) (A), Alexa Fluor 405-labeled B. abortus (blue), and FITC-labeled phospho-JAK2 antibody (green) (B and C). All images shown are representative of three separate experiments. Scale bars, 10 μm.
In F-actin fluorescence intensity, the deficiency of TLR4 resulted in a significant decline of up to 1.66-fold (TLR4−/−) and 1.62-fold (TLR4-siRNA) changes at 0 min p.i. and 1.44-fold (TLR4−/−) and 1.72-fold (TLR4-siRNA) changes at 15 min p.i., as well as the inhibition of JAK2 (Fig. 6D to F), with up to 1.37- and 1.52-fold changes at 0 and 15 min p.i., respectively, for comparisons of B. abortus-infected control cells but not noninfected cells. Moreover, F-actin fluorescence intensity in cells stimulated with E. coli LPS also showed a significantly increased value, up to a 2.19-fold change in WT BMDMs compared to the level for untreated cells and a slightly, but not significantly, increased value, up to 1.13-fold, in TLR4−/− cells (Fig. 6A to C).
Fig 6.

Intensification of F-actin polymerization dependent on TLR4 and JAK2 upon phagocytosis of B. abortus. WT or TLR4−/− BMDMs (A to C), RAW 264.7 cells transfected with TLR4- or JAK2-siRNA (D to F), or E. coli LPS-stimulated cells (A to F) were stained with 1 μM TRITC-phalloidin at the indicated times p.i. and subjected to the FACS analysis for F-actin content. The values are means ± SD of triplicate trials from three independent experiments. Statistically significant differences with untreated control are indicated by asterisks (*, P < 0.05; **, P < 0.01).
DISCUSSION
TLR signaling and phagocytosis are attributes of macrophage-mediated innate immune responses to bacterial infection. Following direct pathogen uptake, TLRs play critical roles in activating signal transduction pathways, including the JAK2, p38α, ERK1/2, and PI3K pathways, leading to the activation of inflammatory target genes (12, 35, 41). Here, we investigated the interaction of B. abortus with TLR4 as well as the correlation between phagocytosis of B. abortus and signal transduction in macrophages, focusing on the intracellular JAK2-associated signaling. Hence, we expected to determine the impact of the distinct signaling pathways associated with TLR4/JAK2 on phagocytic mechanisms of Brucella. Some studies have revealed that TLR4 is important for Brucella uptake and cellular responses in macrophages (14, 24). In this study, we showed that the internalization of B. abortus is strongly suppressed by the disruption of TLR4.
The role of TLR4 in the pathogenic mechanisms of B. abortus to establish chronic infection in macrophages is still a controversial subject. Some reports demonstrated that the TLR4 mutant mice (C3H/HeJ) had enhanced susceptibility to virulent B. abortus compared to the control (24, 42), while other authors observed the independence of TLR4 in host resistance against infection with B. abortus S19 and S2308 (43, 44). Furthermore, a TLR4-dependent production of proinflammatory cytokines elicited by the outer membrane protein 16 (OMP16) of B. abortus has been verified recently (45). In our study, we examined the effect of TLR4 blocking on B. abortus uptake and the involvement of the intracellular replication of bacteria in cultured murine macrophages. The results showed that the blocking of TLR4 in cultured macrophages caused no changes in the patterns of intracellular replication of B. abortus over a longer time period (24 to 48 h) compared to the control, while TLR4-blocked cells allowed the numbers of intracellular bacteria to diminish. We suggested that TLR4 has a more important role in bacterial internalization than in intracellular replication at the stage of pathogenicity of B. abortus. Although mice are not natural Brucella hosts, they have been widely used in brucellosis research, and different strains may vary in their responses to Brucella infection (46, 47). There have been some contradictory results, and conclusions were due to different genetic backgrounds of TLR4 knockout mice used in several studies for the involvement of TLR4 in the host resistance to infection by B. abortus. (24, 42–44). The genetic background of TLR4 knockout mice (C3H/HeJ) used in our study is that of C3H mice, which is the same as the one used by two groups that demonstrated the prominent involvement of TLR4 in B. abortus infection (24, 42); correspondingly, we suggest a role for this receptor as a receptor and signal initiator for B. abortus internalization. Meanwhile, other groups that described the inability of TLR4 to control B. abortus infection used a different mouse strain (C57BL/6 mice) (43, 44), and we believe that the differences between the mouse strains contribute to the differences in the results.
After ligand-induced receptor oligomerization, the local aggregation of JAKs and their subsequent activation induces signal transduction events (17, 19). Because JAK2 can be a crucial target of intracellular signaling transduction that is triggered by the phagocytosis of bacteria, we attempted to examine the possible role of JAK2 as a trigger of TLR4-mediated phagocytosis following B. abortus infection. We first found that activation of JAK2 was induced by B. abortus infection, and the inhibition of JAK2 prevented the internalization of B. abortus by macrophages. Thus, these results confirmed that JAK2 is required for the internalization of B. abortus by macrophages.
Recent studies have demonstrated a linkage of TLR signaling with bacteria phagocytosis (28, 40), which is promoted by the rearrangement of F-actin. The role of TLR4 in cytoskeleton reorganization has been previously determined by using macrophages from TLR4−/− or WT mice that were stimulated with E. coli LPS (48). In the present study, we determined that the induction of phagocytosis and F-actin reorganization caused by viable B. abortus infection was mediated by both TLR4 and JAK2 signaling. Fluorescence microscopy revealed that the colocalization of F-actin and activated JAK2 were increased and redistributed to the sites of filopodia and lamellipodia and the cytoplasm during B. abortus phagocytosis. In contrast, the inhibition of TLR4 and JAK2 markedly attenuated F-actin polymerization and JAK2 redistribution, resulting in the reduction of B. abortus phagocytosis. These findings suggest that the TLR4-dependent sensing of B. abortus is linked with intracellular JAK2 activation, contributing to an increase in actin polymerization and assisting the entry of B. abortus into macrophages. Although the mechanisms of TLR4-mediated F-actin reorganization remain hypothetical, we thought that it could be associated with the activation of JAK2 in B. abortus-infected macrophages.
By interacting with cytoskeletal regulators, such as the small GTPases of the Rho subfamily, intracellular and extracellular bacteria, such as Salmonella, Shigella, Listeria, Neisseria, Yersinia, and Escherichia, have advanced effective ways to evoke cytoskeletal rearrangements (49–51). It has been reported that the mechanism by which the signals from TLR4 influence the phagocytosis of bacteria is the activation of GTPases regulating TLR-mediated phagocytosis through actin assembly (41). Furthermore, available evidence indicates that the Rho subfamily GTPases are associated with the receptor-mediated JAK2 signaling events (52). Together with previous evidence, our result clearly revealed that TLR4-mediated phagocytosis of B. abortus is involved in the activation of Cdc42 GTPase. Accordingly, we suggest that TLR4 cooperated with JAK2 signaling during the phagocytosis of B. abortus into macrophage, which may have contributed to the activation of GTPases as signal linkage mediators between TLR4 and JAK2.
In signal transduction studies on activated macrophages, the stimulation of surface TLR4 induces an intracellular JAK2 phosphorylation cascade, which marks the initiation of diverse cellular responses (35, 53). We observed that entry of B. abortus into macrophages induced a rapid rise of phosphorylation of JAK2, PI3K, and MAPKs (ERK1/2, p38α, and JNK), while interfering with JAK2 suppressed these events. In the case of viable B. abortus cells as TLR4 ligands, the phosphorylation of JAK2 in TLR4−/− macrophages was reduced compared to that of the WT, which enhanced the phosphorylation of JAK2 upon B. abortus uptake. These data principally are in accord with the molecular mechanism by which activated JAK2 transduces extracellular signals via TLR4 (35).
Consequently, we propose that B. abortus induces an elaborate intracellular signal for phagocytosis via TLR4. It is likely that the interaction of B. abortus with TLR4 induces the activation of JAK2, and the subsequent activation of PI3K and MAPKs promotes actin polymerization that helps the phagocytosis of B. abortus by macrophages (Fig. 7). To the best of our knowledge, this is the first demonstration of TLR4-linked JAK2 involvement in the early cellular cascade signaling events associated with the phagocytic process, implying the pathogenic significance of JAK2-mediated entry and that the interaction of viable B. abortus with macrophages is dependent on TLR4-mediated intracellular JAK2 signaling. Importantly, from the viewpoint of the mechanistic entry route correlated with the pathogen's virulence, this work indicates that the cooperation of TLR4 with JAK2 in microbial virulence contributes to the possibility for receptor/kinase-oriented strategies as a means to counteract bacterial infection. Furthermore, this specific phagocytic mechanism of B. abortus could provide new insights into novel approaches to establish the receptor-pathogen interaction and signal transduction in host cells infected by intracellular pathogens.
Fig 7.
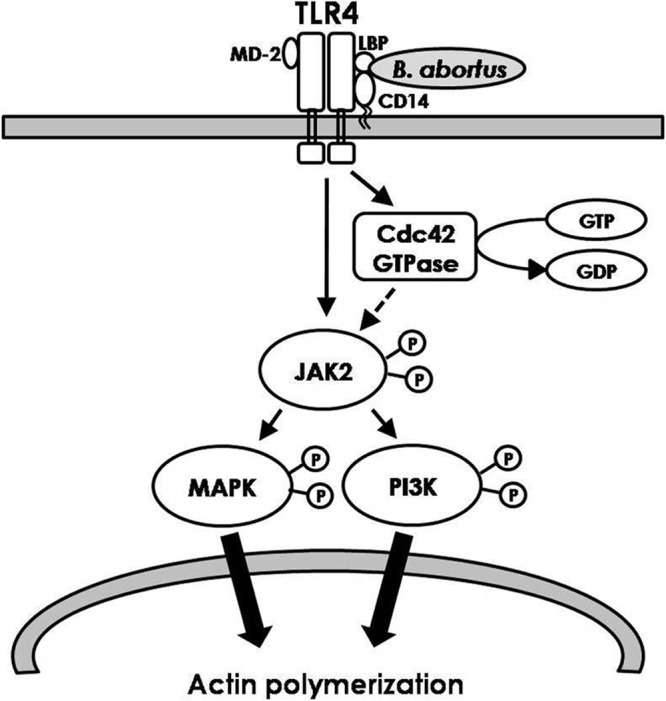
Diagram illustrating the phagocytic signaling pathway initiated by TLR4-linked JAK2 activation during the internalization of B. abortus into macrophage. The interaction of B. abortus with TLR4 induces the activation of Cdc42 GTPase and JAK2, and the subsequent activation of PI3K and MAPKs promotes actin polymerization. This event contributes to the phagocytosis of B. abortus by macrophage. Lines with arrows denote an activating reaction, and the dotted line denotes uncertainty of the reaction.
Supplementary Material
ACKNOWLEDGMENTS
The present work was supported by grants from the Ministry of Education, Basic Science Research Program, National Research Foundation (2010-0009080), and from the National Veterinary Research and Quarantine Service (0468-2010002), South Korea.
Footnotes
Published ahead of print 29 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00403-13.
REFERENCES
- 1. Acha P, Szylres B. 1980. Zoonoses and communicable diseases common to man and animals. Pan American Health Organization, Washington, DC [Google Scholar]
- 2. Baldwin CL, Winter AJ. 1994. Macrophages and Brucella. Immunol. Ser. 60:363–380 [PubMed] [Google Scholar]
- 3. Detilleux PG, Deyoe BL, Cheville NF. 1990. Penetration and intracellular growth of Brucella abortus in nonphagocytic cells in vitro. Infect. Immun. 58:2320–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liautard JP, Gross A, Dornand J, Kohler S. 1996. Interactions between professional phagocytes and Brucella spp. Microbiologia 12:197–206 [PubMed] [Google Scholar]
- 5. Sangari FJ, Aguero J. 1996. Molecular basis of Brucella pathogenicity: an update. Microbiologia 12:207–218 [PubMed] [Google Scholar]
- 6. Gruenheid S, Finlay BB. 2003. Microbial pathogenesis and cytoskeletal function. Nature 422:775–781 [DOI] [PubMed] [Google Scholar]
- 7. Ackermann MR, Cheville NF, Deyoe BL. 1988. Bovine ileal dome lymphoepithelial cells: endocytosis and transport of Brucella abortus strain 19. Vet. Pathol. 25:28–35 [DOI] [PubMed] [Google Scholar]
- 8. Watarai M, Makino S, Fujii Y, Okamoto K, Shirahata T. 2002. Modulation of Brucella-induced macropinocytosis by lipid rafts mediates intracellular replication. Cell. Microbiol. 4:341–355 [DOI] [PubMed] [Google Scholar]
- 9. Guzman-Verri C, Chaves-Olarte E, von Eichel-Streiber C, Lopez-Goni I, Thelestam M, Arvidson S, Gorvel JP, Moreno E. 2001. GTPases of the Rho subfamily are required for Brucella abortus internalization in nonprofessional phagocytes: direct activation of Cdc42. J. Biol. Chem. 276:44435–44443 [DOI] [PubMed] [Google Scholar]
- 10. Detilleux PG, Deyoe BL, Cheville NF. 1991. Effect of endocytic and metabolic inhibitors on the internalization and intracellular growth of Brucella abortus in Vero cells. Am. J. Vet. Res. 52:1658–1664 [PubMed] [Google Scholar]
- 11. Kusumawati A, Cazevieille C, Porte F, Bettache S, Liautard JP, Sri Widada J. 2000. Early events and implication of F-actin and annexin I associated structures in the phagocytic uptake of Brucella suis by the J-774A.1 murine cell line and human monocytes. Microb. Pathog. 28:343–352 [DOI] [PubMed] [Google Scholar]
- 12. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384 [DOI] [PubMed] [Google Scholar]
- 13. Fernandez-Prada CM, Zelazowska EB, Nikolich M, Hadfield TL, Roop RM, Jr, Robertson GL, Hoover DL. 2003. Interactions between Brucella melitensis and human phagocytes: bacterial surface O-polysaccharide inhibits phagocytosis, bacterial killing, and subsequent host cell apoptosis. Infect. Immun. 71:2110–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oliveira SC, de Oliveira FS, Macedo GC, de Almeida LA, Carvalho NB. 2008. The role of innate immune receptors in the control of Brucella abortus infection: toll-like receptors and beyond. Microbes Infect. 10:1005–1009 [DOI] [PubMed] [Google Scholar]
- 15. Schnyder B, Meunier PC, Car BD. 1998. Inhibition of kinases impairs neutrophil activation and killing of Staphylococcus aureus. Biochem. J. 331(Pt 2):489–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ridley AJ. 2001. Rho proteins, PI 3-kinases, and monocyte/macrophage motility. FEBS Lett. 498:168–171 [DOI] [PubMed] [Google Scholar]
- 17. Rane SG, Reddy EP. 2000. Janus kinases: components of multiple signaling pathways. Oncogene 19:5662–5679 [DOI] [PubMed] [Google Scholar]
- 18. Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J, Kubo T, Yoshikawa H. 2005. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 24:6406–6417 [DOI] [PubMed] [Google Scholar]
- 19. Ghoreschi K, Laurence A, O'Shea JJ. 2009. Janus kinases in immune cell signaling. Immunol. Rev. 228:273–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pena G, Cai B, Deitch EA, Ulloa L. JAK2 inhibition prevents innate immune responses and rescues animals from sepsis. J. Mol. Med. (Berlin) 88:851–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Imai K, Kurita-Ochiai T, Ochiai K. 2003. Mycobacterium bovis bacillus Calmette-Guerin infection promotes SOCS induction and inhibits IFN-gamma-stimulated JAK/STAT signaling in J774 macrophages. FEMS Immunol. Med. Microbiol. 39:173–180 [DOI] [PubMed] [Google Scholar]
- 22. Rojas M, Olivier M, Garcia LF. 2002. Activation of JAK2/STAT1-alpha-dependent signaling events during Mycobacterium tuberculosis-induced macrophage apoptosis. Cell. Immunol. 217:58–66 [DOI] [PubMed] [Google Scholar]
- 23. Bar-Sagi D, Hall A. 2000. Ras and Rho GTPases: a family reunion. Cell 103:227–238 [DOI] [PubMed] [Google Scholar]
- 24. Pei J, Turse JE, Ficht TA. 2008. Evidence of Brucella abortus OPS dictating uptake and restricting NF-kappaB activation in murine macrophages. Microbes Infect. 10:582–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Watarai M, Andrews HL, Isberg RR. 2001. Formation of a fibrous structure on the surface of Legionella pneumophila associated with exposure of DotH and DotO proteins after intracellular growth. Mol. Microbiol. 39:313–329 [DOI] [PubMed] [Google Scholar]
- 26. Benard V, Bohl BP, Bokoch GM. 1999. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 274:13198–13204 [DOI] [PubMed] [Google Scholar]
- 27. Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr 2001. Akt stimulates the transactivation potential of the RelA/p65 subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 276:18934–18940 [DOI] [PubMed] [Google Scholar]
- 28. Doyle SE, O'Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, Suzuki S, Suzuki N, Modlin RL, Yeh WC, Lane TF, Cheng G. 2004. Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 199:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thoma-Uszynski S, Stenger S, Takeuchi O, Ochoa MT, Engele M, Sieling PA, Barnes PF, Rollinghoff M, Bolcskei PL, Wagner M, Akira S, Norgard MV, Belisle JT, Godowski PJ, Bloom BR, Modlin RL. 2001. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science 291:1544–1547 [DOI] [PubMed] [Google Scholar]
- 30. Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. U. S. A. 97:13766–13771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miller SI, Ernst RK, Bader MW. 2005. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 3:36–46 [DOI] [PubMed] [Google Scholar]
- 32. Lu YC, Yeh WC, Ohashi PS. 2008. LPS/TLR4 signal transduction pathway. Cytokine 42:145–151 [DOI] [PubMed] [Google Scholar]
- 33. Fortin CF, Lesur O, Fulop T., Jr 2007. Effects of TREM-1 activation in human neutrophils: activation of signaling pathways, recruitment into lipid rafts and association with TLR4. Int. Immunol. 19:41–50 [DOI] [PubMed] [Google Scholar]
- 34. Arts RJ, Joosten LA, Dinarello CA, Kullberg BJ, van der Meer JW, Netea MG. 2011. TREM-1 interaction with the LPS/TLR4 receptor complex. Eur. Cytokine Netw. 22:11–14 [DOI] [PubMed] [Google Scholar]
- 35. Okugawa S, Ota Y, Kitazawa T, Nakayama K, Yanagimoto S, Tsukada K, Kawada M, Kimura S. 2003. Janus kinase 2 is involved in lipopolysaccharide-induced activation of macrophages. Am. J. Physiol. Cell Physiol. 285:C399–C408 [DOI] [PubMed] [Google Scholar]
- 36. De Nardo D, De Nardo CM, Nguyen T, Hamilton JA, Scholz GM. 2009. Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J. Immunol. 183:8110–8118 [DOI] [PubMed] [Google Scholar]
- 37. Wu W, Alexis NE, Chen X, Bromberg PA, Peden DB. 2008. Involvement of mitogen-activated protein kinases and NFkappaB in LPS-induced CD40 expression on human monocytic cells. Toxicol. Appl. Pharmacol. 228:135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tachado SD, Samrakandi MM, Cirillo JD. 2008. Non-opsonic phagocytosis of Legionella pneumophila by macrophages is mediated by phosphatidylinositol 3-kinase. PLoS One 3:e3324. 10.1371/journal.pone.0003324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blander JM, Medzhitov R. 2004. Regulation of phagosome maturation by signals from toll-like receptors. Science 304:1014–1018 [DOI] [PubMed] [Google Scholar]
- 40. Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, Li J, Cetin S, Ford H, Schreiber A, Hackam DJ. 2006. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol. 176:3070–3079 [DOI] [PubMed] [Google Scholar]
- 41. Kong L, Ge BX. 2008. MyD88-independent activation of a novel actin-Cdc42/Rac pathway is required for Toll-like receptor-stimulated phagocytosis. Cell Res. 18:745–755 [DOI] [PubMed] [Google Scholar]
- 42. Campos MA, Rosinha GM, Almeida IC, Salgueiro XS, Jarvis BW, Splitter GA, Qureshi N, Bruna-Romero O, Gazzinelli RT, Oliveira SC. 2004. Role of Toll-like receptor 4 in induction of cell-mediated immunity and resistance to Brucella abortus infection in mice. Infect. Immun. 72:176–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barquero-Calvo E, Chaves-Olarte E, Weiss DS, Guzman-Verri C, Chacon-Diaz C, Rucavado A, Moriyon I, Moreno E. 2007. Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS One 2:e631. 10.1371/journal.pone.0000631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weiss DS, Takeda K, Akira S, Zychlinsky A, Moreno E. 2005. MyD88, but not toll-like receptors 4 and 2, is required for efficient clearance of Brucella abortus. Infect. Immun. 73:5137–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pasquevich KA, Garcia Samartino C, Coria LM, Estein SM, Zwerdling A, Ibanez AE, Barrionuevo P, Oliveira FS, Carvalho NB, Borkowski J, Oliveira SC, Warzecha H, Giambartolomei GH, Cassataro J. 2010. The protein moiety of Brucella abortus outer membrane protein 16 is a new bacterial pathogen-associated molecular pattern that activates dendritic cells in vivo, induces a Th1 immune response, and is a promising self-adjuvanting vaccine against systemic and oral acquired brucellosis. J. Immunol. 184:5200–5212 [DOI] [PubMed] [Google Scholar]
- 46. Grillo MJ, Blasco JM, Gorvel JP, Moriyon I, Moreno E. 2012. What have we learned from brucellosis in the mouse model? Vet. Res. 43:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Silva TM, Costa EA, Paixao TA, Tsolis RM, Santos RL. 2011. Laboratory animal models for brucellosis research. J. Biomed. Biotechnol. 2011:518323. 10.1155/2011/518323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wonderling RS, Ghaffar A, Mayer EP. 1996. Lipopolysaccharide-induced suppression of phagocytosis: effects on the phagocytic machinery. Immunopharmacol. Immunotoxicol. 18:267–289 [DOI] [PubMed] [Google Scholar]
- 49. Hall A. 1998. Rho GTPases and the actin cytoskeleton. Science 279:509–514 [DOI] [PubMed] [Google Scholar]
- 50. Caron E, Hall A. 1998. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science 282:1717–1721 [DOI] [PubMed] [Google Scholar]
- 51. Cox D, Chang P, Zhang Q, Reddy PG, Bokoch GM, Greenberg S. 1997. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 186:1487–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pelletier S, Duhamel F, Coulombe P, Popoff MR, Meloche S. 2003. Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol. Cell. Biol. 23:1316–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hu X, Chen J, Wang L, Ivashkiv LB. 2007. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J. Leukoc. Biol. 82:237–243 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.