

Abstract
Heligmosomoides bakeri is a nematode with parasitic development exclusively in the small intestine of infected mice that induces a potent STAT6-dependent Th2 immune response. We previously demonstrated that host protective expulsion of adult H. bakeri worms from a challenge infection was delayed in selenium (Se)-deficient mice. In order to explore mechanisms associated with the delayed expulsion, 3-week-old female BALB/c mice were placed on a torula yeast-based diet with or without 0.2 ppm Se, and after 5 weeks, they were inoculated with H. bakeri infective third-stage larvae (L3s). Two weeks after inoculation, the mice were treated with an anthelmintic and then rested, reinoculated with L3s, and evaluated at various times after reinoculation. Analysis of gene expression in parasite-induced cysts and surrounding tissue isolated from the intestine of infected mice showed that the local-tissue Th2 response was decreased in Se-deficient mice compared to that in Se-adequate mice. In addition, adult worms recovered from Se-deficient mice had higher ATP levels than worms from Se-adequate mice, indicating greater metabolic activity in the face of a suboptimal Se-dependent local immune response. Notably, the process of worm expulsion was restored within 2 to 4 days after feeding a Se-adequate diet to Se-deficient mice. Expulsion was associated with an increased local expression of Th2-associated genes in the small intestine, intestinal glutathione peroxidase activity, secreted Relm-β protein, anti-H. bakeri IgG1 production, and reduced worm fecundity and ATP-dependent metabolic activity.
INTRODUCTION
Selenium (Se) is a trace element with widely differing levels of soil availability that are associated with local differences in incorporation into plants and animals (1). Reduced Se tissue levels result from inadequate Se in the diet but have also been associated with certain diseases, including inflammatory bowel disease (2, 3). Selenium was shown to be deficient in critically ill patients and pediatric burn patients (4, 5), and benefits have been observed when supplementing with Se in critically ill patients (6, 7). These data suggest that Se status may be altered in the acute-phase response to stress or infection. Due partially to its incorporation as selenocysteine in proteins, Se has an important role in cytotoxic T-lymphocyte responses and natural killer cell activity (8). Protection against endotoxin-induced oxidative stress has also been demonstrated (9). Several studies indicated that Se status affects NF-κB activation (10–12) and that Se deficiency impaired respiratory burst (13, 14). Additional studies showed that chemokine and cytokine responses to viral and bacterial infections were Se dependent and that Se deficiency increased the pathology associated with these infections (15, 16).
Approximately 3.5 billion people worldwide are infected by parasites that cause extensive morbidity, especially in children (17, 18). Inadequate nutrition is often associated with populations at risk for parasitic infections. In many underdeveloped countries, poor nutritional status is correlated with the severity of and susceptibility to infection (reviewed in reference 19). The clearance of H. bakeri, a gastrointestinal nematode parasite that naturally infects rodents, has been used to study intestinal immunity, which has also been shown to be nutritionally dependent (20–22).
Infection with nematode parasites stimulates a potent STAT6-dependent Th2 immune response (reviewed in reference 23). Th2-dependent alternatively activated macrophages have been implicated in intestinal immunity to H. bakeri (24), as has the goblet cell protein Relm-β (Retnlb) (25). A number of additional genes associated with alternatively activated macrophages and the Th2 response, including Il13rα2, Chi3l3 (YM1), Chi3l4 (YM2), Retnla (Relm-α), and Arg-1, are upregulated by H. bakeri infection (23). We previously observed that Se deficiency inhibited the intestinal clearance of adult H. bakeri worms following a secondary challenge infection (26, 27). Here, we show that Se deficiency dampens local Th2-dependent gene expression during an infection with H. bakeri in the small intestine and that feeding a Se-adequate diet rapidly restores immunological functions associated with adult worm expulsion.
MATERIALS AND METHODS
Mice and infections.
Three-week-old female BALB/c mice were obtained from NCI-Frederick. All animal protocols were approved by the USDA/ARS Beltsville Animal Care and Use Committee. Mice were fed a torula yeast-based diet (28) containing 50 mg/kg vitamin E as d-α-tocopherol acetate with 0.0 or 0.2 ppm Se added as sodium selenite. After 5 to 6 weeks on the diet, mice were inoculated per os with 200 H. bakeri (29) infective third-stage larvae (L3s) (U.S. National Helminthological Collection no. 81930) obtained by the modified Baermann method from charcoal/peat moss cultures of feces from infected mice and stored at 4°C until used (24, 30). After 2 weeks, mice were treated with the anthelmintic drug pyrantel pamoate (0.1 ml of a 50 mg/ml solution) to cure the infection. Mice were then rested for at least 4 weeks before being reinoculated with 200 H. bakeri L3s. Mice on Se-adequate or Se-deficient diets were assayed at various times during the infection. The larval and worm burdens and worm fecundity (measured by direct counting of trichostrongyle eggs per gram of feces) were determined depending on the experimental design. In some experiments, Se-deficient mice were fed a Se-adequate diet starting 10 days after reinoculation. Replicate sets of mice were assayed on days 12 and 14 after reinoculation (2 and 4 days, respectively, after restoration of the Se-adequate diet), and small intestine tissues approximately 4 to 10 cm from the stomach were isolated for gene expression and measurement of glutathione peroxidase (GPX) activity. Gene expression was also evaluated for the cysts derived from the invasion of parasitic L3s in the duodenum, which are rich in infiltrating immune cells, and surrounding noncyst tissue adjacent to the cysts. For this procedure, the duodenum of the small intestine was removed and cut open longitudinally, and either individual cysts or non-cyst-containing tissue was localized using a dissecting microscope and excised using glass microcapillary tubes (31), placed into RNAlater, and then snap-frozen in liquid N2 until processed for RNA extraction.
Molecular assay, enzyme-linked immunosorbent assay (ELISA), and Western blot assay.
RNA was extracted from the small intestine using TRIzol reagent (Invitrogen, Carlsbad, CA) and processed (32). Following DNase treatment, RNA integrity and quantity and the detection of genomic DNA were determined using the Experion automated electrophoresis system (Bio-Rad, Hercules, CA). cDNA was synthesized using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA), and 25 ng cDNA/reaction mixture volume was then used for quantitative PCR amplification using a commercially available kit (Abgene, Rochester, NY). Amplification was measured on an ABI 7500 (Applied Biosystems, Foster City, CA) using 5′-tetrachloro-6-carboxyfluorescein- and 3′-Black Hole Quencher 1-labeled probes. The threshold cycle (CT) was calculated using the manufacturer's software. Data were normalized to the results for the housekeeping gene RPL32 (L32 ribosomal protein gene), the mean of CTcontrol was subtracted from the mean of CTtreatment, and the relative fold increase or decrease was then calculated as 2−ΔΔCT (33). Fold changes were expressed as the change in expression compared to the expression in the group of uninfected mice fed a Se-adequate diet and nominally designated 1.0-fold. Data for each of the two gene expression studies came from two independent experiments that were merged to increase the sample number and the power of the analysis.
Anti-H. bakeri excretory/secretory protein (ESP) IgG1, IgG(H+L), and IgG2a antibodies were detected by coating 96-well flat-bottom MediSorp plates (catalog number 12-565-730; Thermo Scientific) with 5 μg/ml H. bakeri ESP, derived from adult worms after 48 h of culture and diluted in 100 mM sodium carbonate buffer (pH 9.6), and incubating overnight at 4°C. The plates were blocked with 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS)–0.05% Tween 20. After 1 h of incubation at room temperature (RT), the wells were washed 6 times with PBS–0.05% Tween 20 and then incubated with diluted serum samples (1/10,000 to 1/30,000 for detecting IgG1 and IgG and 1/1,000 to 1/3,000 for detecting IgG2a) in triplicate for 1 h at 37°C. For detecting specific antibody isotypes, the plates were then incubated with either 1:50,000 biotinylated anti-mouse IgG(H+L) (BA-9200; Vector laboratories), 1:5,000 biotinylated anti-mouse IgG1 (item number 48-370-2B; Antibodies Incorporated), or 1:500 biotinylated anti-mouse IgG2a (item number 48-370-3B; Antibodies Incorporated). After 1 h of incubation at 37°C, the plates were washed and incubated with a 1:50,000 or 1:10,000 dilution of streptavidin-horseradish peroxidase (HRP) concentrate (catalog number SA-5004; Vector Laboratories) added to IgG1 and IgG(H+L) or IgG2a, respectively. The plates were incubated for 1 h at 37°C, washed, and developed using TMB One substrate solution (catalog number G7431; Promega) that was added to each well. The color reaction was terminated by the addition of 0.1 M HCl, and the plates were read at 450 nm on a Molecular Devices SpectraMax-M5 plate reader.
Small intestine tissues from control uninfected and H. bakeri-infected mice were homogenized in PBS, pH 7.4, containing a 1× concentration of protease cocktail (catalog number P8340; Sigma, St. Louis, MO) and then centrifuged at 14,000 × g for 30 min at 4°C. The protein content of the resulting supernatants was determined using the BCA protein assay (Pierce, Chicago, IL) with BSA as the standard. Twenty micrograms of each sample was subjected to SDS-PAGE using Novex 10% Tris–glycine gels (Invitrogen, Carlsbad, CA) and electroblotted onto polyvinylidene difluoride (PVDF) membranes. The resulting blot was blocked with PBS, pH 7.4, containing 1% Hammerstein casein or SuperBlock blocking buffer in Tris-buffered saline (Pierce, Chicago, IL) containing 5% healthy goat serum and then incubated with the same solution containing 0.05% Tween 20 and rabbit anti-mouse Relm-β (Peprotech, Rocky Hill, NJ) and anti-mouse β-actin (Biolegend, San Diego, CA) antibodies. Detection of bound antibodies was accomplished by using biotinylated anti-rabbit IgG, streptavidin-HRP (Vector laboratories, Burlingame, CA), and Thermo SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL). Bands were quantified using NIH ImageJ software, and the levels of Relm-β were normalized to those of β-actin.
Measurement of GPX activity (Se status) and worm ATP levels (metabolic stress).
Glutathione peroxidase activity, an indirect enzymatic indicator of Se status, was measured (34) in supernatants prepared from homogenates of mouse small intestine and liver by centrifugation at 14,000 × g. Adult H. bakeri worm ATP levels were measured using the ATPlite luminescence ATP detection assay system (PerkinElmer, Boston, MA) (35) to determine the ATP-dependent metabolic activities of worms recovered from Se-adequate and Se-deficient mice (36). Briefly, groups of five adult female worms from each of five Se-adequate or Se-deficient mice at 12 or 14 days after reinoculation were morphologically identified under a dissecting microscope and then micromanipulated into tubes, homogenized in lysis buffer with a hand-held motorized pestle, and assayed according to the manufacturer's instructions. The ATP levels in the samples were expressed as relative luminescent units (RLU). An average RLU was determined for five mice per group.
Data analysis.
Data were expressed as the mean ± standard deviation (SD) or standard error of the mean (SEM). Statistical analyses were performed using the t test and one-way analysis of variance (ANOVA) with post hoc analysis for multiple comparisons using the Hom-Sidak method (SigmaPlot 11; SigmaPlot, Richmond, CA). Data were transformed as necessary to achieve normality and equal variance, or a Mann-Whitney rank sum test was performed.
RESULTS
Se-deficient mice have delayed adult worm expulsion associated with a reduced local Th2 response.
Parasitic H. bakeri L3s initially invade the small intestine and develop into adult worms that emerge into the intestinal lumen at approximately 8 days after inoculation. A visible nodular cyst develops at the site of intestinal invasion (24). Parasitic larvae recovered from the tissue by the Baermann procedure were in significantly higher numbers at day 4 and equivalent numbers at day 7 after reinoculation in mice on a Se-deficient diet compared to the numbers in mice on a Se-adequate diet on the same days (Fig. 1A). However, mice fed a Se-deficient diet had significantly more adult worms at day 12 through day 21 after reinoculation than mice fed a Se-adequate diet, which had expelled worms by day 21 after reinoculation (Fig. 1B). Expulsion is normally linked to a coordinated interleukin-4 (IL-4)/IL-13 STAT6-dependent immune response (24, 37). Cysts and noncyst tissues were separately dissected from the small intestines of infected mice fed a Se-adequate or Se-deficient diet to determine if local Th2-associated gene expression was altered. Cyst tissue isolated from mice fed a Se-adequate diet had high levels of expression of Il4, Il13, and Il13ra2, as well as of genes associated with alternatively activated macrophages, including Chi3l3, Chi3l4, and Retnla (Table 1). In contrast, cyst tissue obtained from H. bakeri-infected mice fed a Se-deficient diet had reduced expression of Il4 and Il13, as well as of genes associated with alternatively activated macrophages. The expression of the Th1 cytokine Ifng was equally low in both noncyst and cyst tissues isolated from infected mice fed the Se-deficient or Se-adequate diet (data not shown). With the exception of Chi3l3, gene expression was lower in noncyst tissue isolated from infected mice fed a Se-deficient diet than in mice fed a Se-adequate diet (Table 1; also data not shown). The expression of Retnlb was significantly higher in cyst and noncyst tissues from H. bakeri-infected mice on a Se-adequate diet. Taken together, these results suggested that Se deficiency impaired the local Th2-dependent response in small intestine tissue from H. bakeri-infected mice.
Fig 1.

Selenium (Se) deficiency did not affect the tissue stage but delayed clearance of H. bakeri infection in mice. Mice fed either a Se-adequate or Se-deficient diet were primed and reinoculated with 200 H. bakeri infective larvae. (A) At days 4 and 7 after reinoculation, mice were killed and worms recovered and counted. Results are means ± SEM (n = 5). *, results for Se-deficient group are significantly different from results for Se-adequate group; P < 0.05. (B) At days 12 and 21 after reinoculation, mice were killed and adult worms recovered and counted. Results are means ± SD (n = 5). *, results for Se-deficient group were significantly different from results for Se-adequate control group; P < 0.05. nd, no adult worms detected.
Table 1.
Intestinal gene expression was altered in H. bakeri-infected mice fed a selenium-deficient diet
| Tissue | Gene | Result in infected mice with indicated Se level |
|||
|---|---|---|---|---|---|
| Adequate |
Deficient |
||||
| Mean CT ± SEMc | Fold changed | Mean CT ± SEM | Fold change | ||
| Cysta | Il4 | 8.7 ± 0.2 | 116 | 9.9 ± 0.2 | 50* |
| Il13 | 8.8 ± 0.3 | 136 | 10.5 ± 0.3 | 43* | |
| Chi3L3 | −2.1 ± 0.2 | 24,845 | −0.7 ± 0.2 | 9,276* | |
| Chi3L4 | 5.5 ± 0.3 | 871 | 8.3 ± 0.2 | 125* | |
| Retnla | −0.6 ± 0.2 | 5,463 | 0.5 ± 0.1 | 2,521* | |
| Retnlb | 3.4 ± 0.2 | 390 | 4.3 ± 0.2 | 200* | |
| Il13ra2 | 6.6 ± 0.2 | 503 | 8.1 ± 0.2 | 186* | |
| Noncystb | Il4 | 11.8 ± 0.2 | 13 | 12.8 ± 0.3 | 7* |
| Il13 | 10.1 ± 0.5 | 56 | 11.7 ± 0.4 | 18* | |
| Chi3L3 | 3.2 ± 0.3 | 634 | 4.2 ± 0.4 | 320 | |
| Retnlb | 2.5 ± 0.2 | 705 | 3.8 ± 0.3 | 288* | |
Cysts that formed during invasion and development of parasitic larvae were mechanically removed from the intestine.
Noncyst tissue is the mucosa surrounding the cysts.
CT indicates the threshold cycle method; the lower the number, the higher the gene expression.
Fold change in gene expression 12 days postinfection in comparison to the expression in uninfected mice fed a Se-adequate diet, which is assigned a fold change of 1.0; n = 13 to 15. Within uninfected mice fed a Se-deficient diet, the fold changes in the expression of two genes, Chi3L4 and Retnlb, varied significantly (<5-fold change) in comparison to their expression in uninfected mice on Se-adequate diet (data not shown). The fold change in expression for infected mice on either diet was significantly higher than that in in uninfected mice fed either diet. Results with an asterisk are significantly different at a P value of <0.05.
Adult H. bakeri worms isolated from mice fed a Se-deficient diet had enhanced metabolic activity.
In the face of an adaptive immune response, parasitic worms become metabolically stressed, with reduced levels of ATP, smaller size, and decreased egg production. Relm-β may be an important factor in this process (25, 36). To determine the metabolic activities of worms isolated from mice fed Se-adequate or Se-deficient diets, the ATP levels were measured in worms isolated at 12 days after reinoculation when egg production was nearly undetectable in mice fed the Se-adequate diet, but remained elevated in mice fed the Se-deficient diet (Fig. 2A). The results showed that worms from Se-adequate mice were less metabolically active (i.e., reduced ATP levels) than those from Se-deficient mice, and these decreased ATP levels correlated with lower numbers of worms (Fig. 1) and egg production (Fig. 2B). Decreased ATP in H. bakeri was observed after exposure of adult worms to Relm-β (25). Gene expression for Relmb was lower in the small intestine of H. bakeri-infected Se-deficient mice than in Se-adequate mice (Table 1), and there was significantly less Relm-β protein (P < 0.001) expressed in the small intestine of infected Se-deficient mice (Fig. 3), suggesting that Relm-β is one factor responsible for decreased worm ATP levels and worm expulsion from Se-deficient mice.
Fig 2.
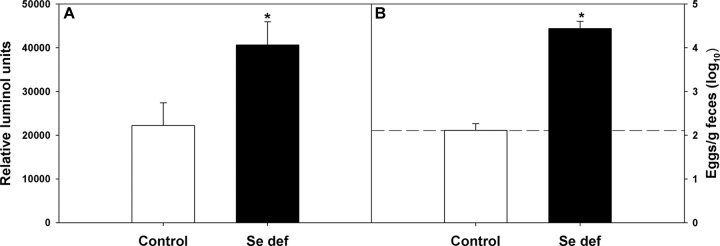
Adult worm ATP levels were dependent on host selenium (Se) status. Mice fed a Se-adequate or Se-deficient diet were primed and reinoculated with 200 H. bakeri infective larvae. (A) At 12 days postinfection, mice were killed, and adult female worms were isolated and their ATP levels determined. *, results for ATP content of worms from Se-deficient group were significantly different from results in Se-adequate group; P < 0.001. Results are means ± SD (n = 5). (B) At 12 days postinfection, the number of eggs excreted per gram of feces was determined. Results are expressed as means ± SD. *, number of eggs excreted per gram of feces was significantly higher in Se-deficient mice; P < 0.001 (n = 5). Dashed line indicates limit of detection.
Fig 3.
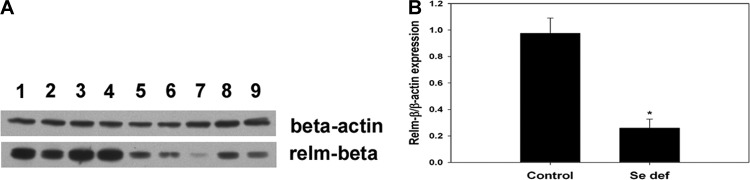
Parasite-induced Relm-β protein in small intestine was dependent on host selenium (Se) status. (A) Small intestine tissue from Se-adequate (lanes 1 to 4) and Se-deficient (lanes 5 to 9) H. bakeri-infected mice were obtained 12 days after reinoculation and used for Western immunoblot analysis for Relm-β and β-actin expression. (B) Bands were quantified using NIH ImageJ software. The amount of Relm-β was normalized to the amount of β-actin and plotted as relative expression. *, P < 0.001.
Feeding Se-adequate diet rapidly restores Th2 antiworm immunity.
Selenium-adequate mice had little to no measurable egg production at 10, 12, and 14 days after reinoculation with H. bakeri, while egg production in infected Se-deficient mice increased over that time period (Fig. 4). In contrast, Se-deficient mice fed a Se-adequate diet starting on day 10 after reinoculation had significantly lower egg production on day 12 than Se-deficient mice, and egg production decreased further by day 14 after reinoculation (Fig. 4). Thus, by 48 h after switching from a Se-deficient to a Se-adequate diet, mice restored the anti-fecundity response. In addition, adult worm numbers (Fig. 5A) and ATP activities (Fig. 5B) were reduced in Se-deficient mice fed adequate Se for 96 h compared to the results for worms from mice maintained on a Se-deficient diet, and worms were cleared by day 21 from Se-deficient mice fed adequate Se but not from mice on a Se-deficient diet (Fig. 1; also data not shown).
Fig 4.

Adult worm egg production was dependent on host selenium (Se) status. Mice fed a Se-adequate or Se-deficient (Se def) diet were primed and reinoculated with 200 H. bakeri infective larvae. At 10 days postinfection, one group of Se-deficient mice was fed a Se-adequate diet (Refeed). The numbers of eggs excreted per gram of feces were monitored at days 10, 12, and 14 (0, 2, and 4 days, respectively, after feeding Se-adequate diet) after reinoculation. Only within-day comparisons were made. Results are expressed as means ± SD. Groups (n = 5) with different letters had significantly different results; P < 0.05. Dashed line indicates limit of detection. Con, control.
Fig 5.
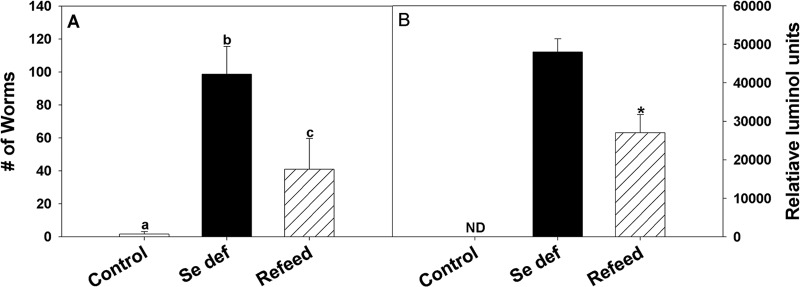
Feeding selenium (Se) activated adult worm expulsion and reduced worm ATP levels. Mice fed a Se-adequate or Se-deficient diet were reinoculated with H. bakeri infective larvae, and one set of infected Se-deficient mice were fed a Se-adequate diet at 10 days after reinoculation and sacrificed 4 days later. (A) The number of adult worms recovered from the small intestine was determined. Bars with different letters show significantly different results; P < 0.001. (B) The ATP levels of adult worms recovered were significantly reduced when mice were fed a Se-adequate diet; *, P < 0.001. Data are expressed as the means ± SD (n = 5). ND, not determined.
Small intestine tissue was obtained from uninfected and H. bakeri-infected mice fed a Se-adequate or Se-deficient diet and Se-deficient mice fed adequate Se for 2 days to determine if feeding Se-deficient mice a Se-adequate diet restored Th2-dependent gene expression. Mice fed a Se-deficient diet had reduced Th2 gene expression following reinoculation compared to that in mice fed a Se-adequate diet, but Th2-depenent gene expression increased within 2 days after feeding reinoculated Se-deficient mice a Se-adequate diet (Table 2). The fold change in gene expression of the infected Se-deficient mice fed adequate Se approached that seen in infected Se-adequate mice by 48 h, and this coincided with decreased egg production.
Table 2.
Expression of Th2-associated genes increased after feeding adequate Se to H. bakeri-infected Se-deficient mice
| Gene | Result in infected mice with indicated Se level |
Result when Se-deficient mice were fed adequate Se |
||||
|---|---|---|---|---|---|---|
| Adequate |
Deficient |
|||||
| Mean CTa ± SEM | Fold changeb | Mean CTa ± SEM | Fold changeb | Mean CTa ± SEM | Fold changeb | |
| Il4 | 10.4 ± 0.2 | 39 | 11.6 ± 0.2 | 16* | 10.6 ± 0.2 | 32 |
| Il13 | 8.5 ± 0.3 | 61 | 10.9 ± 0.3 | 12* | 8.9 ± 0.4 | 48 |
| Chi3L3 | 0.4 ± 0.2 | 571 | 1.7 ± 0.2 | 224* | 0.9 ± 0.3 | 398 |
| Chi3L4 | 6.4 ± 0.3 | 486 | 11.1 ± 0.2 | 19* | 6.9 ± 0.5 | 353 |
| Retnla | 0.9 ± 0.2 | 325 | 2.5 ± 0.3 | 108* | 1.3 ± 0.2 | 251 |
| Retnlb | 2.4 ± 0.3 | 325 | 4.5 ± 0.2 | 75* | 2.8 ± 0.3 | 255 |
| Arg1 | 4.9 ± 0.4 | 120 | 6.5 ± 0.3 | 39* | 5.2 ± 0.3 | 97 |
CT indicates the threshold cycle method; the lower the number, the higher the gene expression.
Fold change in gene expression 12 days postinfection in comparison to the expression in uninfected mice fed a Se-adequate diet; n = 9 to 10. Within uninfected mice, gene expression was not significantly different between Se-adequate and Se-deficient mice, but gene expression in infected mice fed either diet was significantly different from that in uninfected mice fed either diet (data not shown). Results with an asterisk are significantly different at a P value of ≤0.05.
A role for memory B cells and anti-H. bakeri-specific antibodies in protective immunity to infection has been demonstrated (31, 38). To determine if Se deficiency affected parasite-specific antibody production, serum IgG1, IgG, and IgG2a anti-H. bakeri ESP levels in uninfected and H. bakeri-infected mice fed a Se-adequate or Se-deficient diet or Se-deficient mice fed adequate Se for 4 days (day 14 postinfection) were analyzed by ELISA. A 1/10,000 dilution of serum obtained from Se-deficient infected mice had significantly lower levels of anti-H. bakeri IgG1(Fig. 6) (P < 0.05) and IgG antibodies (data not shown) than Se-adequate mice; the same pattern was observed using serum dilutions of 1/20,000 and 1/30,000 (data not shown). Anti-H. bakeri IgG2a antibodies were also increased after challenge infection but at a level 10-fold lower that of IgG1 and not significantly dependent on Se in the diets (data not shown). Similar to the local gene expression response in the intestine, Se-deficient mice fed adequate Se for 4 days had rebounds in serum IgG1 and IgG anti-H. bakeri antigen response to levels comparable to those of infected Se-adequate mice.
Fig 6.

Selenium deficiency reduces and feeding deficient mice a Se-adequate diet restores the anti-H. bakeri IgG1 antibody response. The anti-H. bakeri IgG1 antibody response was measured using H. bakeri excretory/secretory proteins and serum obtained 14 days postinfection. Bound serum antibody at a 1/10,000 dilution was detected using biotinylated anti-mouse IgG1 followed by streptavidin-HRP complex. Dashed line indicates absorbance of serum from uninfected mice. Results are expressed as means ± SEM (n = 4 to 5), and groups with different letters had significantly different results; P < 0.05.
GPX activity is affected by Se status and parasitic infection.
Selenium deficiency reduces the activity of selenocysteine-containing proteins, including glutathione peroxidases and thioredoxin reductases (39, 40). To determine if there was a correlation between GPX activity in liver and small intestine and parasite egg production, GPX activity was measured in H. bakeri-infected Se-adequate or Se-deficient mice and Se-deficient mice fed adequate Se for 2 or 4 days. Feeding a Se-deficient diet to uninfected mice reduced the glutathione peroxidase activity in both liver (primarily GPX1) and small intestine (primarily GPX1 and GPX2) (Fig. 7A and B) by 99% and 93%, respectively. Notably, liver GPX activity was decreased at 14 days after reinoculation with H. bakeri L3s in Se-adequate mice, while small intestine GPX activity increased at day 12 and returned to normal at day 14 after reinoculation of Se-adequate mice. These data raise the possibility that Se may be mobilized from the liver to the small intestine in response to infection. In addition, GPX activity recovered to a greater extent in the small intestine than in the liver in response to feeding Se-deficient infected mice a Se-adequate diet for 2 or 4 days. These results suggested that Se levels are slower to fall in the small intestine in response to deficiency and are restored more quickly in response to feeding adequate Se.
Fig 7.

GPX activity is altered by Se deficiency, parasite infection, and Se repletion. Replicate groups of mice fed a Se-adequate or Se-deficient diet were primed and reinoculated with 200 H. bakeri infective larvae. At 10 days after reinoculation, one group of Se-deficient mice was fed a Se-adequate diet. Mice were killed at days 12 (D12) and 14 (D14) after reinoculation and 2 (D+2) and 4 (D+4) days after feeding the Se-adequate diets, and GPX activity in the liver (A) or small intestine (SI) (B) was determined. Selenium-adequate and Se-deficient mice were analyzed separately. Results are expressed as means ± SEM (n = 5 to 10), and groups with different letters had significantly different results; P < 0.05. munits, milliunits.
DISCUSSION
We previously demonstrated that dietary deficiencies in either vitamin E or selenium (Se) delayed intestinal clearance of adult H. bakeri worms from a secondary challenge infection (26, 27). However, vitamin E deficiency but not Se deficiency prevented the worm-induced decreases in intestinal epithelial cell glucose absorption and smooth muscle hypercontractility that are associated with worm expulsion (26, 37). We also observed that the circulating serum levels of IL-4 induced by reinoculation with H. bakeri L3s were not affected by deficiencies in vitamin E or Se (27), suggesting that one of the signature cytokines of the Th2 response was largely intact when either micronutrient was deficient in the diet following a nematode infection. The more-comprehensive evaluation of expression for multiple Th2-dependent genes presented here, however, showed that Se deficiency resulted in significantly decreased local gene expression in the small intestine of H. bakeri-infected mice that included Il4 and Il13 and several genes associated with alternatively activated macrophages, including Chi3l3, Chi3l4, Retnla, Retnlb, and Arg1. Alternatively activated macrophages support a protective response against the parasitic stages of H. bakeri in intestinal tissue (24), and our gene expression data suggested that alternatively activated macrophage function was altered by Se deficiency. Notably, we did not find significantly altered recovery of parasitic larvae from intestinal tissue at 7 days after reinoculation of Se-adequate or Se-deficient mice, suggesting that this protective response was Se independent. However, there was a Se-dependent effect on adult worm ATP (metabolic activity) and fecundity (egg production) and the kinetics of expulsion from the intestine.
Th2-related genes and genes associated with alternatively activated macrophages were generally increased in intestinal cyst tissue compared to their levels in adjacent noncyst tissue at 12 days after reinoculation, when most parasitic larvae have emerged from the cysts as luminal-dwelling adults. The levels of gene expression were reduced in infected Se-deficient mice compared to the levels in Se-adequate mice, and there were perceptible differences in the intensity and size of cysts from Se-adequate versus Se-deficient mice (personal observation). In addition, feeding adequate Se to Se-deficient mice restored local Th2 gene expression. This suggested that Se-dependent mechanisms regulate the activity of alternatively activated macrophages that are important for tissue repair and collagen expression (41) driven by parasite products in and around the cyst (42).
The host protective response to gastrointestinal nematode parasites creates an inhospitable environment for the worm that facilitates expulsion (37). Previous studies showed that adult H. bakeri worms exhibit immune-dependent metabolic stress associated with decreased ATP levels (25, 36). Adult worms recovered from Se-adequate mice had lower ATP levels than worms from Se-deficient mice, and feeding adequate Se to Se-deficient mice lowered adult worm ATP levels and decreased egg production and worm burden. The factor(s) responsible for the increased metabolic stress on worms isolated from Se-adequate mice have not been resolved, but Relm-β protein was demonstrated as important for the clearance of adult H. bakeri by, presumably, interfering with worm feeding (25). The decreased expression of Retnlb mRNA and reduced Relm-β protein in extracts of intestinal tissue from Se-deficient mice indicated that decreased expression of this gene product may be causally related to the delayed clearance in Se-deficient mice.
Similarly, the host memory IgG1 and IgG antibody response to H. bakeri ESP, which was significantly lower in Se-deficient mice, was restored by feeding adequate dietary Se. This response mimics the recovery seen in cytokine production and worm expulsion and may contribute to the restoration of worm expulsion in Se-deficient mice. This is consistent with earlier work where an adoptive transfer of memory B cells or injection of immune serum from previously infected wild-type mice into JHD mice prior to a challenge infection reduced the adult worm burden and fecundity (31). Other studies in mice infected with viruses that induced a largely Th1 response showed similar levels of virus-specific antibody titers in Se-adequate and Se-deficient mice (43, 44).
Although feeding mice a Se-deficient diet reduced both liver and small intestine GPX activity substantially, the effect was more pronounced in liver. This may be due to the fact that GPX1 is more sensitive to Se deficiency than GPX2 (45) and is the primary GPX isozyme present in liver (46), while both GPX1 and GPX2 are present in the small intestine (47). GPX activity rebounded faster in the small intestine than in the liver when Se-deficient mice were fed a Se-adequate diet. Nearly 50% of the activity was recovered in the small intestine within 4 days, compared to only 12% in the liver. It is not clear, however, whether one or both GPX isozymes were increased equally because the enzyme assay used did not differentiate between the two isozymes, but it has been reported that GPX2 activity is restored more quickly upon refeeding Se (48). These results showed that Se-dependent activities rebounded rapidly in the small intestine, and this correlated with the recovery of Th2-associated gene expression, increased worm metabolic stress, and decreased egg production and worm burden in Se-deficient mice fed a Se-adequate diet.
The response to parasitic infection is complex, and it is likely that multiple features contribute to protective immunity. Local immune-activated oxidative stress has been associated with protection against gastrointestinal nematodes (49), along with macrophages (24) and IgG antibody response (38). The recovery of Se-dependent GPX activity was associated with adult worm expulsion in the intestine and reflects the increase in Se content of the tissue after refeeding a Se-adequate diet, as GPX activity is dependent on Se status (39, 40). Selenium has been shown to affect multiple aspects of immunity, including the oxidative bursts from neutrophils (13, 14) and T-cell (50, 51) and macrophage (52) function. Selenium also may affect the redox status of the host through selenocysteine-containing proteins, including GPX and thioredoxin. There are many studies demonstrating a role for intracellular thiols in the regulation of T- and B-cells, dendritic cells, and macrophages. Monick et al. (53) demonstrated that increasing intracellular thiol levels with N-acetyl-cysteine blocked the induction of gamma interferon (IFN-γ) and increased the IL-4 production in αCD3 or αCD3- and IL-12-stimulated splenocytes (53). Selenium and IL-4 appear to synergize in the modulation of redox potential of alternatively activated macrophages (54). Additional studies will determine whether altered cellular thiol levels contribute to the decreased Th2 response in Se-deficient mice.
Here, we showed that delayed clearance of adult H. bakeri worms from Se-deficient mice was associated with a decreased Th2 response in the small intestine. The expression of several genes associated with alternatively activated macrophages and goblet cell function was also downregulated in Se-deficient mice. Feeding Se-deficient mice a Se-adequate diet rapidly restored immune function, and this correlated with decreased adult worm metabolic activity, fecundity, and expulsion from the intestine that were associated with increased Relm-β protein and oxidative enzyme activity in the small intestine. There was also a Se-dependent increase in alternatively activated macrophage gene expression in cyst tissue induced by parasite invasion that implied a role for Se in tissue repair. Additional studies will determine whether delayed clearance in Se-deficient mice and the altered intensity of cyst formation and resolution result from a particular Se-dependent gene product or the combined decrease of multiple factors associated with a Th2 response and alternatively activated macrophages.
ACKNOWLEDGMENTS
A.D.S., T.S.-D., and J.F.U. designed the research, analyzed the data, and wrote the manuscript, A.D.S., L.C., and J.F.U. conducted the research, and A.D.S. had primary responsibility for the final content. All authors have read and approved the final manuscript.
Funding for this research was provided by the United States Department of Agriculture, Agricultural Research Service, project number 1235-51000-055.
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
Published ahead of print 6 May 2013
REFERENCES
- 1. Combs GF., Jr 2001. Selenium in global food systems. Br. J. Nutr. 85:517–547 [DOI] [PubMed] [Google Scholar]
- 2. Rannem T, Ladefoged K, Hylander E, Hegnhoj J, Staun M. 1998. Selenium depletion in patients with gastrointestinal diseases: are there any predictive factors? Scand. J. Gastroenterol. 33:1057–1061 [DOI] [PubMed] [Google Scholar]
- 3. Andoh A, Hirashima M, Maeda H, Hata K, Inatomi O, Tsujikawa T, Sasaki M, Takahashi K, Fujiyama Y. 2005. Serum selenoprotein-P levels in patients with inflammatory bowel disease. Nutrition 21:574–579 [DOI] [PubMed] [Google Scholar]
- 4. Forceville X, Vitoux D, Gauzit R, Combes A, Lahilaire P, Chappuis P. 1998. Selenium, systemic immune response syndrome, sepsis, and outcome in critically ill patients. Crit. Care Med. 26:1536–1544 [DOI] [PubMed] [Google Scholar]
- 5. Sakr Y, Reinhart K, Bloos F, Marx G, Russwurm S, Bauer M, Brunkhorst F. 2007. Time course and relationship between plasma selenium concentrations, systemic inflammatory response, sepsis, and multiorgan failure. Br. J. Anaesth. 98:775–784 [DOI] [PubMed] [Google Scholar]
- 6. Angstwurm MW, Engelmann L, Zimmermann T, Lehmann C, Spes CH, Abel P, Strauss R, Meier-Hellmann A, Insel R, Radke J, Schuttler J, Gartner R. 2007. Selenium in Intensive Care (SIC): results of a prospective randomized, placebo-controlled, multiple-center study in patients with severe systemic inflammatory response syndrome, sepsis, and septic shock. Crit. Care Med. 35:118–126 [DOI] [PubMed] [Google Scholar]
- 7. Manzanares W, Biestro A, Torre MH, Galusso F, Facchin G, Hardy G. 2011. High-dose selenium reduces ventilator-associated pneumonia and illness severity in critically ill patients with systemic inflammation. Intensive Care Med. [DOI] [PubMed] [Google Scholar]
- 8. Kiremidjian-Schumacher L, Roy M, Wishe HI, Cohen MW, Stotzky G. 1994. Supplementation with selenium and human immune cell functions. II. Effect on cytotoxic lymphocytes and natural killer cells. Biol. Trace Elem. Res. 41:115–127 [DOI] [PubMed] [Google Scholar]
- 9. Sakaguchi S, Iizuka Y, Furusawa S, Tanaka Y, Takayanagi M, Takayanagi Y. 2000. Roles of selenium in endotoxin-induced lipid peroxidation in the rats liver and in nitric oxide production in J774A.1 cells. Toxicol. Lett. 118:69–77 [DOI] [PubMed] [Google Scholar]
- 10. Kim IY, Stadtman TC. 1997. Inhibition of NF-kappaB DNA binding and nitric oxide induction in human T cells and lung adenocarcinoma cells by selenite treatment. Proc. Natl. Acad. Sci. U. S. A. 94:12904–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim SH, Johnson VJ, Shin TY, Sharma RP. 2004. Selenium attenuates lipopolysaccharide-induced oxidative stress responses through modulation of p38 MAPK and NF-kappaB signaling pathways. Exp. Biol. Med. (Maywood) 229:203–213 [DOI] [PubMed] [Google Scholar]
- 12. Maehira F, Miyagi I, Eguchi Y. 2003. Selenium regulates transcription factor NF-kappaB activation during the acute phase reaction. Clin. Chim. Acta 334:163–171 [DOI] [PubMed] [Google Scholar]
- 13. Baker SS, Cohen HJ. 1983. Altered oxidative metabolism in selenium-deficient rat granulocytes. J. Immunol. 130:2856–2860 [PubMed] [Google Scholar]
- 14. Serfass RE, Ganther HE. 1975. Defective microbicidal activity in glutathione peroxidase-deficient neutrophils of selenium-deficient rats. Nature 255:640–641 [DOI] [PubMed] [Google Scholar]
- 15. Beck MA, Matthews CC. 2000. Micronutrients and host resistance to viral infection. Proc. Nutr. Soc. 59:581–585 [DOI] [PubMed] [Google Scholar]
- 16. Smith AD, Cheung L, Botero S. 2011. Long-term selenium deficiency increases the pathogenicity of a Citrobacter rodentium infection in mice. Biol. Trace Elem Res. 144:965–982 [DOI] [PubMed] [Google Scholar]
- 17. Al-Shammari S, Khoja T, El-Khwasky F, Gad A. 2001. Intestinal parasitic diseases in Riyadh, Saudi Arabia: prevalence, sociodemographic and environmental associates. Trop. Med. Int. Health 6:184–189 [DOI] [PubMed] [Google Scholar]
- 18. Berkman DS, Lescano AG, Gilman RH, Lopez SL, Black MM. 2002. Effects of stunting, diarrhoeal disease, and parasitic infection during infancy on cognition in late childhood: a follow-up study. Lancet 359:564–571 [DOI] [PubMed] [Google Scholar]
- 19. Rodriguez L, Cervantes E, Ortiz R. 2011. Malnutrition and gastrointestinal and respiratory infections in children: a public health problem. Int. J. Environ. Res. Public Health 8:1174–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ing R, Su Z, Scott ME, Koski KG. 2000. Suppressed T helper 2 immunity and prolonged survival of a nematode parasite in protein-malnourished mice. Proc. Natl. Acad. Sci. U. S. A. 97:7078–7083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scott ME, Koski KG. 2000. Zinc deficiency impairs immune responses against parasitic nematode infections at intestinal and systemic sites. J. Nutr. 130:1412S–1420S [DOI] [PubMed] [Google Scholar]
- 22. Koski KG, Su Z, Scott ME. 1999. Energy deficits suppress both systemic and gut immunity during infection. Biochem. Biophys. Res. Commun. 264:796–801 [DOI] [PubMed] [Google Scholar]
- 23. Patel N, Kreider T, Urban JF, Jr, Gause WC. 2009. Characterisation of effector mechanisms at the host:parasite interface during the immune response to tissue-dwelling intestinal nematode parasites. Int. J. Parasitol. 39:13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anthony RM, Urban JF, Jr, Alem F, Hamed HA, Rozo CT, Boucher JL, Van Rooijen N, Gause WC. 2006. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat. Med. 12:955–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herbert DR, Yang JQ, Hogan SP, Groschwitz K, Khodoun M, Munitz A, Orekov T, Perkins C, Wang Q, Brombacher F, Urban JF, Jr, Rothenberg ME, Finkelman FD. 2009. Intestinal epithelial cell secretion of RELM-beta protects against gastrointestinal worm infection. J. Exp. Med. 206:2947–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Au Yeung KJ, Smith A, Zhao A, Madden KB, Elfrey J, Sullivan C, Levander O, Urban JF, Shea-Donohue T. 2005. Impact of vitamin E or selenium deficiency on nematode-induced alterations in murine intestinal function. Exp. Parasitol. 109:201–208 [DOI] [PubMed] [Google Scholar]
- 27. Smith A, Madden KB, Yeung KJ, Zhao A, Elfrey J, Finkelman F, Levander O, Shea-Donohue T, Urban JF., Jr 2005. Deficiencies in selenium and/or vitamin E lower the resistance of mice to Heligmosomoides polygyrus infections. J. Nutr. 135:830–836 [DOI] [PubMed] [Google Scholar]
- 28. Beck MA, Kolbeck PC, Rohr LH, Shi Q, Morris VC, Levander OA. 1994. Vitamin E deficiency intensifies the myocardial injury of coxsackievirus B3 infection of mice. J. Nutr. 124:345–358 [DOI] [PubMed] [Google Scholar]
- 29. Behnke J, Harris PD. 2010. Heligmosomoides bakeri: a new name for an old worm? Trends Parasitol. 26:524–529 [DOI] [PubMed] [Google Scholar]
- 30. Persson L. 1974. A modified Baermann apparatus for the recovery of infective nematode larvae from herbage and manure. Zentralbl. Veterinarmed. B 21:483–488 [DOI] [PubMed] [Google Scholar]
- 31. Liu Q, Kreider T, Bowdridge S, Liu Z, Song Y, Gaydo AG, Urban JF, Jr, Gause WC. 2010. B cells have distinct roles in host protection against different nematode parasites. J. Immunol. 184:5213–5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dawson HD, Beshah E, Nishi S, Solano-Aguilar G, Morimoto M, Zhao A, Madden KB, Ledbetter TK, Dubey JP, Shea-Donohue T, Lunney JK, Urban JF., Jr 2005. Localized multigene expression patterns support an evolving Th1/Th2-like paradigm in response to infections with Toxoplasma gondii and Ascaris suum. Infect. Immun. 73:1116–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 34. Smith AD, Morris VC, Levander OA. 2001. Rapid determination of glutathione peroxidase and thioredoxin reductase activities using a 96-well microplate format: comparison to standard cuvette-based assays. Int. J. Vitam. Nutr. Res. 71:87–92 [DOI] [PubMed] [Google Scholar]
- 35. Kangas L, Gronroos M, Nieminen AL. 1984. Bioluminescence of cellular ATP: a new method for evaluating cytotoxic agents in vitro. Med. Biol. 62:338–343 [PubMed] [Google Scholar]
- 36. Ishiwata K, Watanabe N. 2007. Nippostrongylus brasiliensis: reversibility of reduced-energy status associated with the course of expulsion from the small intestine in rats. Exp. Parasitol. 117:80–86 [DOI] [PubMed] [Google Scholar]
- 37. Shea-Donohue T, Urban JF., Jr 2004. Gastrointestinal parasite and host interactions. Curr. Opin. Gastroenterol. 20:3–9 [DOI] [PubMed] [Google Scholar]
- 38. McCoy KD, Stoel M, Stettler R, Merky P, Fink K, Senn BM, Schaer C, Massacand J, Odermatt B, Oettgen HC, Zinkernagel RM, Bos NA, Hengartner H, Macpherson AJ, Harris NL. 2008. Polyclonal and specific antibodies mediate protective immunity against enteric helminth infection. Cell Host Microbe 4:362–373 [DOI] [PubMed] [Google Scholar]
- 39. Forstrom JW, Zakowski JJ, Tappel AL. 1978. Identification of the catalytic site of rat liver glutathione peroxidase as selenocysteine. Biochemistry 17:2639–2644 [DOI] [PubMed] [Google Scholar]
- 40. Tamura T, Stadtman TC. 1996. A new selenoprotein from human lung adenocarcinoma cells: purification, properties, and thioredoxin reductase activity. Proc. Natl. Acad. Sci. U. S. A. 93:1006–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dong L, Wang SJ, Camoretti-Mercado B, Li HJ, Chen M, Bi WX. 2008. FIZZ1 plays a crucial role in early stage airway remodeling of OVA-induced asthma. J. Asthma 45:648–653 [DOI] [PubMed] [Google Scholar]
- 42. Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, Van Rooijen N, Urban JF, Jr, Wynn TA, Gause WC. 2012. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat. Med. 18:260–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beck MA, Nelson HK, Shi Q, Van Dael P, Schiffrin EJ, Blum S, Barclay D, Levander OA. 2001. Selenium deficiency increases the pathology of an influenza virus infection. FASEB J. 15:1481–1483 [PubMed] [Google Scholar]
- 44. Beck MA, Kolbeck PC, Shi Q, Rohr LH, Morris VC, Levander OA. 1994. Increased virulence of a human enterovirus (coxsackievirus B3) in selenium-deficient mice. J. Infect. Dis. 170:351–357 [DOI] [PubMed] [Google Scholar]
- 45. Brigelius-Flohe R, Muller C, Menard J, Florian S, Schmehl K, Wingler K. 2001. Functions of GI-GPx: lessons from selenium-dependent expression and intracellular localization. Biofactors 14:101–106 [DOI] [PubMed] [Google Scholar]
- 46. Esworthy RS, Ho YS, Chu FF. 1997. The Gpx1 gene encodes mitochondrial glutathione peroxidase in the mouse liver. Arch. Biochem. Biophys. 340:59–63 [DOI] [PubMed] [Google Scholar]
- 47. Esworthy RS, Swiderek KM, Ho YS, Chu FF. 1998. Selenium-dependent glutathione peroxidase-GI is a major glutathione peroxidase activity in the mucosal epithelium of rodent intestine. Biochim. Biophys. Acta 1381:213–226 [DOI] [PubMed] [Google Scholar]
- 48. Wingler K, Bocher M, Flohe L, Kollmus H, Brigelius-Flohe R. 1999. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur. J. Biochem. 259:149–157 [DOI] [PubMed] [Google Scholar]
- 49. Brophy PM, Ben-Smith A, Brown A, Behnke JM, Pritchard DI. 1995. Differential expression of glutathione S-transferase (GST) by adult Heligmosomoides polygyrus during primary infection in fast and slow responding hosts. Int. J. Parasitol. 25:641–645 [DOI] [PubMed] [Google Scholar]
- 50. Hoffmann FW, Hashimoto AC, Shafer LA, Dow S, Berry MJ, Hoffmann PR. 2010. Dietary selenium modulates activation and differentiation of CD4+ T cells in mice through a mechanism involving cellular free thiols. J. Nutr. 140:1155–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shrimali RK, Irons RD, Carlson BA, Sano Y, Gladyshev VN, Park JM, Hatfield DL. 2008. Selenoproteins mediate T cell immunity through an antioxidant mechanism. J. Biol. Chem. 283:20181–20185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carlson BA, Yoo MH, Sano Y, Sengupta A, Kim JY, Irons R, Gladyshev VN, Hatfield DL, Park JM. 2009. Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol. 10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Monick MM, Samavati L, Butler NS, Mohning M, Powers LS, Yarovinsky T, Spitz DR, Hunninghake GW. 2003. Intracellular thiols contribute to Th2 function via a positive role in IL-4 production. J. Immunol. 171:5107–5115 [DOI] [PubMed] [Google Scholar]
- 54. Nelson SM, Lei X, Prabhu KS. 2011. Selenium levels affect the IL-4-induced expression of alternative activation markers in murine macrophages. J. Nutr. 141:1754–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]