

Abstract
During growth in the environment, bacteria encounter stresses which can delay or inhibit their growth. To defend against these stresses, bacteria induce both resistance and repair mechanisms. Many bacteria regulate these resistance mechanisms using a group of alternative σ factors called extracytoplasmic function (ECF) σ factors. ECF σ factors represent the largest and most diverse family of σ factors. Here, we demonstrate that the activation of a member of the ECF30 subfamily of ECF σ factors, σV in Bacillus subtilis, is controlled by the proteolytic destruction of the anti-σ factor RsiV. We will demonstrate that the degradation of RsiV and, thus, the activation of σV requires multiple proteolytic steps. Upon exposure to the inducer lysozyme, the extracellular domain of RsiV is removed by an unknown protease, which cleaves at site 1. This cleavage is independent of PrsW, the B. subtilis site 1 protease, which cleaves the anti-σ factor RsiW. Following cleavage by the unknown protease, the N-terminal portion of RsiV requires further processing, which requires the site 2 intramembrane protease RasP. Our data indicate that the N-terminal portion of RsiV from amino acid 1 to 60, which lacks the extracellular domain, is constitutively degraded unless RasP is absent, indicating that RasP cleavage is constitutive. This suggests that the regulatory step in RsiV degradation and, thus, σV activation are controlled at the level of the site 1 cleavage. Finally, we provide evidence that increased resistance to lysozyme decreases σV activation. Collectively, these data provide evidence that the mechanism for σV activation in B. subtilis is controlled by regulated intramembrane proteolysis (RIP) and requires the site 2 protease RasP.
INTRODUCTION
Stress response systems are important for bacteria to adapt and survive in changing environments. Extracytoplasmic function (ECF) σ factors are alternative σ factors that interact with RNA polymerase and transcribe sets of genes required for environmental adaption in response to specific signal(s) (1). Most known ECF σ factors are held in an inactive state during normal growth conditions by an anti-σ factor that prevents the ECF σ factor from interacting with RNA polymerase (1, 2). Exposure to specific cell envelope stresses induces a proteolytic cascade termed regulated intramembrane proteolysis (RIP), which results in the destruction of a membrane-tethered anti-σ factor (3, 4). Upon sensing cell envelope stress, a protease cleaves the anti-σ factor at site 1 in the extracellular domain of the anti-σ factor (3). This is followed by a second cleavage (site 2) within the transmembrane domain of the anti-σ factor (3). The remainder of the freed anti-σ factor is degraded by cytosolic proteases, resulting in the release of free σ factor, which is able to interact with RNA polymerase to transcribe the appropriate target genes (4).
The best-studied example of ECF σ factor activation is that of Escherichia coli σE. Normally, σE is held inactive by the membrane-bound anti-σ factor RseA, which sequesters σE to the cytoplasmic membrane. In response to cell envelope stress, in this case unfolded outer membrane β-barrel proteins, RseA is cleaved by the protease DegS at site 1 (see Fig. S1 in the supplemental material) (5–8). The RseA site 1 cleavage product becomes a substrate for RseP, a site 2 protease (9–11). RseP then clips RseA within the transmembrane domain, releasing the N-terminal portion of RseA to the cytosol where it can be further degraded by multiple cytosolic proteases (12). This proteolytic cascade leads to the destruction of the anti-σ factor RseA and results in free σE, which can transcribe genes required to respond to cell envelope stress (13).
Bacillus subtilis encodes seven ECF σ factors (14). Of those ECF σ factors, only the mechanism of σW activation has been well characterized. The activity of σW is induced by a variety of cell envelope stresses, including salt stress, pH stress (base stress), and antimicrobial peptides (15, 16). The regulation of σW activity is similar to but distinct from the regulation of σE activity in E. coli (4, 9, 17–19). In response to cell envelope stress, PrsW, a type II CAAX prenyl endopeptidase, cleaves the extracellular domain of the anti-σ factor RsiW at site 1 (see Fig. S1 in the supplemental material) (18, 19). A second, as-yet-unidentified protease further trims the extracellular portion of RsiW (17). Following truncation of the extracellular domain of RsiW by PrsW and the unknown protease(s), RsiW is cleaved by the site 2 metalloprotease RasP (17). The activation of σW requires degradation of the released σ factor binding domain of RsiW by the cytosolic proteases ClpXP (20). While PrsW is structurally unrelated to DegS, the E. coli site 1 protease, RasP shares homology to the E. coli site 2 protease RseP. In both regulatory systems, it has been shown that site 1 cleavage is required before site 2 cleavage can occur (17, 21).
Site 2 proteases are thought to have an ancient origin and can be found in the genome sequences of archaea, bacteria, and eukaryotes (22). Although there is great substrate diversity, site 2 proteases cleave within the transmembrane helices of proteins which contain helix-destabilizing residues (22). In Mycobacterium tuberculosis, it has been shown that a site 2 protease homolog, Rv2869c (Rip1), is required for the cleavage of three different anti-σ factors, each of which inhibits the activity of a different ECF σ factor (23). In humans, the site 2 protease, S2P, has four different substrates, some of which regulate cholesterol biosynthesis and the endoplasmic reticulum stress response (24, 25). In B. subtilis, the site 2 protease RasP is known to cleave the cell division protein FtsL, in addition to the σW anti-σ factor RsiW (26). Recent evidence suggests that site 2 proteases, including RasP, are required for clearing signal peptides from the membrane (27). Here, we identify another substrate for RasP in B. subtilis, namely, the anti-σ factor RsiV, which inhibits σV activity.
σV belongs to a unique subfamily of ECF σ factors (ECF30) which are primarily found in the firmicutes (low-GC, Gram-positive bacteria) (2). In B. subtilis, σV is encoded with three other genes, rsiV (the anti-σ factor), oatA (O-acetyltransferase), and yrhK (unknown function) (28, 29). We previously found that the activity of B. subtilis σV was induced by lysozyme (28). Lysozyme is an important component of the innate immune system which cleaves the peptidoglycan of bacteria between the β-(1,4)-linked N-acetylglucosamine and N-acetylmuramic acid residues (30). The activation of σV induces the expression of oatA (28, 31). OatA increases lysozyme resistance, likely by transferring an acetyl group to the C-6-OH position of N-acetylmuramic acid and thereby blocking lysozyme cleavage (28, 31–33).
Homologs of the σV system have been identified in Enterococcus faecalis and Clostridium difficile, where they are also induced in response to lysozyme (34–36). In E. faecalis, it has been demonstrated that σV is required for resistance to lysozyme and is important for virulence in mouse models of systemic and urinary tract infection (35). It is clear that in all of these cases, σV activity is inhibited by a single-pass transmembrane protein, RsiV (29, 34, 36). However, little is known about the mechanisms controlling the activation of σV in any organism. Here, we present evidence that the σV anti-σ factor RsiV is degraded in a RIP-dependent manner in response to subinhibitory concentrations of lysozyme. Furthermore, our results show that RsiV degradation is independent of the RsiW site 1 protease PrsW. In contrast, the site 2 protease RasP is required for RsiV degradation once site 1 cleavage has occurred. Using truncated forms of RsiV, we also demonstrate that RasP constitutively degrades the site 1 cleavage product.
MATERIALS AND METHODS
Strain construction.
All of B. subtilis strains used are isogenic derivatives of PY79, a prototrophic derivative of B. subtilis strain 168 (37). All cloning plasmids were constructed in the E. coli cloning strain Omnimax-2 (Invitrogen). Protein purification was done using E. coli BL21λDE3. Strain genotypes and plasmid descriptions are listed in Table 1. B. subtilis strains were constructed by transformation using competent cells prepared by the one-step method as previously described (38). All DNA oligomers were synthesized by IDT (Coralville, IA) and are listed in Table S1 in the supplemental material. The sequences of all plasmid constructs were confirmed by DNA sequencing (Iowa State University).
Table 1.
| Strain or plasmid | Genotypea | Referenceb |
|---|---|---|
| Plasmids | ||
| pDR111 | amyE Phs Ampr Specr | 56 |
| pCE292 | pDR111 amyE Phs Specr Ampr Camr Phs-RfA | 34 |
| pCE351 | pENTR/D-TOPO rsiV+ Kanr | |
| pJH158 | pCE292 amyE Phs-rsiV+ Specr Ampr | |
| pCE458 | pENTR/D-TOPO rsiV59-285 Kanr | |
| pKBW202 | pDEST17 PT7-6×His-rsiV59-285 Ampr | |
| pCE235 | pDR111 amyE Phs Specr Ampr | |
| pCE236 | pCE235 amyE Phs-GFP Specr Ampr | |
| pJH183 | pCE235 amyE Phs-GFP-RfA Specr Camr Ampr | |
| pJH198 | pENTR/D-TOPO rsiV1-60 Kanr | |
| pJH204 | pJH183 amyE Phs-gfp-rsiV1-60 Specr Ampr | |
| pJH101 | pENTR/D-TOPO oatA+ Kanr | 28 |
| pJH102 | pCE292 amyE Phs-oatA+ Specr Ampr | 28 |
| pJH107 | pENTR/D-TOPO pdaC+ Kanr | 44 |
| pJH166 | pCE292 amyE Phs-pdaC+ Specr Ampr | |
| pCE170 | pDR111 amyE Phs-rasP+ Specr Ampr | |
| pEP20 | TnHyJump kan amp mls mariner-Himar1ori(Ts)Bs | |
| Escherichia coli strains | ||
| Omnimax-2 T1R | F′ proAB+ lacIq lacZΔM15 Tn10(Tetr) ΔccdAB mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 Δ(lacZYA-argF) U169 endA1 recA1 supE44 thi-1 gyrA96 relA1 tonA panD | |
| BL21(DE3) | fhuA2 [lon] ompTgal(λDE3) [dcm] ΔhsdS(λDE3) [sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5] | |
| Bacillus subtilis strains | ||
| PY79 | Prototrophic derivative of B. subtilis 168 | 37 |
| CDE1546 | PY79 pyrD::PsigV-lacZ (cat) | 28 |
| CDE1563 | PY79 Δ(sigV rsiV)::kan | |
| CDE379 | PY79 ΔprsW::erm | 18 |
| CDE1549 | PY79 pyrD::PsigV-lacZ (cat) ΔprsW::erm | |
| CDE1557 | PY79 pyrD::PsigV-lacZ (cat) ΔsigW::kan | |
| JLH632 | PY79 ΔrasP::tet | |
| JLH653 | PY79 pyrD::PsigV-lacZ (cat) ΔrasP::tet | |
| JLH640 | PY79 pyrD::PsigV-lacZ (cat) ΔrasP::tet amyE::Phs-rasP+ (spec) | |
| JLH402 | PY79 amyE::Phs-rsiV+ (spec) Δ(sigV rsiV)::kan | |
| JLH416 | PY79 amyE::Phs-rsiV+ (spec) Δ(sigV rsiV)::kan ΔrasP::cat | |
| JLH415 | PY79 amyE::Phs-rsiV+ (spec) Δ(sigV rsiV)::kan ΔprsW::erm | |
| JLH453 | PY79 amyE::Phs-gfp-rsiV+ (spec) Δ(sigV rsiV)::kan | |
| JLH466 | PY79 amyE::Phs-gfp-rsiV+ (spec) Δ(sigV rsiV)::kan ΔrasP::cat | |
| JLH465 | PY79 amyE::Phs-gfp-rsiV+ (spec) Δ(sigV rsiV)::kan ΔprsW::erm | |
| JLH467 | PY79 amyE::Phs-gfp-rsiV+ (spec) Δ(sigV rsiV)::kan ΔprsW::erm rasP::cat | |
| JLH516 | PY79 amyE::Phs-gfp-rsiV1-60 (spec) Δ(sigV rsiV)::kan | |
| JLH536 | PY79 amyE::Phs-gfp-rsiV1-60 (spec) Δ(sigV rsiV)::kan ΔrasP::cat | |
| TPM1263 | PY79 amyE::Phs-gfp (spec) | |
| JLH345 | PY79 pryD::PsigV-lacZ (cat) amyE::Phs-oatA+ | |
| JLH176 | PY79 pyrD::PsigV-lacZ (cat) amyE::Phs-pdaC+ | |
| JLH121 | PY79 pyrD::PsigV-lacZ (cat) amyE::PsigV-lacZ (spec) |
Unless otherwise noted, all strains are isogenic derivatives of PY79.
This study, unless otherwise noted.
Native rsiV+ with an optimized ribosome binding site was PCR amplified from B. subtilis using oligonucleotides CDEP1430 and CDEP952 and cloned into pENTR/D-TOPO (Invitrogen), resulting in pCE351. For isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible expression, the rsiV+ gene was moved into pCE292 (34) using LR Clonase II (Invitrogen). The resulting plasmid, pJH158, places the IPTG-inducible hyperspac promoter (Phs) upstream from rsiV+. An IPTG-inducible pdaC+ was constructed similarly, by PCR amplifying pdaC from B. subtilis using oligonucleotides CDEP1245 and CDEP1246. The PCR product was cloned into pENTR/D-TOPO, resulting in pJH107, and moved into pCE292 with LR Clonase II, resulting in pJH166.
The rasP complementation plasmid was constructed by PCR amplifying rasP from B. subtilis using oligonucleotides CDEP568 and CDEP569. The PCR product was digested with SalI and SphI and cloned into pDR111 digested with SalI and SphI, resulting in pCE170. In this construct, the expression of rasP+ is IPTG inducible; however, the basal levels of expression were sufficient to complement a rasP null mutant (data not shown).
We constructed a derivative of pDR111, pCE235, in which the EcoRI and BamHI sites had been sequentially destroyed by digestion with the appropriate enzyme, blunt ended with the Klenow fragment, and religated. The Phs-green fluorescent protein (GFP) vector was constructed by PCR amplifying superfolder gfp from pCM29 (39) using oligonucleotides CDEP615 and CDEP618. The resulting PCR product was digested with SalI and SphI and cloned into pDR111, which had been digested with HindIII and NheI, resulting in pCE236. A Gateway destination vector was constructed to build N-terminal gfp-rsiV fusions (Invitrogen). This was generated by cloning the RfA cassette (Invitrogen) into pCE236, which had been digested with SphI and EcoRI and blunt ended with the Klenow fragment to generate pJH183. N-terminally GFP-tagged RsiV+ (GFP-RsiV+) was constructed by PCR amplifying rsiV from B. subtilis using oligonucleotides CDEP1460 and CDEP952 and cloning it into pENTR/D-TOPO, resulting in pCE351. To construct a plasmid producing GFP-RsiV+, rsiV+ was moved from pCE352 onto pJH183 using LR Clonase II (Invitrogen), resulting in plasmid pJH181. The GFP-RsiV1-60 vector was generated by PCR amplifying the portion of rsiV that encodes the N-terminal amino acids 1 to 60 (rsiV1-60) from B. subtilis using CDEP1460 and CDEP978 and cloning the product into pENTR/D-TOPO, resulting in pJH198. To construct the plasmid producing GFP-RsiV1-60, rsiV1-60 was moved from pJH198 onto pJH183 using LR Clonase II (Invitrogen), resulting in plasmid pJH204.
To generate antibodies to the extracellular domain of RsiV, the portion of rsiV encoding the C-terminal extracellular domain (codons 59 to 285 [rsiV59-285]) was PCR amplified using CDEP1007 and CDEP952 and cloned into pENTR/D-TOPO, resulting in pCE458. To construct the recombinant 6×His-RsiV59-285, rsiV59-285 from pCE458 was moved into the T7-inducible 6×His destination vector, pDEST17 (Invitrogen), using LR Clonase II, resulting in pKBW202.
Long flanking homology PCR was used to create ΔrasP::tet as previously described (40). The rasP 5′ flanking region was PCR amplified using CDEP209 and CDEP1575. The rasP 3′ flanking region was PCR amplified using CDEP1576 and CDEP212. The resulting PCR products were used as primers to amplify the tet cassette from pDG1515 (41). The resulting PCR product was directly transformed into PY79, resulting in strain JLH632.
Bacterial growth conditions.
All E. coli and B. subtilis strains were grown in LB broth or on LB agar. Antibiotics were used at the following concentrations for B. subtilis: chloramphenicol, 5 μg/ml; erythromycin plus lincomycin (MLS), 1 μg/ml and 25 μg/ml, respectively; kanamycin, 5 μg/ml; spectinomycin, 100 μg/ml; and tetracycline, 10 μg/ml. The β-galactosidase chromogenic indicator 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-Gal; RPI) was used at a concentration of 100 μg/ml. Isopropyl β-d-1-thiogalactopyranoside (IPTG; RPI) was used at a final concentration of 1 mM unless otherwise noted. Antibiotics were used at the following concentrations for E. coli: ampicillin, 100 μg/ml; chloramphenicol, 10 μg/ml; and kanamycin, 50 μg/ml.
β-Galactosidase activity assays.
Cultures were grown overnight in LB broth at 30°C, and 20-μl amounts were spotted in triplicate onto LB agar. The plates were incubated at 37°C for 6 h. Samples were harvested from the plates and resuspended in 500 μl of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, pH 7.0). Resuspended cells (250 μl) were transferred to a 96-well plate, and measurements of the optical density at 600 nm (OD600) were taken using a Tecan F50 (Tecan). The remaining cells were permeabilized by mixing with chloroform and 2% Sarkosyl (28, 42). The permeabilized cells (100 μl) were added to 96-well plates with 10 mg/ml ortho-nitrophenyl-β-galactoside (ONPG, 50 μl; RPI), and the OD405 was measured every 1 min for 45 min. β-Galactosidase activity units were determined using the following calculation (μmol of ONP formed min−1) × 103/(OD600 × ml of cell suspension) as previously described (43). Experiments were performed in triplicate. Means and standard deviations are shown.
Determination of MIC.
The MIC was determined by diluting overnight cultures 1:100 and growing cells to an OD600 of 1.5 in LB containing 1 mM IPTG. The cultures were corrected to an OD600 of 0.8 and diluted 1:50 in LB containing 1 mM IPTG. The cell mixtures (50 μl) were inoculated into 200 μl of LB containing 1 mM IPTG with hen egg white lysozyme ranging from 100 μg/ml to 0.10 μg/ml in a round-bottom 96-well plate. The cells were grown for 16 h at 37°C. Growth was defined as an OD600 greater than 0.05. All assays were performed in triplicate on the same day, and each MIC determination was performed on three different days.
Expression of 6×His-tagged RsiV.
Overnight cultures of E. coli BL21λDE3 containing pKBW202 (pDEST17-6×His-rsiV59-285) were grown at 30°C in LB with 100 μg/ml ampicillin. The following day, cell cultures were diluted 1:100 into 500 ml of fresh LB with ampicillin in 2-liter baffled flasks and incubated at 30°C until reaching an OD600 of 0.5 to 0.6. IPTG was added to a final concentration of 1 mM to induce protein production, and the cultures incubated for an additional 4 h. At that time, the cells were chilled on ice and collected by centrifugation at 5,000 × g. The cell pellets were stored at −80°C until time for purification.
Purification of 6×His-tagged RsiV.
Cell pellets were thawed on ice and resuspended in 5 ml lysis buffer (50 mM sodium phosphate, 250 mM NaCl, 10 mM imidazole, pH 8.0) per 500 ml of initial culture volume. The cells were lysed by passaging through a Microfluidics LV1 high-shear microfluidizer (Newton, MA) twice. Lysate was centrifuged at 15,000 × g for 30 min at 4°C to pellet cellular debris. The cleared lysate was applied to a nickel affinity column to bind 6×His-tagged protein (Qiagen). The column was washed with 10 column volumes of wash buffer (50 mM sodium phosphate, 250 mM NaCl, 20 mM imidazole, pH 8.0). Protein was eluted with elution buffer (50 mM sodium phosphate, 250 mM NaCl, 250 mM imidazole, pH 8.0) and collected in 0.5-ml fractions. Samples from each fraction were analyzed by SDS-PAGE, and elution fractions containing the desired protein were combined. The combined fractions were then dialyzed into lysis buffer to remove the excess imidazole. The protein was further purified with a GE Healthcare ÄKTA fast protein liquid chromatography (FPLC) system (GE Healthcare Sciences, Pittsburg, PA) using a HisTrap HP nickel affinity column. The fractions containing 6×His-RsiV59-285 were again combined, dialyzed into a storage buffer (25 mM Tris, 200 mM NaCl, 5% glycerol, pH 8.0), and frozen at −80°C until use.
Production of anti-RsiV polyclonal antibodies.
Purified 6×His-RsiV59-285 protein was sent for production of polyclonal antibodies in New Zealand White rabbits (ProSci Inc., San Diego, CA). Serum from the final bleed showed high specificity in the detection of RsiV at dilutions of 1:10,000.
Immunoblot analysis.
For immunoblot analysis, cultures were grown overnight at 30°C and subcultured at a dilution of 1:50 into 5 ml LB broth supplemented with 1 mM IPTG or 0.5 mM IPTG (GFP constructs). Cells were grown at 37°C with rotation to mid-log phase (OD600 of 0.8 to 1.0) and were left untreated as a control or were treated for 10 min with hen egg white lysozyme (2 μg/ml unless otherwise noted). The cells (1.5 ml) were then pelleted by centrifugation, resuspended in 100 μl of 2× sample buffer (65.8 mM Tris-HCl, pH 6.8, 2.1% SDS, 26.3% [wt/vol] glycerol, 0.01% bromophenol blue, and 5% β-mercaptoethanol), and lysed by sonication. Samples were electrophoresed on a 15% SDS–polyacrylamide gel (Bio-Rad). The proteins were then blotted onto nitrocellulose and incubated with one of the following primary antibodies: anti-RsiV (1:10,000), anti-GFP (1:10,000), or anti-σA (1:15,000) antibodies. The primary antibody was detected by incubation with goat anti-rabbit IRDye 800CW antibody (1:10,000, LiCor), and the immunoblots were imaged on an Odyssey CLx (LiCor).
Transposon mutagenesis of a strain carrying a PsigV-lacZ reporter fusion.
Transposon mutagenesis was performed to identify mutants which blocked lysozyme induction of PsigV-lacZ. The transposon TnHyJump (44) was used to mutagenize JLH121, which contains two copies of PsigV-lacZ (amyE::PsigV-lacZ pyrD::PsigV-lacZ), in order to avoid isolating simple lacZ null mutants. pEP20 (44) was transformed into JLH121, and cells were grown at 30°C overnight. The strains were subcultured 1:100 into 2 ml LB plus MLS, grown at 30°C for 3 h, shifted to 37°C for 4 h, and then frozen at −80°C after the addition of 400 μl 50% glycerol to create pools of cells. Each pool was serially diluted, plated for single colonies on LB plus kanamycin, and grown overnight. The resulting colonies were replicate plated to LB plus X-Gal (100 μg/ml), IPTG (1 mM), and lysozyme (20 μg/ml) and incubated at 37°C for 6 h. Cells which were unable to induce the expression of PsigV-lacZ were isolated, and the location of the mutation determined by sequencing as previously described (44).
RESULTS
RsiV is degraded in response to lysozyme stress.
We previously demonstrated that B. subtilis σV activity was induced in the presence of lysozyme (28). The activity of σV is inhibited by a membrane-tethered anti-σ factor, RsiV (29). Several ECF σ factors are activated by proteolytic destruction by anti-σ factors (17, 21, 45). To better understand the activation of σV, we sought to examine the effect of increasing lysozyme concentrations on a reporter of σV activity (PsigV-lacZ) and on the stability of the anti-σ factor RsiV. We first determined the effect of increasing concentrations of lysozyme on the expression of a PsigV-lacZ transcriptional reporter. We found that increasing concentrations of lysozyme resulted in increased expression of the PsigV-lacZ reporter (Fig. 1A).
Fig 1.
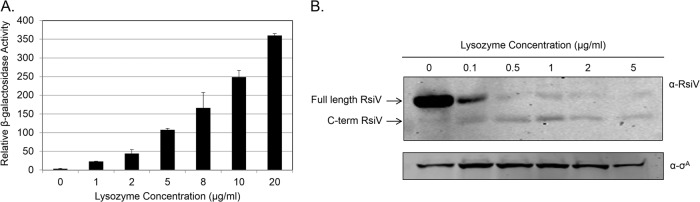
RsiV degradation and σV induction occur in a lysozyme concentration-dependent manner. (A) B. subtilis with transcriptional fusion PsigV-lacZ (CDE1546) was grown overnight at 30°C and spotted (20 μl) in triplicate on LB agar plates with various concentrations of lysozyme (0, 2, 5, 10, or 20 μg/ml). Plates were incubated at 37°C for 6 h. Cells were collected and resuspended in Z buffer. β-Galactosidase activity was calculated as described in Materials and Methods. (B) B. subtilis expressing an IPTG-inducible rsiV in a ΔsigV ΔrsiV mutant (JLH402 ΔsigV ΔrsiV::kan Phs-rsiV+) was grown to mid-log phase and then incubated with increasing concentrations of hen egg white lysozyme (0, 0.1, 0.5, 1, 2, and 5 μg/ml) for 10 min. The immunoblot was probed with antibodies against the extracellular domain of RsiV (α-RsiV) and the cytoplasmic protein σA (α-σA).
The sigV and rsiV genes are encoded in an operon, and the expression of this operon is σV dependent (29). Therefore, to determine the effects of lysozyme stress on RsiV stability, we sought to uncouple rsiV expression from σV activity. We constructed a strain which lacked the chromosomal copy of sigV and rsiV and expressed rsiV+ from an IPTG-inducible promoter (JLH402). After growing the cells to mid-log phase, we treated them with increasing concentrations of lysozyme for 10 min. The levels of RsiV were monitored by immunoblotting with polyclonal antibodies that had been raised against the extracellular C-terminal domain of RsiV. We detected a 32-kDa protein in the absence of lysozyme, which corresponds to the size of full-length RsiV (Fig. 1B). We found that at the lowest concentrations of lysozyme (0.1 μg/ml), we observed partial loss of full-length RsiV. However, at higher concentrations of lysozyme, 0.5 μg/ml and greater, RsiV was almost completely degraded (Fig. 1B). We found that, as the 32-kDa protein decreased, a smaller product of ∼25 kDa appeared (Fig. 1B). This band is presumably the extracellular domain of RsiV alone (Fig. 1B). Decreasing the amount of time cells were incubated with lysozyme to 2 min resulted in similar levels of degradation (data not shown). These data suggest that the σV anti-σ factor RsiV is rapidly degraded following lysozyme exposure and that this degradation is dependent upon the lysozyme concentration.
The site 2 protease RasP is required for σV activation.
In B. subtilis, the only anti-σ factor known to be degraded by a proteolytic cleavage cascade is RsiW, which controls the activation of σW. After the degradation of RsiW is initiated by PrsW, it is trimmed by an unknown protease and then cleaved by the site 2 protease RasP (17–19). We sought to determine if the site 1 protease PrsW and site 2 protease RasP were required for σV activity. We examined the effects of prsW and rasP null mutations on σV activity in response to lysozyme by monitoring the β-galactosidase activity from a PsigV-lacZ reporter. In wild-type cells, PsigV-lacZ expression is induced ∼65-fold in the presence of lysozyme (Fig. 2). As expected, in the absence of σV, PsigV is not induced in response to lysozyme (Fig. 2). In the rasP mutant background, PsigV induction is limited to low basal levels even in the presence of lysozyme (Fig. 2). The defect in PsigV-lacZ expression in the rasP mutant could be complemented by expressing rasP+ from an ectopic locus (Fig. 2), suggesting that RasP is required for σV activation. In contrast, the expression of PsigV-lacZ was induced ∼68-fold in response to lysozyme in the prsW mutant (Fig. 2), which suggests that PrsW is not required for σV activation. It appears that the expression of PsigV-lacZ is induced to a much greater level in the prsW mutant than in the wild type when lysozyme is present (Fig. 2). However, the basal level of PsigV-lacZ is also significantly higher in the prsW mutant, and thus, the fold induction of PsigV-lacZ in both the wild type and the prsW mutants is ∼65-fold (Fig. 2). We found that a sigW mutation does not result in an increase similar to that in the prsW mutant (Fig. 2). This suggests that the effect of the prsW mutation is not due to an inability to activate σW. It was recently demonstrated that PrsW altered the levels of several membrane proteins in a σW-independent manner, suggesting that PrsW has additional roles in the cell (46). Our results do demonstrate that RasP is required for the lysozyme induction of σV activity and that PrsW is dispensable.
Fig 2.
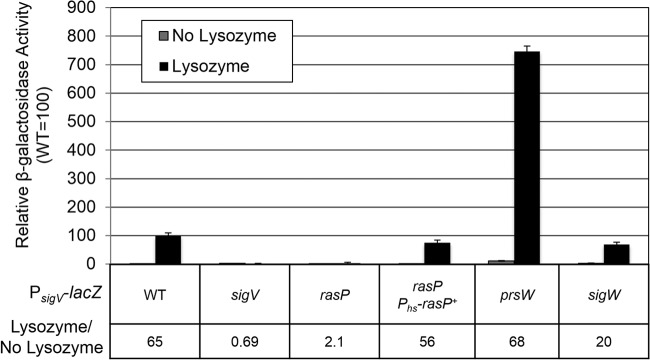
Activation of σV is dependent upon the site 2 protease RasP but not the site 1 protease PrsW. B. subtilis PsigV-lacZ (CDE1546), ΔsigV::kan PsigV-lacZ (CDE1547), ΔprsW::erm PsigV-lacZ (CDE1549), ΔrasP::tet PsigV-lacZ (JLH653), and ΔrasP::tet Phs-rasP+ PsigV-lacZ (JLH640) strains were grown overnight at 30°C and spotted (20 μl) in triplicate on plates containing LB supplemented with IPTG (1 mM) and LB supplemented with IPTG (1 mM) plus lysozyme (10 μg/ml). Plates were incubated at 37°C for 6 h. Cells were collected and resuspended in Z buffer. β-Galactosidase activity was calculated as described in Materials and Methods. Wild type (WT) induction in the presence of lysozyme was set as 100.
RasP is required for site 2 cleavage of RsiV.
Since RasP is required for σV activity and is a known site 2 protease for other ECF anti-σ factors, we tested the effects of a rasP mutant on RsiV degradation. Cells expressing rsiV from an IPTG-inducible promoter were grown to mid-log phase and were left untreated or treated with lysozyme (2 μg/ml) for 10 min. The state of RsiV was probed using anti-RsiV antibodies which detect only the C-terminal 230 amino acids of RsiV. As expected, RsiV was cleaved to near completion in both the wild-type and prsW mutant strain (Fig. 3A). Similarly, we found that a rasP mutation did not inhibit cleavage of RsiV in the presence of lysozyme (Fig. 3A). This is not surprising, since the C-terminal product would be the result of successful site 1 cleavage and our antibodies were raised against the C-terminal portion of RsiV.
Fig 3.
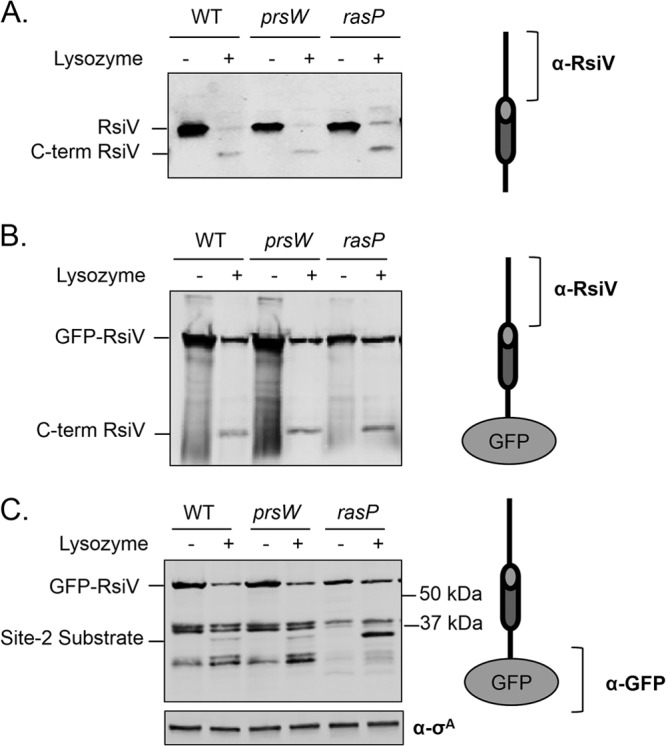
RsiV degradation is dependent upon the site 2 protease RasP. B. subtilis containing an IPTG-inducible copy of rsiV+ (JLH402) or gfp-rsiV+ (JLH453) was subcultured 1:100 into LB supplemented with IPTG (1 mM or 0.5 mM, respectively). Strains with a site 1 prsW mutation (JLH415 and JLH465) were grown in the same manner. Strains with a site 2 rasP mutation (JLH416 and JLH466) were subcultured 1:50 but otherwise grown similarly. At mid-log phase, cultures were incubated for 10 min without (−) or with (+) lysozyme (2 μg/ml) at 37°C. The immunoblot was probed with anti-RsiV antibodies (A and B) or anti-GFP antibodies (C).
RasP is known to perform site 2 cleavage of RsiW (47). To determine if RasP is required for site 2 cleavage of RsiV, we constructed a strain producing an N-terminal GFP-RsiV+ fusion protein which localizes to the cell membrane (see Fig. S2 in the supplemental material). Cells producing GFP-RsiV+ were treated as described above and then probed with either anti-RsiV or anti-GFP antibodies. When using anti-RsiV antibodies to follow GFP-RsiV+ degradation, we found that GFP-RsiV+ was degraded in response to lysozyme similarly to wild-type RsiV (Fig. 3B). Consistent with previous results, neither the absence of PrsW nor of RasP altered the degradation of RsiV (Fig. 3B). However, when the same samples were probed using anti-GFP antibodies to detect the N-terminal portion of the GFP-RsiV fusion protein, we were able to detect further cleavage events (Fig. 3C). In the presence of lysozyme, a faint band corresponding to a truncated form of GFP-RsiV+ could be observed in both wild-type and prsW mutant cells (Fig. 3C). In the absence of RasP, we observed an increase in this truncated product of GFP-RsiV only when the cells had been treated with lysozyme (Fig. 3C). This suggests that RasP is required for site 2 cleavage of RsiV.
PT inhibits degradation of GFP-RsiV at site 2.
Although B. subtilis RasP is not essential for growth, a rasP mutation has pleiotropic effects. Mutants of rasP grow more slowly in liquid, are not competent, cannot activate σW, have cell division defects, and have decreased long-term survival (26, 47, 48). Since the deletion of rasP affects several other cellular processes, we sought to temporarily inhibit RasP function by using an inhibitor to avoid the secondary effects due to long-term growth without RasP. The site 2 intramembrane metalloproteases are dependent upon the coordination of zinc for enzymatic activity (49). The chelator 1,10-phenanthroline (PT) inhibits site 2 protease function by chelating the zinc molecule (49). Using the GFP-RsiV+ construct to follow RsiV degradation, cells were grown to mid-log phase and then treated for 30 min with increasing concentrations of PT. The cells were then incubated with lysozyme (2 μg/ml) for 10 min. At the highest concentration of PT, a truncated product of GFP-RsiV accumulates that is the same size as the product that accumulates in the rasP mutant (Fig. 4). As a control, we also compared the effects of PT on the degradation of GFP-RsiV in a rasP mutant. We did not detect any further accumulation of the site 1 product in the rasP mutant even with the highest levels of PT. This suggests that the effect of PT is likely via inhibition of the site 2 protease RasP. This provides further evidence that RasP is required for site 2 cleavage of RsiV and indicates that, like other site 2 proteases, zinc is likely necessary for RasP function.
Fig 4.
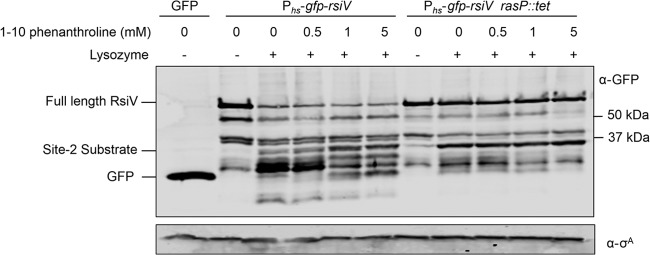
1,10-phenanthroline inhibits site 1 protease cleavage. B. subtilis cells of Phs-gfp-rsiV (JLH453) and Phs-gfp-rsiV ΔrasP::cat (JLH466) strains were grown overnight and then subcultured 1:100 (JLH453) or 1:50 (JLH466) into 5 ml LB supplemented with IPTG (0.5 mM). When cultures reached mid-log phase, samples were treated with increasing concentrations of 1,10-phenanthroline (0, 0.5, 1, and 5 mM) for 30 min and then treated (+) with lysozyme (2 μg/ml) for 10 min at 37°C. The immunoblot was probed with anti-GFP antibodies or anti-σA antibody, which was used as a loading control.
RasP constitutively degrades an RsiV intermediate.
In other systems, site 2 cleavage is preceded by and dependent upon site 1 cleavage to remove the extracellular domain of the target anti-σ factor (17, 20, 48, 50). To mimic site 1 cleavage of RsiV, we constructed a strain producing GFP-RsiV1-60, which lacks the extracellular domain. Wild-type cells expressing full-length GFP-RsiV+ showed lysozyme-dependent degradation of RsiV (Fig. 5). In the absence of RasP, full-length GFP-RsiV+ develops a truncated form of GFP-RsiV only in the presence of lysozyme (Fig. 5). When we examined wild-type cells producing GFP-RsiV1-60, lacking the extracellular domain, the fusion protein was undetectable in the presence and absence of lysozyme (Fig. 5). Note that free GFP accumulates in all strains producing the RsiV1-60 truncation, suggesting that the truncated fusion protein is inherently unstable and may be degraded by other proteases as well (Fig. 5). However, in the rasP mutant, we observed accumulation of GFP-RsiV1-60 regardless of whether cells had been exposed to lysozyme or not (Fig. 5). This suggests that, similar to other ECF σ factors, which are regulated by intramembrane proteases, RasP cleavage of RsiV occurs after site 1 cleavage and is constitutive once site 1 processing has occurred.
Fig 5.
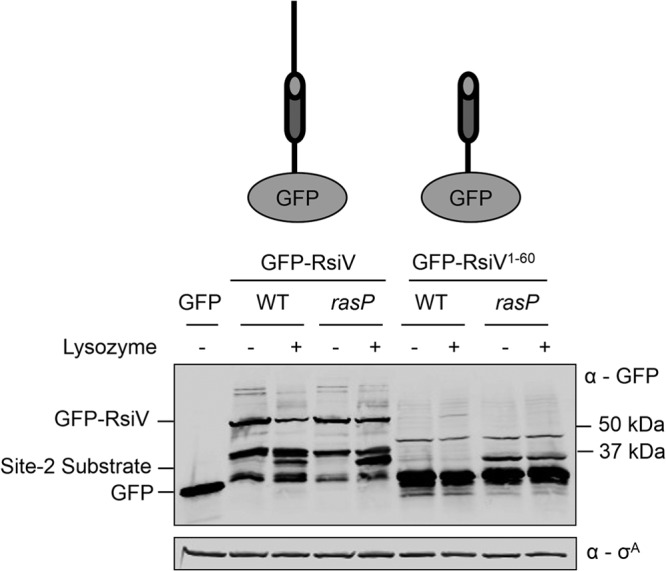
Truncation of RsiV results in constitutive degradation in a RasP-dependent manner. Immunoblot comparing full-length RsiV from B. subtilis Phs-gfp-rsiV (JLH453) and Phs-gfp-rsiV ΔrasP::cat (JLH466) strains and RsiV without the extracellular domain from Phs-gfp-rsiV1-60 (JLH516) and Phs-gfp-rsiV1-60 ΔrasP::cat (JLH536) strains. WT strains (JLH453 and JLH516) were subcultured 1:100 into 5 ml LB supplemented with IPTG (0.5 mM). Strains containing rasP mutations (JLH466 and JLH536) were subcultured 1:50 into 5 ml LB supplemented with IPTG (0.5 mM). Cultures were grown to mid-log phase and then left untreated (−) or treated (+) with lysozyme (2 μg/ml) for 10 min at 37°C. The immunoblot was probed with anti-GFP antibodies or anti-σA antibody, which was used as a loading control. Cytoplasmic GFP was used as a size control (TPM1263).
Increased lysozyme resistance decreases σV activation.
We performed a transposon screen to identify mutations which altered σV activation in response to lysozyme. We utilized the transposon TnHyJump, a TnYLB transposon with an outward-facing constitutive promoter, which allowed us to identify both null mutations and insertions which resulted in the overexpression of a gene downstream of the insertion site (44). A strain containing two copies of a PsigV-lacZ reporter was mutagenized with TnHyJump, and more than 20,000 colonies were screened for either increased or decreased PsigV-lacZ expression in the presence of lysozyme. As predicted, we isolated a number of mutations in sigV which block PsigV-lacZ expression in the presence of lysozyme (data not shown). We also isolated an insertion immediately upstream from pdaC (yjeA), which encodes a peptidoglycan deacetylase (51). We hypothesized that the constitutive promoter on the transposon led to increased pdaC expression. To determine whether overexpression of pdaC was responsible for the decreased PsigV-lacZ expression, we constructed a B. subtilis strain in which pdaC expression could be induced from an IPTG-inducible promoter. We found that the overexpression of pdaC led to a 2-fold decrease in the expression of PsigV-lacZ compared to its expression in the wild type (Fig. 6A). This confirmed that increased expression of pdaC caused a decrease in PsigV-lacZ expression.
Fig 6.
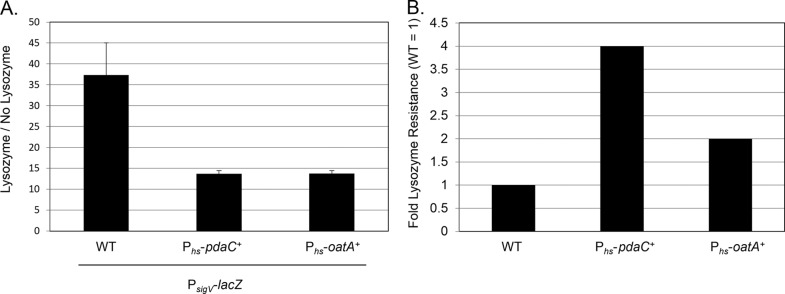
Peptidoglycan modifications decrease σV activation by increasing lysozyme resistance. (A) B. subtilis wild-type (CDE1546 PsigV-lacZ), Phs-pdaC+ (JLH176 PsigV-lacZ Phs-pdaC+), and Phs-oatA+ (JLH345 PsigV-lacZ Phs-oatA+) strains were grown overnight at 30°C and spotted onto LB plates supplemented with IPTG (1 mM) or LB plates supplemented with IPTG (1 mM) plus hen egg white lysozyme (10 μg/ml). Plates were incubated at 37°C for 6 h. Cells were collected and resuspended in Z buffer. β-Galactosidase activity was calculated as described in Materials and Methods. (B) MICs were determined for wild-type (CDE1546 PsigV-lacZ), Phs-pdaC+ (JLH176 PsigV-lacZ Phs-pdaC+), and Phs-oatA+ (JLH345 PsigV-lacZ Phs-oatA+) strains. Cells were grown to mid-log phase, and the OD600 adjusted to 0.8. The cells were diluted 1:50 into LB supplemented with IPTG (1 mM). Bacteria were added to plates in which lysozyme (200 μg) had been serially diluted 2-fold in a microtiter plate. The plates were incubated overnight at 30°C, and the OD600 was read. Wells with an OD600 of <0.05 were considered to have no growth. The MIC of the WT (PY79) is set to 1.
PdaC deacetylates N-acetylmuramic acid, and pdaC null mutants have increased lysozyme sensitivity (51). Therefore, we wanted to determine whether the overexpression of pdaC increased lysozyme resistance. We found that pdaC overexpression causes a 4-fold increase in lysozyme resistance (Fig. 6B). Since we had shown that increasing concentrations of lysozyme led to increased PsigV-lacZ expression (Fig. 1), we hypothesized that by increasing resistance to lysozyme, we decreased σV activation by decreasing the effectiveness of lysozyme for cleaving peptidoglycan. To determine whether this held true for other mechanisms of lysozyme resistance, we also compared the overexpression of oatA, which encodes an O-acetyltransferase activated by σV that increases lysozyme resistance (28, 31, 32). When oatA was overexpressed, lysozyme resistance was increased 2-fold (Fig. 6B). We found that, similar to the overexpression of pdaC, the overexpression of oatA led to a 2-fold decrease in σV activation (Fig. 6A). This suggests that the activation of σV decreases as lysozyme resistance increases.
DISCUSSION
Here, we have demonstrated that RsiV, the anti-σ factor which inhibits σV, is degraded in response to lysozyme stress. Furthermore, our data show that this degradation requires the site 2 protease RasP but not the site 1 protease PrsW. Our data indicate that the activation of σV requires at least two proteases to degrade RsiV. This is similar to other ECF σ factors that are activated by RIP, including σE from E. coli and σW from B. subtilis (52). Based on the similarity to analogous systems, we hypothesize that cytosolic proteases are required for degrading the cytoplasmic portion of RsiV after RasP cleavage (17, 20, 53, 54). Using antibodies specific to the C-terminal extracellular domain of RsiV or GFP antibodies and an N-terminal GFP-RsiV fusion protein, we found that full-length RsiV is rapidly and almost completely degraded following brief lysozyme exposure. Additionally, the RsiV extracellular domain is still detectible in lysozyme-treated samples. This raises the possibility that the majority of the RsiV extracellular domain is removed in a single step by cleavage at site 1 in B. subtilis. This would be more similar to E. coli, where activation of σE requires a single cleavage event to allow for the site 2 protease to cleave RseA (13). In contrast, σW activation requires PrsW cleavage and then further processing by an unknown protease(s) before the site 2 protease RasP efficiently recognizes RsiW as a substrate (17).
The site 2 protease RasP is required for σV activation.
Our data indicate that RasP, the site 2 protease for RsiW, is also required for site 2 cleavage of RsiV. In the absence of site 2 cleavage, either through a rasP mutant or by inhibition of RasP activity using a zinc chelator, a truncated form of RsiV accumulates that cannot be degraded. Our data suggest RasP is the site 2 protease, although we cannot rule out the possibility that additional factors may be involved in site 2 processing. In contrast to the site 1 proteases, which appear highly specific for each substrate, it is becoming increasingly clear that the site 2 proteases can recognize a wide range of substrates. In addition to cleaving anti-σ factors RsiV and RsiW, RasP also cleaves the cell division protein FtsL (26). Furthermore, it is thought that RasP aids in clearing signal peptides from the cell membrane (27). RsiV and RsiW do not share any homology at the amino acid level; however, both are single-pass transmembrane anti-σ factors. Although it is still unclear how site 2 proteases recognize their substrate, one hypothesis is that site 2 proteases sense membrane proteins with helix-destabilizing amino acids rather than recognizing a specific amino acid sequence, which is consistent with the ability of RasP to act on multiple membrane proteins (26, 47).
Our work also suggests that RasP activity is constitutive. We constructed a putative site 2 substrate which lacked the extracellular domain of RsiV. This substrate was degraded by RasP even in the absence of lysozyme. Thus, while lysozyme is required to initiate the proteolytic cascade leading to RsiV destruction, lysozyme is not required to induce RasP activity.
Site 1 cleavage is the regulated step in σV activation.
Site 1 cleavage is the rate-limiting step in the activation of both σE in E. coli and σW in B. subtilis (18, 19, 50). Our data suggest that the activation of σV is also controlled at the site 1 cleavage step. First, we found that the accumulation of the truncated GFP-RsiV product was only detected in rasP mutants upon treatment with lysozyme (Fig. 3C), suggesting that the degradation of RsiV in response to lysozyme is tightly controlled. Second, when we mimicked the site 1-cleaved RsiV by constructing GFP-RsiV1-60, which lacks the extracellular domain, GFP-RsiV1-60 was unstable and was degraded even in the absence of lysozyme. However, in the absence of RasP, the truncated GFP-RsiV1-60 accumulates. This mimics the putative site 2 substrate buildup observed when rasP mutant cells producing full-length GFP-RsiV+ are treated with lysozyme (Fig. 3C and 5). Taken together, these data suggest that site 2 cleavage of RsiV by RasP is not regulated by the presence of lysozyme but simply occurs as a consequence of the appearance of the appropriate substrate. This suggests that the regulation of σV activity, like several other ECF σ factors, is controlled at the level of the site 1 cleavage of the anti-σ factor RsiV.
The site 1 protease for RsiV is unknown.
We tested whether the known B. subtilis site 1 protease, PrsW, which is required for σW activation, was involved in RsiV degradation. Our data indicate that PrsW, the site 1 protease for RsiW, is not required for site 1 cleavage of RsiV. In the absence of PrsW, both PsigV expression and RsiV degradation remain lysozyme inducible. It is interesting to note that the overall level of PsigV-lacZ expression was greater in the absence of PrsW. The molecular mechanism for this is currently unclear. It was previously reported that prsW mutation leads to significant changes in the composition and levels of several membrane proteins (46). Some of these differences were due to an inability of the prsW mutant to activate σW (46). However, Zweers et al. also found that the absence of PrsW had effects on membrane proteins which could not be accounted for by an inability to activate σW, suggesting that PrsW may have substrates in the cell other than RsiW (46). One possible explanation for increased PsigV induction in the absence of PrsW is that the levels of the site 1 protease responsible for cleaving RsiV may be increased. However, we were unable to detect an increase in the degradation of RsiV in the absence of lysozyme. In addition, we did not detect an increase of the RsiV site 2 substrate in a prsW rasP double mutant in the absence of lysozyme (data not shown). We also observed that the levels of RsiV appeared lower in the absence of RasP. In addition, it appears that the amount of RsiV site 1 cleavage is also lower in the absence of RasP. We hypothesize that both of these may be linked to the effect the absence of RasP can have on the accumulation of membrane proteins (46).
In an attempt to identify the site 1 protease, we performed multiple variations on a transposon screen to identify mutants which could not respond to lysozyme, and as yet, we have been unable to identify the site 1 protease. It is possible that multiple proteases are able to perform the initial site 1 cleavage of RsiV or that the site 1 protease is essential. However, if the site 1 protease is essential, it must have other roles in the cell, as σV itself is not essential. It is interesting that the site 1 proteases have been more difficult to identify in several cases, including ECF σL, σK, and σM in M. tuberculosis (23). In addition, while PrsW is absolutely required for site 1 cleavage of RsiW, there is another protease(s) that is required to trim RsiW before the site 2 protease RasP can cleave RsiW within the transmembrane domain (17). This protease(s) has not yet been identified. However, our data suggest that cleavage of RsiV at site 1 is the rate limiting step in σV activation, and thus, identifying the protease(s) required for this processing step is of interest.
Increased lysozyme resistance decreases σV activation.
Many stress response signal transduction systems detect a specific environmental stress and, in response, induce changes in gene expression to deal with this stress. By increasing the expression of repair mechanisms to correct damage associated with these stresses, cells downregulate the activation of the stress response system (55). The activation of σV causes an increase in lysozyme resistance through mechanisms that modify the structure of peptidoglycan (OatA) or lipoteichoic acid (Dlt), thus impairing the ability of lysozyme to cause damage and increasing bacterial survival (28, 31, 33). Our screen for mutants which block σV activation included an insertion that led to increased pdaC expression and increased lysozyme resistance (51). We found that increased lysozyme resistance when either pdaC (peptidoglycan deacetylase) or oatA (peptidoglycan O-acetyltransferase) was overexpressed caused a marked decrease in σV activation. This suggests that, like other cell stress response systems, σV is activated and induces genes which can decrease the damage generated by lysozyme, and in response, σV activity is then decreased.
Activation of σV in other organisms.
The ECF sigma factor σV has been identified and shown to be lysozyme inducible in two other Gram-positive organisms, C. difficile and E. faecalis (34, 35). This suggests that σV may be important for controlling resistance to lysozyme in a number of Gram-positive bacteria. In the accompanying article, Varahan et al. (57) show that Eep (a protease required for peptide pheromone production) is required for the σV activation and site 2 processing of E. faecalis RsiV in response to lysozyme. It is not currently known whether a site 2 protease is required for the activation of σV in C. difficile. However, together, these results suggest that the site 2 proteases are probably involved in the activation of σV in other organisms as well.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01AI087834 from the National Institute of Allergy and Infectious Diseases to C.D.E. and American Heart Association grant 13PRE14650053 to J.L.H.
We thank members of the Ellermeier laboratory for helpful comments and Theresa D. Ho (University of Iowa) for comments on the manuscript.
Footnotes
Published ahead of print 17 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00292-13.
REFERENCES
- 1. Butcher BG, Mascher T, Helmann JD. 2008. Environmental sensing and the role of extracytoplasmic function (ECF) σ factors, p 233–261 In El-Sharoud WM. (ed), Bacterial physiology: a molecular approach. Springer, Berlin, Germany [Google Scholar]
- 2. Staron A, Sofia HJ, Dietrich S, Ulrich LE, Liesegang H, Mascher T. 2009. The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol. Microbiol. 74:557–581 [DOI] [PubMed] [Google Scholar]
- 3. Lal M, Caplan M. 2011. Regulated intramembrane proteolysis: signaling pathways and biological functions. Physiology (Bethesda) 26:34–44 [DOI] [PubMed] [Google Scholar]
- 4. Ho TD, Ellermeier CD. 2012. Extra cytoplasmic function σ factor activation. Curr. Opin. Microbiol. 15:182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ades SE, Connolly LE, Alba BM, Gross CA. 1999. The Escherichia coli sigma(E)-dependent extracytoplasmic stress response is controlled by the regulated proteolysis of an anti-sigma factor. Genes Dev. 13:2449–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alba BM, Zhong HJ, Pelayo JC, Gross CA. 2001. degS (hhoB) is an essential Escherichia coli gene whose indispensable function is to provide sigma (E) activity. Mol. Microbiol. 40:1323–1333 [DOI] [PubMed] [Google Scholar]
- 7. Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT. 2003. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 113:61–71 [DOI] [PubMed] [Google Scholar]
- 8. Wilken C, Kitzing K, Kurzbauer R, Ehrmann M, Clausen T. 2004. Crystal structure of the DegS stress sensor: how a PDZ domain recognizes misfolded protein and activates a protease. Cell 117:483–494 [DOI] [PubMed] [Google Scholar]
- 9. Alba BM, Gross CA. 2004. Regulation of the Escherichia coli sigma-dependent envelope stress response. Mol. Microbiol. 52:613–619 [DOI] [PubMed] [Google Scholar]
- 10. Kanehara K, Ito K, Akiyama Y. 2002. YaeL (EcfE) activates the sigma(E) pathway of stress response through a site-2 cleavage of anti-sigma(E), RseA. Genes Dev. 16:2147–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanehara K, Ito K, Akiyama Y. 2003. YaeL proteolysis of RseA is controlled by the PDZ domain of YaeL and a Gln-rich region of RseA. EMBO J. 22:6389–6398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chaba R, Grigorova IL, Flynn JM, Baker TA, Gross CA. 2007. Design principles of the proteolytic cascade governing the sigmaE-mediated envelope stress response in Escherichia coli: keys to graded, buffered, and rapid signal transduction. Genes Dev. 21:124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA. 2002. DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma(E)-dependent extracytoplasmic stress response. Genes Dev. 16:2156–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo Y, Asai K, Sadaie Y, Helmann JD. 2010. Transcriptomic and phenotypic characterization of a Bacillus subtilis strain without extracytoplasmic function sigma factors. J. Bacteriol. 192:5736–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wiegert T, Homuth G, Versteeg S, Schumann W. 2001. Alkaline shock induces the Bacillus subtilis sigma(W) regulon. Mol. Microbiol. 41:59–71 [DOI] [PubMed] [Google Scholar]
- 16. Cao M, Wang T, Ye R, Helmann JD. 2002. Antibiotics that inhibit cell wall biosynthesis induce expression of the Bacillus subtilis sigma(W) and sigma(M) regulons. Mol. Microbiol. 45:1267–1276 [DOI] [PubMed] [Google Scholar]
- 17. Heinrich J, Hein K, Wiegert T. 2009. Two proteolytic modules are involved in regulated intramembrane proteolysis of Bacillus subtilis RsiW. Mol. Microbiol. 74:1412–1426 [DOI] [PubMed] [Google Scholar]
- 18. Ellermeier CD, Losick R. 2006. Evidence for a novel protease governing regulated intramembrane proteolysis and resistance to antimicrobial peptides in Bacillus subtilis. Genes Dev. 20:1911–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heinrich J, Wiegert T. 2006. YpdC determines site-1 degradation in regulated intramembrane proteolysis of the RsiW anti-sigma factor of Bacillus subtilis. Mol. Microbiol. 62:566–579 [DOI] [PubMed] [Google Scholar]
- 20. Zellmeier S, Schumann W, Wiegert T. 2006. Involvement of Clp protease activity in modulating the Bacillus subtilis sigma(W) stress response. Mol. Microbiol. 61:1569–1582 [DOI] [PubMed] [Google Scholar]
- 21. Li X, Wang B, Feng L, Kang H, Qi Y, Wang J, Shi Y. 2009. Cleavage of RseA by RseP requires a carboxyl-terminal hydrophobic amino acid following DegS cleavage. Proc. Natl. Acad. Sci. U. S. A. 106:14837–14842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kinch LN, Ginalski K. 2006. Site-2 protease regulated intramembrane proteolysis: sequence homologs suggest an ancient signaling cascade. Protein Sci. 15:84–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sklar JG, Makinoshima H, Schneider JS, Glickman MS. 2010. M. tuberculosis intramembrane protease Rip1 controls transcription through three anti-sigma factor substrates. Mol. Microbiol. 77:605–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen G, Zhang X. 2010. New insights into S2P signaling cascades: regulation, variation, and conservation. Protein Sci. 19:2015–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. 2006. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124:587–599 [DOI] [PubMed] [Google Scholar]
- 26. Wadenpohl I, Bramkamp M. 2010. DivIC stabilizes FtsL against RasP cleavage. J. Bacteriol. 192:5260–5263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saito A, Hizukuri Y, Matsuo E-i, Chiba S, Mori H, Nishimura O, Ito K, Akiyama Y. 2011. Post-liberation cleavage of signal peptides is catalyzed by the site-2 protease (S2P) in bacteria. Proc. Natl. Acad. Sci. U. S. A. 108:13740–13745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ho TD, Hastie JL, Intile PJ, Ellermeier CD. 2011. The Bacillus subtilis extracytoplasmic function sigma factor σV is induced by lysozyme and provides resistance to lysozyme. J. Bacteriol. 193:6215–6222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zellmeier S, Hofmann C, Thomas S, Wiegert T, Schumann W. 2005. Identification of sigma (v)-dependent genes of Bacillus subtilis. FEMS Microbiol. Lett. 253:221–229 [DOI] [PubMed] [Google Scholar]
- 30. Callewaert L, Michiels CW. 2010. Lysozymes in the animal kingdom. J. Biosci. 35:127–160 [DOI] [PubMed] [Google Scholar]
- 31. Guariglia-Oropeza V, Helmann JD. 2011. Bacillus subtilis σV confers lysozyme resistance by activation of two cell wall modification pathways, peptidoglycan O-acetylation and d-alanylation of teichoic acids. J. Bacteriol. 193:6223–6232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bernard E, Rolain T, Courtin P, Guillot A, Langella P, Hols P, Chapot-Chartier MP. 2011. Characterization of O-acetylation of N-acetylglucosamine: a novel structural variation of bacterial peptidoglycan. J. Biol. Chem. 286:23950–23958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laaberki M-H, Pfeffer J, Clarke AJ, Dworkin J. 2011. O-Acetylation of peptidoglycan is required for proper cell separation and S-layer anchoring in Bacillus anthracis. J. Biol. Chem. 286:5278–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ho TD, Ellermeier CD. 2011. PrsW is required for colonization, resistance to antimicrobial peptides, and expression of extracytoplasmic function sigma factors in Clostridium difficile. Infect. Immun. 79:3229–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Le Jeune A, Torelli R, Sanguinetti M, Giard JC, Hartke A, Auffray Y, Benachour A. 2010. The extracytoplasmic function sigma factor SigV plays a key role in the original model of lysozyme resistance and virulence of Enterococcus faecalis. PLoS One 5:e9658. 10.1371/journal.pone.0009658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benachour A, Muller C, Dabrowski-Coton M, Le Breton Y, Giard JC, Rince A, Auffray Y, Hartke A. 2005. The Enterococcus faecalis SigV protein is an extracytoplasmic function sigma factor contributing to survival following heat, acid, and ethanol treatments. J. Bacteriol. 187:1022–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Youngman P, Perkins JB, Losick R. 1984. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid 12:1–9 [DOI] [PubMed] [Google Scholar]
- 38. Wilson GA. 1968. Nutritional factors influencing the development of competence in the Bacillus subtilis transformation system. J. Bacteriol. 95:1439–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. 2010. agr-dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J. Innate Immun. 2:546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259–265 [DOI] [PubMed] [Google Scholar]
- 41. Guerout-Fleury AM, Shazand K, Frandsen N, Stragier P. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335–336 [DOI] [PubMed] [Google Scholar]
- 42. Griffith KL, Wolf RE. 2002. Measuring beta-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem. Biophys. Res. Commun. 290:397–402 [DOI] [PubMed] [Google Scholar]
- 43. Slauch JM, Silhavy TJ. 1991. cis-acting ompF mutations that result in OmpR-dependent constitutive expression. J. Bacteriol. 173:4039–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pozsgai ER, Blair KM, Kearns DB. 2012. Modified mariner transposons for random inducible-expression insertions and transcriptional reporter fusion insertions in Bacillus subtilis. Appl. Environ. Microbiol. 78:778–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kazmierczak MJ, Wiedmann M, Boor KJ. 2005. Alternative sigma factors and their roles in bacterial virulence. Microbiol. Mol. Biol. Rev. 69:527–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zweers JC, Wiegert T, van Dijl JM. 2009. Stress-responsive systems set specific limits to the overproduction of membrane proteins in Bacillus subtilis. Appl. Environ. Microbiol. 75:7356–7364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schobel S, Zellmeier S, Schumann W, Wiegert T. 2004. The Bacillus subtilis sigma(W) anti-sigma factor RsiW is degraded by intramembrane proteolysis through YluC. Mol. Microbiol. 52:1091–1105 [DOI] [PubMed] [Google Scholar]
- 48. Heinrich J, Lunden T, Kontinen VP, Wiegert T. 2008. The Bacillus subtilis ABC transporter EcsAB influences intramembrane proteolysis through RasP. Microbiology 154:1989–1997 [DOI] [PubMed] [Google Scholar]
- 49. Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, Shi Y. 2007. Structure of a site-2 protease family intramembrane metalloprotease. Science 318:1608–1612 [DOI] [PubMed] [Google Scholar]
- 50. Grigorova IL, Chaba R, Zhong HJ, Alba BM, Rhodius V, Herman C, Gross CA. 2004. Fine-tuning of the Escherichia coli sigmaE envelope stress response relies on multiple mechanisms to inhibit signal-independent proteolysis of the transmembrane anti-sigma factor, RseA. Genes Dev. 18:2686–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kobayashi K, Sudiarta IP, Kodama T, Fukushima T, Ara K, Ozaki K, Sekiguchi J. 2012. Identification and characterization of a novel polysaccharide deacetylase C (PdaC) from Bacillus subtilis. J. Biol. Chem. 287:9765–9776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heinrich J, Wiegert T. 2009. Regulated intramembrane proteolysis in the control of extracytoplasmic function sigma factors. Res. Microbiol. 160:696–703 [DOI] [PubMed] [Google Scholar]
- 53. Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11:671–683 [DOI] [PubMed] [Google Scholar]
- 54. Flynn JM, Levchenko I, Sauer RT, Baker TA. 2004. Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 18:2292–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hayden JD, Ades SE. 2008. The extracytoplasmic stress factor, sigmaE, is required to maintain cell envelope integrity in Escherichia coli. PLoS One 3:e1573. 10.1371/journal.pone.0001573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ben-Yehuda S, Rudner DZ, Losick R. 2003. RacA, a bacterial protein that anchors chromosomes to the cell poles. Science 299:532–536 [DOI] [PubMed] [Google Scholar]
- 57. Varahan S, Iyer VS, Moore WT, Hancock LE. 2013. Eep confers lysozyme resistance to Enterococcus faecalis via the activation of the extracytoplasmic function sigma factor SigV. J. Bacteriol. 195:3125–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.