

Abstract
The strictly anaerobic Syntrophus aciditrophicus is a fermenting deltaproteobacterium that is able to degrade benzoate or crotonate in the presence and in the absence of a hydrogen-consuming partner. During growth in pure culture, both substrates are dismutated to acetate and cyclohexane carboxylate. In this work, the unknown enzymes involved in the late steps of cyclohexane carboxylate formation were studied. Using enzyme assays monitoring the oxidative direction, a cyclohex-1-ene-1-carboxyl-CoA (Ch1CoA)-forming cyclohexanecarboxyl-CoA (ChCoA) dehydrogenase was purified and characterized from S. aciditrophicus and after heterologous expression of its gene in Escherichia coli. In addition, a cyclohexa-1,5-diene-1-carboxyl-CoA (Ch1,5CoA)-forming Ch1CoA dehydrogenase was characterized after purification of the heterologously expressed gene. Both enzymes had a native molecular mass of 150 kDa and were composed of a single, 40- to 45-kDa subunit; both contained flavin adenine dinucleotide (FAD) as a cofactor. While the ChCoA dehydrogenase was competitively inhibited by Ch1CoA in the oxidative direction, Ch1CoA dehydrogenase further converted the product Ch1,5CoA to benzoyl-CoA. The results obtained suggest that Ch1,5CoA is a common intermediate in benzoate and crotonate fermentation that serves as an electron-accepting substrate for the two consecutively operating acyl-CoA dehydrogenases characterized in this work. In the case of benzoate fermentation, Ch1,5CoA is formed by a class II benzoyl-CoA reductase; in the case of crotonate fermentation, Ch1,5CoA is formed by reversing the reactions of the benzoyl-CoA degradation pathway that are also employed during the oxidative (degradative) branch of benzoate fermentation.
INTRODUCTION
The strictly anaerobic deltaproteobacterium Syntrophus aciditrophicus SB is a fermenting organism capable of using various carboxylic acids, including benzoic acid and crotonate, as growth substrates. During fermentation of these substrates in the presence of a syntrophic partner like Methanospirillum hungatei JF1 or Desulfovibrio strain G11, the reducing equivalents can be released as H2 or formate as they are readily consumed by the coculture for CO2 or sulfate reduction, respectively. Recent studies revealed that S. aciditrophicus is also able to grow with benzoate or crotonate in the absence of an H2/formate-consuming syntrophic partner (1, 2). Without the syntrophic partner, the release of reducing equivalents as H2 or formate is not thermodynamically favorable. For this reason, the redox equivalents produced during oxidation of benzoate or crotonate to acetate are recycled by the concomitant reduction of benzoate- and crotonate-derived metabolites to cyclohexane carboxylate. The formation of cyclohexane carboxylate is a unique fermentation process that has been described only for S. aciditrophicus so far.
In the case of benzoate fermentation, benzoate is first activated to benzoyl-coenzyme A (CoA), which is then converted to acetate by enzymes of the benzoyl-CoA degradation pathway of aromatic compounds (Fig. 1). Experimental support for this pathway includes the identification of the corresponding genes in the genome (3) and the characterization of the enzymes and/or enzyme activities in S. aciditrophicus (4–8). The reactions of the benzoyl-CoA degradation pathway comprise the following: (i) activation of benzoate by an AMP-forming benzoate CoA ligase, (ii) reductive dearomatization of benzoyl-CoA by an ATP-independent class II benzoyl-CoA reductase, most probably to cyclohexa-1,5-diene-1-carboxyl-CoA (Ch1,5CoA) (two-electron reduction) rather than to cyclohex-1-ene-1-carboxyl-CoA (Ch1CoA) (four-electron reduction), (iii) modified β-oxidation reactions resulting in ring cleavage catalyzed by an enoyl-CoA hydratase (BamR), an alcohol dehydrogenase (BamQ), and a hydrolase (BamA), and (iv) a series of subsequent β-oxidation-like reactions and a decarboxylation reaction to yield three acetyl-CoAs that may be used for ATP synthesis (Fig. 1). The reducing equivalents formed in this oxidative branch of the pathway are recycled by the six-electron reduction of benzoate to cyclohexane carboxylate, most probably via CoA-ester substrates. During the fermentation of benzoate, approximately 1.5 mol acetate and 0.5 mol cyclohexane carboxylate are formed (1).
Fig 1.

Fermentation of crotonate (A) or benzoate (B) to acetate and cyclohexane carboxylate. Fermentation substrates are shown by solid-line rectangles with a gray fill; fermentation products are surrounded by solid-line rectangles without a fill. Both benzoate and crotonate are dismutated to acetate and cyclohexane carboxylate. (A) During crotonate fermentation, the reducing equivalents produced during oxidative acetyl-CoA formation are regenerated by reverse (upwards) reaction of the benzoyl-CoA degradation pathway, yielding Ch1,5CoA; the latter is then further reduced to ChCoA by the two enzymes studied in this work. (B) During benzoate fermentation, enzymes of the benzoyl-CoA degradation pathway are employed in the oxidative direction (downwards). The enzymes involved in the formation of cyclohexane carboxylate from their respective CoA-ester (dashed lines) are unknown. Reactions 1 and 2 were investigated in this work; both are necessary for redox cofactor recycling. Reaction 3 is catalyzed by class II benzoyl-CoA reductase.
When S. aciditrophicus grows alone, it ferments crotonate to acetate and cyclohexane carboxylate rather than to acetate and butyrate as reported for crotonate-fermenting clostridial species (see reference 2 and references therein). The reducing equivalents derived from the oxidation of crotonate to acetate are used to form cyclohexane carboxylate. The formation of cyclohexane carboxylate from crotonate is believed to involve many of the enzymes used for the oxidative degradation of benzoyl-CoA to acetyl-CoA (Fig. 1), since similar metabolites were detected in crotonate-grown and benzoate-grown S. aciditrophicus pure cultures (1, 2). It is assumed that these reactions operate reversibly under physiological conditions.
While several of the enzymes of the benzoyl-CoA degradation pathway involved in benzoate and crotonate fermentation have been studied, the enzymes involved in cyclohexane carboxyl-CoA (ChCoA) and cyclohexane carboxylate formation are not known. Intermediates of the benzoyl-CoA degradation pathway that could serve as electron-accepting precursors for cyclohexane carboxylate formation are benzoyl-CoA or Ch1,5CoA. ChCoA could be formed by three consecutive two-electron transfer reactions to benzoyl-CoA by class II benzoyl-CoA reductase (9). However, this enzyme is assumed to require a reduced ferredoxin as an electron donor and is supposed to strictly couple electron transfer to an energy-consuming process (10). For these reasons, it is unlikely that benzoyl-CoA reductase catalyzes the six-electron reduction. An alternative and less energy-consuming scenario is that the class II benzoyl-CoA reductase is involved only in the reduction of benzoyl-CoA to Ch1,5CoA. The further reduction of the Ch1,5CoA would then be catalyzed by additional oxidoreductases during both benzoate and crotonate fermentations. In this work, the enzymes involved in ChCoA fermentation were identified and characterized as two substrate-specific FAD-containing acyl-CoA dehydrogenases. The results reveal the previously unknown enzymatic steps involved in the formation of the unusual fermentation product cyclohexane carboxylate.
MATERIALS AND METHODS
Materials.
Chemicals and biochemicals were obtained from Roche Diagnostics (Mannheim, Germany), Fluka (Neu-Ulm, Germany), Merck (Darmstadt, Germany), Roth (Karlsruhe, Germany), Sigma-Aldrich (Deisenhofen, Germany), Applichem (Darmstadt, Germany), and Bio-Rad (Munich, Germany). Coenzyme A was obtained from Applichem. Materials and equipment for protein purification were obtained from GE Healthcare (Munich, Germany), Millipore (Bedford, MA), and Sigma-Aldrich (Deisenhofen, Germany). CoA esters were synthesized as described earlier (11, 12).
Computational analysis.
Plots were prepared and regression coefficients were calculated using the Graphpad Prism 4 software package. BLAST searches were performed using the NCBI BLAST server (http://www.ncbi.nlm.nih.gov/BLAST/). Phylogenetic alignments were performed using the Mega4 software package (www.megasoftware.net).
Strains and culture conditions.
Syntrophus aciditrophicus SB was obtained from Mike McInerney (Norman, OK) and cultivated anaerobically in a 200-liter fermenter at 37°C in a mineral medium (pH 7.3) with crotonate (20 mM) as the sole carbon source without addition of rumen fluid (13). Cells were harvested at an optical density at 578 nm (OD578) of approximately 1.0 under anaerobic conditions and stored in liquid nitrogen.
Enzyme assays.
ChCoA dehydrogenase and Ch1CoA dehydrogenase activities were measured in a continuous assay at 30°C in a cuvette (1-cm diameter) containing 200 mM morpholinepropanesulfonic acid (MOPS)-KOH (pH 7.0), 15 mM MgCl2, 0.2 mM ferricenium hexafluorophosphate, and 1 to 10 μg enzyme; the reaction was started by addition of 200 μM ChCoA or Ch1CoA to the mixture (total volume, 450 μl). The time-dependent reduction of ferricenium hexafluorophosphate was monitored at 300 nm (Δε300 = 3,600 M−1 cm−1, self-determined) (7). For the oxidation of 1 mol ChCoA, Ch1CoA, or Ch1,5CoA, 2 mol ferricenium was required. Ch1,5CoA, 6-hydroxy-cyclohex-1-ene-carboxyl-CoA, butyryl-CoA, glutaryl-CoA, succinyl-CoA, and cyclohexane carboxylate (each at 0.5 mM) were tested as substrates. Oxygen sensitivity was tested by comparison of the activities of air-exposed and anaerobic (cuvettes sealed with rubber stoppers under nitrogen atmosphere) enzyme preparations. To measure the rate in the reduction direction, titanium(III) citrate (5 mM) and methyl viologen instead of ferricenium hexafluorophosphate were added under anaerobic conditions to the reaction mixture with Ch1CoA or Ch1,5CoA for the ChCoA dehydrogenase and Ch1CoA dehydrogenase, respectively.
Determination of Km and Ki.
Purified enzymes were used to determine the initial rate at different substrate concentrations (0.015 to 0.4 mM). The apparent Km and Ki values were obtained after plotting using the Graphpad Prism 4 software package. Benzoyl-CoA, Ch1,5CoA, and Ch1CoA were tested for their inhibitory effects (0.5 mM each) by adding them to the assay mixture in the presence of the substrate. For Ki determination, the concentration of Ch1CoA was varied from 0.005 to 0.1 mM at 0.05 and 0.1 mM ChCoA (Dixon plot).
Product formation assay.
In a discontinuous assay (150 μl), 25 μl samples of the assay mixture were taken at different time points (see results) and immediately mixed with 50 μl methanol. After centrifugation, the supernatant was diluted 1:1 with water and analyzed by High-Performance Liquid Chromatography within a 12 min gradient from 5% to 20% acetonitrile in 50 mM phosphate buffer (pH 6.8), followed by a washing step using 30% acetonitrile (1 min) and a recalibration step to 5% acetonitrile (5 min). The products and educts typically eluted after 10.8 min (6-hydroxy-cyclohex-1-ene-carboxyl-CoA), 14.6 min (benzoyl-CoA), 15.1 min (Ch1,5CoA), 16.1 min (Ch1CoA), 17.2 min (ChCoA). The products were identified by UV-visible spectra and by coelution with synthesized standards. To test the reductive direction reaction, Ti(III) citrate (5 mM) together with methylviologen (50 μM) was added to the assay mixture instead of ferricenium hexafluorophosphate.
Purification of ChCoA dehydrogenase from S. aciditrophicus SB.
All steps were carried out at 4°C.
(i) Preparation of cell extract.
Three grams of cells (wet mass) grown on crotonate as the sole carbon source were suspended in 6 ml buffer containing 20 mM triethanolamine hydrochloride and 5 mM MgCl2, pH 7.8 (referred to as buffer A), 0.1 mg DNase I, and 0.2 mg lysozyme. Cell extract was obtained by a passage through a French pressure cell at 137 MPa followed by centrifugation at 100,000 × g (1 h at 4°C).
(ii) Enzyme enrichment.
The supernatant (6 ml; referred to as cell extract) was applied to an Octyl-Sepharose column (Fast Flow; volume, 8 ml; diameter, 1.6 cm; GE Healthcare), which had been equilibrated with buffer A plus 1 M ammonium sulfate. The column was washed (1 ml min−1) with 3 bed volumes of the same buffer, followed by 5 volumes of buffer A containing 500 mM ammonium sulfate. The activity was eluted in a step gradient using buffer A without ammonium sulfate within 24 ml. The fractions containing activity were applied onto a hydroxyapatite column (MacroPrep; ceramic hydroxyapatite, 40 μm, 2.5 ml, 1.6-cm diameter; Bio-Rad) at a flow rate of 2 ml min−1 that had been equilibrated with buffer A containing 5 mM potassium phosphate, pH 7.8. After the column was washed with 3 bed volumes of buffer (20 mM potassium phosphate), the activity eluted in a linear gradient from 20 to 400 mM potassium phosphate (30 bed volumes) in a 4-ml fraction at 80 mM phosphate. The pooled fractions (12 ml) were applied onto a Mono Q ion exchange column (Mono Q 5/50 GL, 1 ml; GE Healthcare) equilibrated with buffer A. After a washing step with buffer A containing 100 mM NaCl (3 bed volumes), the activity eluted in a linear 100 to 300 mM sodium chloride gradient in 1-ml fractions at 170 mM (3 ml). The pooled enzyme fractions were slowly diluted with glycerol to a final concentration of 30% glycerol, and the activity was stable for several weeks at −20°C.
Heterologous gene expression and enzyme production in E. coli.
The genes encoding the ChCoA dehydrogenase (gi 85721157; SYN_02586) and Ch1CoA dehydrogenase (gi 85721158; SYN_02587) were amplified from S. aciditrophicus chromosomal DNA by PCR using primers suitable for the Champion pET101 directional Topo expression kit (Life Technologies) and for introducing a C-terminal 6×His tag according to the manufacturer's instructions. For the oligonucleotide sequences used, see the supplemental material (Table S1). The following PCR conditions were used: 30 cycles of 30 s of denaturation at 94°C, 30 s of primer annealing at 57°C, and 2.5 min of elongation at 72°C; Pfu DNA polymerase (Thermo Scientific) was used. Gel elution, ligation, cloning, and expression were performed according to the manufacturer's protocol. Chemically competent Escherichia coli BL 21 Star(DE3) OneShot cells were transformed with the purified vector and grown at 37°C in LB medium containing 100 μg ml−1 of ampicillin and 12.5 mg liter−1 of riboflavin. Cells were induced with 1.0 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density of 0.4. After growth over night at 20°C, the cells were harvested and stored at −20°C until use.
Purification of recombinant proteins from S. aciditrophicus. (i) Preparation of cell extract.
Frozen E. coli cells (2 to 3 g [wet mass]) were suspended, 5 ml per g cells, in buffer B (20 mM potassium phosphate, 200 mM sodium chloride, and 20 mM imidazole, pH 7.9) containing 0.1 mg of DNase I and 0.5 mg of lysozyme. The cell suspension was passed through a French pressure cell at 138 MPa (4°C). The cell lysate was centrifuged at 100,000 × g for 60 min.
(ii) Affinity chromatography.
For nickel affinity chromatography, 6 to 7 ml of the supernatant obtained after ultracentrifugation was applied onto a 1-ml Ni-chelating affinity column (HisTrap HP; GE Healthcare) that had been equilibrated with buffer B. After washing with buffer B was done, the His-tagged proteins eluted in a gradient from 0.02 to 0.5 M imidazole over 10 ml at a flow rate of 1.0 ml min−1. Active fractions (6.5 ml) were pooled, mixed with 20 ml buffer A, and concentrated to 1.5 ml by ultrafiltration (Vivaspin 6, 10,000-molecular-weight cutoff; Sartorius). The proteins were stored at −20°C in 25% glycerol.
Determination of molecular mass.
Native masses were determined by analytical gel filtration via Superdex 200 (10/300 GL column [GE Healthcare]; 25-ml volume, 0.5 ml min−1) using 20 mM triethanolamine-HCl, pH 7.8, 4 mM MgCl2, and 150 mM KCl. For calibration, apoferritin (443 kDa), catalase (245 kDa), bovine serum albumin (BSA) (67 kDa), and carboanhydrase (29 kDa) were used.
Determination of cofactor content.
For flavin determination, 100 μl of purified enzyme was precipitated with 200 μl methanol. The supernatants obtained after centrifugation were analyzed by high-performance liquid chromatography (HPLC) using an acetonitrile gradient (5% to 20% in 12 min) in 50 mM phosphate buffer, pH 6.8, and compared with a calibration curve of 0.1 to 10 μM free FAD. For reconstitution experiments, heterologously expressed and purified enzyme was incubated with 1 mM FAD for 60 min at 30°C. After desalting using a Sephadex G-25 column (PD10; GE Healthcare), the protein was used for determination of activity and of protein and FAD content. UV-visible (UV/vis) spectra of purified enzymes (approximately 3 μM) were recorded using a UV-1650PC Shimadzu spectrophotometer.
Mass spectrometry analysis.
Identification of proteins purified from the wild type or after heterologous expression of its gene was performed by in-gel tryptic digestion followed by ultraperformance liquid chromatography (UPLC) LTQ Orbitrap MS/MS analysis as described previously (14). Mascot searches of the raw data were done against genome entry of Syntrophus aciditrophicus SB (taxonomic identifier [ID] 56780).
Further determinations.
SDS-PAGE (12.5%) was carried out according to the method of Laemmli. Proteins were visualized using the SimplyBlue SafeStain stain (Invitrogen). Protein was routinely determined by the method of Bradford using BSA as a standard.
RESULTS
General remarks.
To identify the enzymes involved in cyclohexane ring formation during the fermentation of benzoate and crotonate, enzyme assays were usually performed in the reverse reaction and followed the reduction of the artificial electron acceptor ferricenium hexafluorophosphate by the acyl-CoA substrate. Starting material for ChCoA dehydrogenase was S. aciditrophicus cells grown on crotonate. Because cyclohexane carboxylate is formed during both benzoate and crotonate fermentation and because benzoate even serves as an electron acceptor during crotonate fermentation (1), the enzyme activities involved in cyclohexane carboxylate formation were expected to be present in both crotonate- and benzoate-grown cells.
Enzyme assay and enrichment of ChCoA dehydrogenase.
Cell extracts of S. aciditrophicus grown on crotonate as the sole carbon source catalyzed the time-, protein- and ferricenium hexafluorophosphate-dependent oxidation of cyclohexanecarboxyl-CoA (ChCoA) to cyclohex-1-ene-1-carboxyl-CoA (Ch1CoA) as evidenced by HPLC analysis of substrate consumption and product formation (2.1 μmol min−1 mg−1) (Fig. 2). Purification of the ChCoA-oxidizing enzyme was accomplished by three chromatographic steps, including hydrophobic octyl-Sepharose chromatography [elution at 0 mM (NH4)2SO4 at pH 7.8], hydroxyapatite chromatography (elution at 80 mM sodium phosphate at pH 7.8), and MonoQ anion-exchange chromatography (elution at 170 mM KCl at pH 7.8). A typical purification protocol is shown in Table 1. A 25-fold enrichment with a yield of 16% was achieved. From 3-g (wet mass) cells, 1.7 mg with a specific activity of 52 μmol min−1 mg−1 was obtained. SDS-PAGE analysis revealed a highly enriched protein band of about 40 kDa (Fig. 3A).
Fig 2.

HPLC analysis of ChCoA dehydrogenase activity. The upper panel shows reactions catalyzed by ChCoA dehydrogenase. Oxidation of ChCoA (a) to Ch1CoA (b) in the presence of ferricenium (FcPF6) as an electron acceptor is shown. HPLC diagrams of CoA ester analysis are shown after 0 min (A), 1 min (B), and 3 min (C).
Table 1.
Enrichment protocol of ChCoA dehydrogenase purified from S. aciditrophicus cells
| Purification step | Vol (ml) | Total activity (μmol min−1) | Amt of protein (mg) | Sp act (μmol min−1 mg−1) | Yield (%) | Enrichment (fold) |
|---|---|---|---|---|---|---|
| Cell extract | 6 | 285 | 138 | 2.1 | 100 | 1.0 |
| Octyl-Sepharose | 24 | 171 | 30 | 5.7 | 60 | 2.8 |
| Hydroxyapatite | 12 | 149 | 13 | 11 | 52 | 5.5 |
| MonoQ | 3 | 44 | 1.7 | 52 | 16 | 25 |
Fig 3.

Enrichment of ChCoA dehydrogenase. SDS-polyacrylamide gels of ChCoA-oxidizing activity (8 μg of protein per lane) are shown. (A) Purification from S. aciditrophicus. The protein band of about 40 kDa is highly enriched. Lanes 1 and 6, molecular mass standards; lane 2, cell extract; lane 3, fractions obtained after chromatography on octyl Sepharose; lane 4, hydroxyapatite; lane 5, MonoQ. (B) Purification after heterologous expression of its gene (SYN_02586) in E. coli BL21. Lane 1, molecular mass standard; lane 2, supernatant after ultracentrifugation; lane 3, pooled activity-containing fractions after Ni-chelating affinity chromatography.
Identification of the gene encoding ChCoA dehydrogenase.
The enriched protein was excised from the gel and analyzed by UPLC-LTQ Orbitrap tandem mass spectrometry (MS/MS) analysis after enzymatic digestion with trypsin. With use of the MASCOT platform, the resulting peptides showed the best match with the deduced peptides of the SYN_02586 gene product (gi 85858066; 48% sequence coverage) from Syntrophus aciditrophicus SB. The gene was annotated as an acyl-CoA dehydrogenase with a theoretical mass of 42 kDa as calculated from the deduced amino acid sequence.
Heterologous expression of the gene encoding ChCoA dehydrogenase in E. coli and purification of its product.
To verify the function of SYN_02586, the gene was heterologously expressed in E. coli BL21 with a C-terminal six-His tag. After purification by Ni-chelating affinity chromatography, a highly enriched protein band was observed on an SDS gel that migrated at a slightly higher mass than calculated, which can be explained by the 6-fold His tag (Fig. 3B). The protein was excised, and the product of SYN_02586 was verified by mass spectrometric analysis of tryptic digests. The specific ChCoA oxidizing activity of the heterologously expressed enzyme was 16 μmol min−1 mg−1 (31% of the activity purified from S. aciditrophicus).
Molecular and catalytic properties of ChCoA dehydrogenase.
Using gel filtration, the apparent molecular masses of 155 ± 10 kDa for the enzyme purified from S. aciditrophicus and 150 ± 10 kDa for the heterologously expressed enzyme were obtained, indicating a homotetrameric composition. The protein purified from S. aciditrophicus was yellow, with a UV/vis spectrum typical for flavin-containing proteins, with maxima at 358 nm and 447 nm (ε447 = 42 mM−1 cm−1 for the tetramer, Fig. 4). The FAD content of the native enzyme was 3.7 FAD/enzyme (0.9 FAD per 42-kDa monomer). In contrast, the heterologously expressed protein contained only 0.31 FAD/subunit. Incubation of the latter with free FAD resulted in an increase in the FAD/enzyme ratio to 0.58 FAD/monomer and a concomitant increase in the specific activity from 14.5 to 28 μmol min−1 mg−1 (54% of wild-type enzyme activity).
Fig 4.
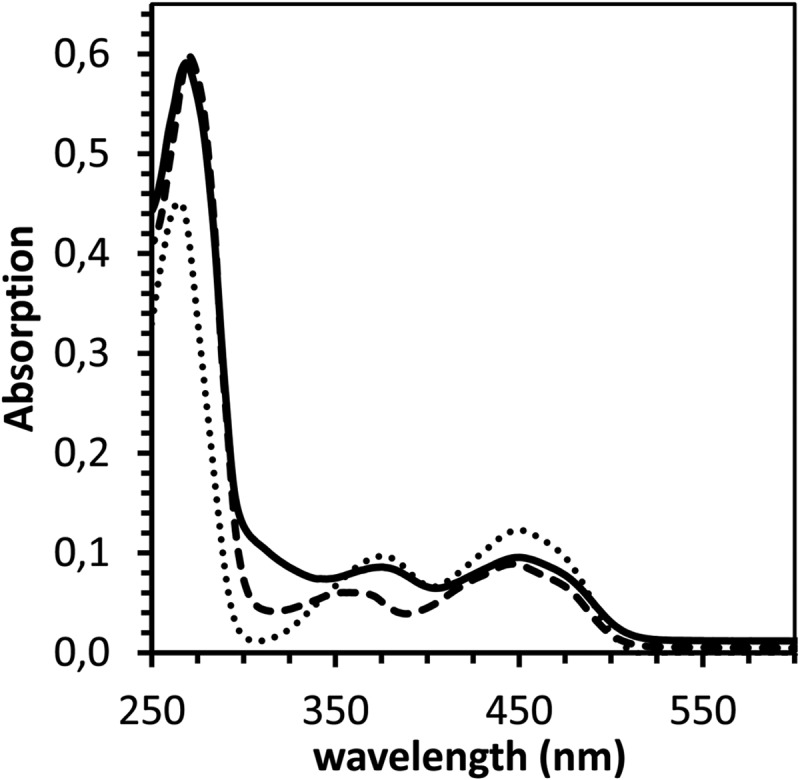
UV-Vis spectra of ChCoA and Ch1CoA dehydrogenase. Free FAD (dotted line), purified S. aciditrophicus ChCoA dehydrogenase (3.4 μM; dashed line), and heterologously expressed Ch1CoA dehydrogenase (3.2 μM; solid line) are shown. The cofactors/enzymes were all in the oxidized state.
The enzyme was highly specific for ChCoA, with an apparent Km value of 22 ± 5 μM. No activity was observed with Ch1CoA, Ch1,5CoA, 6-hydroxycyclohex-1-ene-1-carboxyl-CoA, butyryl-CoA, glutaryl-CoA, succinyl-CoA, or cyclohexane carboxylate. The reaction product Ch1CoA competitively inhibited the ChCoA dehydrogenating activity (Ki, 49 ± 10 μM). The physiologically relevant reduction of Ch1CoA (0.2 mM) was tested using a combination of Ti(III) citrate (5 mM) and methyl viologen (1 mM) as an electron donor system. The activity was 48 μmol min−1 mg−1 (170% of the ChCoA oxidation rate).
Enzyme assay and putative gene coding for Ch1CoA dehydrogenase.
Cell extracts of S. aciditrophicus converted Ch1CoA to Ch1,5CoA (0.6 μmol min−1 mg−1), which was subsequently converted to benzoyl-CoA (0.2 μmol min−1 mg−1) in a time-, enzyme-, and ferricenium hexafluorophosphate-dependent reaction. While the formation of Ch1,5CoA was expected to be catalyzed by a previously unknown Ch1CoA dehydrogenase, the further conversion of Ch1,5CoA to benzoyl-CoA could also be assigned to the reverse activity of the class II benzoyl-CoA reductases in the cell extract, as described previously for the Geobacter metallireducens enzyme (15). Attempts to purify the Ch1CoA-oxidizing enzyme from cell extracts of S. aciditrophicus failed due to highly similar chromatographic binding properties for other proteins (e.g., ChCoA dehydrogenase and subunits of benzoyl-CoA reductase).
Adjacent to the gene for ChCoA dehydrogenase is a gene (gi 85858067; SYN_02587) annotated as an acyl-CoA dehydrogenase; the deduced amino acid sequence of its product is 44% identical to that of S. aciditrophicus ChCoA dehydrogenase, with a similar theoretical mass of 45 kDa (Fig. 5). The genetic context of the two acyl-CoA dehydrogenase genes indicates that they apparently form a transcription unit. For these reasons, attempts to heterologously express SYN_02587 were carried out.
Fig 5.
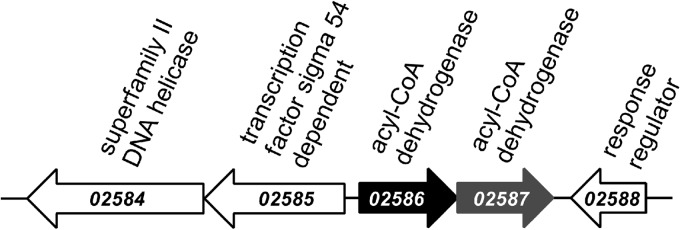
DNA region with the gene encoding ChCoA dehydrogenase (solid black). The numbers indicate the gene locus tag, and the annotations in the database are shown above the gene. Adjacent to SYN_02586, encoding the ChCoA dehydrogenase, a second gene (SYN_02587; solid gray), putatively coding for an acyl-CoA dehydrogenase-like protein, is present.
Heterologous expression, purification, and characterization of Ch1CoA dehydrogenase.
The start of translation of SYN_02587 was assigned to a TTG start codon (encoding leucine) in the annotated genome. Fifty-two base pairs downstream of the putative TTG start codon, an ATG start codon with a conserved upstream Shine-Dalgarno sequence (see Fig. S1 in the supplemental material) was identified. Additional sequence comparisons with related acyl-CoA dehydrogenases and their conserved domains strongly supported that the ATG codon encoded the start of the N-terminal amino acid sequence of the putative acyl-CoA dehydrogenase, and the cloning protocols were adjusted accordingly.
Taking into account the predicted alternative start of translation, the gene SYN_02587 was heterologously expressed in E. coli BL21 with a C-terminal six-His tag (see Table S1 in the supplemental material). The fractions obtained after purification by Ni-chelating affinity chromatography contained two enriched protein bands when analyzed by SDS-polyacrylamide gel electrophoresis (Fig. 6). UPLC-LTQ Orbitrap MS/MS analysis of tryptic digests using the MASCOT platform indicated that both proteins matched peptides from the anticipated SYN_02587 gene product (gi 85858067; 21 and 22% sequence coverage, respectively). The apparent mass of the subunits was slightly higher than expected due to the 6-fold His tag. The presence of two protein bands was not expected but was reproducible with three different enzyme expression preparations. The final pooled fractions obtained after gel filtration had the same ratio of the two protein bands, with the larger subunit being present to approximately 50% less abundance than the smaller one based on staining intensity. In summary, peptide analysis by mass spectrometry shows that both bands are encoded by SYN_05287; the reason for the different migrations remains unknown. The native molecular mass of the protein was determined by size exclusion chromatography and yielded a single protein peak corresponding to 150 ± 10 kDa, suggesting a tetrameric composition. The yellow enzyme showed a typical spectrum for flavin-containing enzymes (Fig. 4), with an FAD content of 4.2 per enzyme (1.05 FAD per monomer).
Fig 6.
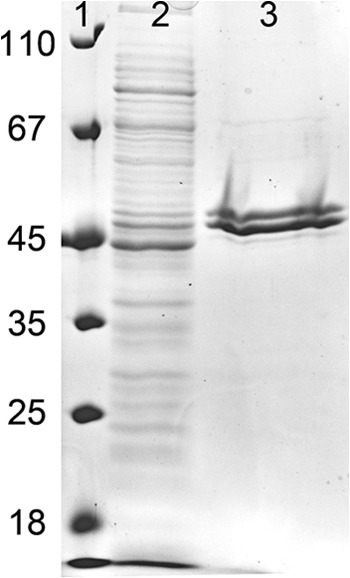
SDS gel of protein fractions during the enrichment of heterologously expressed Ch1CoA dehydrogenase. Lane 1, molecular mass standards; lane 2, supernatant after ultracentrifugation; lane 3, pooled fractions containing Ch1CoA dehydrogenase activity obtained after Ni-chelating affinity chromatography.
HPLC analysis revealed that the purified enzyme catalyzed the ferricenium hexafluorophosphate-dependent oxidation of Ch1CoA to Ch1,5CoA (the rate was 9.5 μmol min−1 mg−1). The formation of Ch1,5CoA was confirmed by comigration and an identical UV/Vis spectrum with an authentic standard. Surprisingly, Ch1,5CoA was further oxidized to benzoyl-CoA by purified Ch1CoA dehydrogenase, which rules out that benzoyl-CoA formation was catalyzed by a class II benzoyl-CoA reductase (Fig. 7). Assays with Ch1,5CoA as the substrate yielded benzoyl-CoA with a 5-fold lower specific activity than Ch1CoA dehydrogenation (2.1 μmol min−1 mg−1). The apparent substrate affinity constants were a Km value of <5 μM (Ch1CoA) and a Km value of 19 ± 7 μM (Ch1,5CoA). None of the following compounds tested served as a substrate for Ch1CoA dehydrogenase: ChCoA, 6-hydroxycyclohex-1-en-1-carboxyl-CoA, butyryl-CoA, glutaryl-CoA, succinyl-CoA, or cyclohexane carboxylate. Ti(III) citrate (5 mM) in the presence of methyl viologen (1 mM) served as the electron donor system for the reduction of Ch1,5CoA (0.2 mM) to Ch1CoA (20 μmol min−1 mg−1). No reduction of benzoyl-CoA to Ch1,5CoA or of Ch1CoA to ChCoA was observed using this assay.
Fig 7.

HPLC analysis of Ch1CoA dehydrogenase assay. The upper panel shows reactions catalyzed by Ch1CoA dehydrogenase. Oxidation of Ch1CoA (a) to Ch1,5CoA (b) in the presence of ferricenium (FcPF6) as an electron acceptor is shown. This product is further oxidized to benzoyl-CoA (c). HPLC diagrams of CoA ester analysis are shown after 0 min (A), 1 min (B), and 5 min (C). AU, absorption units.
DISCUSSION
In S. aciditrophicus, the regeneration of reducing equivalents during the fermentation of benzoate or crotonate to acetate is accomplished by the concomitant formation of cyclohexane carboxylate (1, 2). The newly identified ChCoA dehydrogenase and the Ch1CoA dehydrogenase are proposed to play essential roles in cyclohexane carboxylate formation during benzoate or crotonate formation. The proposal is based on the high specificity and affinity of the two acyl-CoA dehydrogenases for their individual substrates. The properties of these enzymes are summarized and compared in Table 2. The unexpected aromatization of Ch1,5CoA to benzoyl-CoA catalyzed by Ch1CoA dehydrogenase may also occur in vivo to some extent. Notably, benzoate was identified as a minor fermentation product during crotonate fermentation (1, 2) and may result from a side reaction of Ch1CoA dehydrogenase. While ChCoA and Ch1CoA dehydrogenation could be measured in both directions with appropriate electron accepting/donating systems, no benzoyl-CoA reduction by Ch1CoA dehydrogenase was observed. In benzoate fermentation, BCoA reduction to Ch1,5CoA must then be catalyzed by a class II benzoyl-CoA reductase, which was recently identified in the obligate anaerobe Geobacter metallireducens (9, 15) and predicted to be present in S. aciditrophicus (3, 16).
Table 2.
Properties of ChCoA and Ch1CoA dehydrogenases from S. aciditrophicusa
| Property | Profile for enzymeb |
|
|---|---|---|
| ChCoA dehydrogenase | Ch1CoA dehydrogenase | |
| Native molecular mass (kDa) | 150 ± 10 | 150 ± 10 |
| Subunit composition | α4 | α4 |
| Gene | SYN_02586 | SYN_02587 |
| Cofactor per subunit | 0.92 FAD (SB) | 0.41 FAD |
| 0.58 FAD (ht) | 0.67 FAD (after reconstitution) | |
| Substrates/inhibitors | ChCoA (Km, 22 ± 5 μM) | Ch1CoA (Km, <5 μM) |
| Ch1CoA (Ki, 49 ± 10 μM) | 1,5-DCoA (Km, 19 ± 7 μM) | |
| Sp act (U mg−1) | ChCoA, 52 ± 0.7 (SB) | Ch1CoA, 9.5 ± 0.3 |
| ChCoA, 28 ± 1 (ht) | 1,5-DCoA, 2.1 ± 0.4 | |
SB, purified from S. aciditrophicus cells; ht, protein heterologously expressed in E. coli.
Molecular masses, Km and Ki values, and specific activities are expressed as means ± SD.
The results obtained in this work support the earlier hypothesis that formation of cyclohexane carboxylate from crotonate proceeds via CoA-ester intermediates of the reverse benzoyl-CoA degradation pathway (2). The identification of a specific and highly active Ch1CoA dehydrogenase strongly suggests that during crotonate fermentation, crotonyl-CoA is reductively converted to Ch1,5CoA by identical enzymes of the oxidative branch of benzoate fermentation (Fig. 1). An energy-conserving synthetase or a transferase is likely to catalyze the final cyclohexane carboxylate-releasing step. This pathway represents an alternative route to the known cyclohexane carboxylate formation pathway from shikimate, which has been described for Streptomyces species and Alicyclobacillus species (17, 18).
Benzoate fermentation can be summarized as follows: 2 benzoate + 7H2O → 3 acetate + HCO3− + cyclohexane carboxylate. It involves three different natural electron donors/acceptors (Fig. 1): (i) most likely a reduced ferredoxin (Fdred) as an electron donor for benzoyl-CoA reduction, (ii) three NAD+ as acceptors for alcohol dehydrogenases acting on 6-OH-cyclohex-1-ene-1-carboxyl-CoA, 3-OH-pimeloyl-CoA, and 3-OH-butyryl-CoA, and (iii) an oxidized (ox) electron-transferring flavoprotein (ETF) as an acceptor for glutaryl-CoA dehydrogenase. In addition, two reduced ETFs, the typical acceptors for acyl-CoA dehydrogenases, are most probably used for the reductive branch (Fig. 1). The recycling of reducing equivalents formed/consumed during benzoate fermentation could then be summarized as follows: 3NADH + 2Fdox + ETFox → 3NAD+ + 3H+ + 2Fdred2− + ETFred2−.
Consequently, the NADH formed (E°′ = −320 mV) by the three alcohol dehydrogenase reactions has to be recycled both by endergonic redox processes (reduction of Fdox; E°′ = −500 mV) and by exergonic redox processes (reduction of ETFox; E°′ is approximately 0 mV). It is tempting to speculate that endergonic and exergonic electron transfer steps are coupled by an electron bifurcation process as has been described for the ETF from Clostridium kluyveri (19).
The fermentative dismutation of crotonate to acetate and cyclohexane carboxylate follows the equation 6(crotonate) + HCO3− + 5H2O → 9(acetate) + cyclohexane carboxylate and does not necessarily involve a ferredoxin. The recycling of reducing equivalents formed/consumed during crotonate fermentation is expected to proceed via the exergonic reduction of ETFox by NADH: 3NADH + 3ETFox → 3NAD+ + 3H+ + 3ETFred2−.
A Ch1CoA dehydrogenase has been studied in Rhodopseudomonas palustris, where it is involved in cyclohexane carboxylate degradation. In this organism, aliB (gi 39647576) was identified as the encoding gene (20, 21). Interestingly, the AliB and SYN_02586 gene products catalyze identical reactions but share only a little sequence similarity (29%), suggesting that their respective genes evolved independently from the other acyl-CoA dehydrogenase. Sequence analyses of the protein sequences of SYN_02586 and SYN_02587 showed highest similarities (up to 70% amino acid sequence identity) to acyl-CoA dehydrogenase-like deduced gene products from Geobacter species (see Fig. S2 in the supplemental material). Since Geobacter species neither have a fermentative metabolism nor have been reported to utilize cyclohexane carboxylate as a carbon source, the function of the ChCoA- and Ch1CoA-dehydrogenase-like proteins in Geobacter species remains unknown.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the Deutsche Forschungsgemeinschaft (BO 1565/9-2).
We thank Alexander Schmidt (Konstanz) for growing Syntrophus aciditrophicus and M. J. McInerney (Norman) for carefully editing the manuscript.
Footnotes
Published ahead of print 10 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00322-13.
REFERENCES
- 1. Elshahed MS, McInerney MJ. 2001. Benzoate fermentation by the anaerobic bacterium Syntrophus aciditrophicus in the absence of hydrogen-using microorganisms. Appl. Environ. Microbiol. 67:5520–5525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mouttaki H, Nanny MA, McInerney MJ. 2007. Cyclohexane carboxylate and benzoate formation from crotonate in Syntrophus aciditrophicus. Appl. Environ. Microbiol. 73:930–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McInerney MJ, Rohlin L, Mouttaki H, Kim U, Krupp RS, Rios-Hernandez L, Sieber J, Struchtemeyer CG, Bhattacharyya A, Campbell JW, Gunsalus RP. 2007. The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc. Natl. Acad. Sci. U. S. A. 104:7600–7605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elshahed MS, Bhupathiraju VK, Wofford NQ, Nanny MA, McInerney MJ. 2001. Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by “Syntrophus aciditrophicus” strain SB in syntrophic association with H(2)-using microorganisms. Appl. Environ. Microbiol. 67:1728–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peters F, Shinoda Y, McInerney MJ, Boll M. 2007. Cyclohexa-1,5-diene-1-carbonyl-coenzyme A (CoA) hydratases of Geobacter metallireducens and Syntrophus aciditrophicus: evidence for a common benzoyl-CoA degradation pathway in facultative and strict anaerobes. J. Bacteriol. 189:1055–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuntze K, Shinoda Y, Mouttaki H, McInerney MJ, Vogt C, Richnow HH, Boll M. 2008. 6-Oxocyclohex-1-ene-1-carbonyl-coenzyme A hydrolases from obligately anaerobic bacteria: characterization and identification of its gene as a functional marker for aromatic compounds degrading anaerobes. Environ. Microbiol. 10:1547–1556 [DOI] [PubMed] [Google Scholar]
- 7. Wischgoll S, Taubert M, Peters F, Jehmlich N, von Bergen M, Boll M. 2009. Decarboxylating and nondecarboxylating glutaryl-coenzyme A dehydrogenases in the aromatic metabolism of obligately anaerobic bacteria. J. Bacteriol. 191:4401–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mouttaki H, Nanny MA, McInerney MJ. 2009. Metabolism of hydroxylated and fluorinated benzoates by Syntrophus aciditrophicus and detection of a fluorodiene metabolite. Appl. Environ. Microbiol. 75:998–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kung JW, Baumann S, von Bergen M, Müller M, Hagedoorn P, Hagen WR, Boll M. 2010. Reversible biological Birch reduction at an extremely low redox potential. J. Am. Chem. Soc. 132:9850–9856 [DOI] [PubMed] [Google Scholar]
- 10. Fuchs G, Boll M, Heider J. 2011. Microbial degradation of aromatic compounds—from one strategy to four. Nat. Rev. Microbiol. 9:803–816 [DOI] [PubMed] [Google Scholar]
- 11. Thiele B, Rieder O, Jehmlich N, von Bergen M, Müller M, Boll M. 2008. Aromatizing cyclohexa-1,5-diene-1-carboxyl-coenzyme A oxidase. Characterization and its role in anaerobic aromatic metabolism. J. Biol. Chem. 283:20713–20721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gross GG, Zenk MH. 1966. Darstellung und Eigenschaften von Coenzym A-Thiolestern substituierter Zimtsauren. Z. Naturforschg. 21b:683–690 [Google Scholar]
- 13. McInerney MJ, Bryant MP, Pfennig N. 1979. Anaerobic bacterium that degrades fatty acids in syntrophic association with methanogens. Arch. Microbiol. 122:129–135 [DOI] [PubMed] [Google Scholar]
- 14. Kuntze K, Kiefer P, Baumann S, Seifert J, von Bergen M, Vorholt JA, Boll M. 2011. Enzymes involved in the anaerobic degradation of meta-substituted halobenzoates. Mol. Microbiol. 82:758–769 [DOI] [PubMed] [Google Scholar]
- 15. Kung JW, Löffler C, Dörner K, Heintz D, Gallien S, van Dorsselaer A, Friedrich T, Boll M. 2009. Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc. Natl. Acad. Sci. U. S. A. 106:17687–17692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Löffler C, Kuntze K, Vazquez JR, Rugor A, Kung JW, Böttcher A, Boll M. 2011. Occurrence, genes and expression of the W/Se-containing class II benzoyl-coenzyme A reductases in anaerobic bacteria. Environ. Microbiol. 13:696–709 [DOI] [PubMed] [Google Scholar]
- 17. Floss HG, Cho H, Reynolds KA, Kennedy E, Moore BS, Beale JM, Mocek U, Poralla K. 1992. Secondary-metabolite biosynthesis and metabolism. Diversions of the shikimate pathway—the biosynthesis of cyclohexanecarboxylic acid, p 77–88 In Petroski RJ, McCormick SP. (ed), Environmental science research, vol 44 Plenum Press, New York, NY [Google Scholar]
- 18. Moore BS, Walker K, Tornus I, Handa S, Poralla K, Floss HG. 1997. Biosynthesis of ω-cycloheptyl fatty acids in Alicyclobacillus cycloheptanicus. Formation of cycloheptanecarboxylic acid from phenylacetic acids. J. Org. Chem. 62:2173–2185 [DOI] [PubMed] [Google Scholar]
- 19. Li F, Hinderberger J, Seedorf H, Zhang J, Buckel W, Thauer RK. 2008. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190:843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pelletier DA, Harwood CS. 2000. 2-Hydroxycyclohexanecarboxyl coenzyme A dehydrogenase, an enzyme characteristic of the anaerobic benzoate degradation pathway used by Rhodopseudomonas palustris. J. Bacteriol. 182:2753–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Larimer FW, Chain P, Hauser L, Lamerdin J, Malfatti S, Do L, Land ML, Pelletier DA, Beatty JT, Lang AS, Tabita FR, Gibson JL, Hanson TE, Bobst C, Torres Y, Torres JL, Peres C, Harrison FH, Gibson J, Harwood CS. 2004. Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 22:55–61 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.