

Abstract
Flavobacterium johnsoniae cells move rapidly over surfaces by gliding motility. Gliding results from the movement of adhesins such as SprB and RemA along the cell surface. These adhesins are delivered to the cell surface by a Bacteroidetes-specific secretion system referred to as the type IX secretion system (T9SS). GldN, SprE, SprF, and SprT are involved in secretion by this system. Here we demonstrate that GldK, GldL, GldM, and SprA are each also involved in secretion. Nonpolar deletions of gldK, gldL, or gldM resulted in the absence of gliding motility and in T9SS defects. The mutant cells produced SprB and RemA proteins but failed to secrete them to the cell surface. The mutants were resistant to phages that use SprB or RemA as a receptor, and they failed to attach to glass, presumably because of the absence of cell surface adhesins. Deletion of sprA resulted in similar but slightly less dramatic phenotypes. sprA mutant cells failed to secrete SprB and RemA, but cells remained susceptible to some phages and retained some limited ability to glide. The phenotype of the sprA mutant was similar to those previously described for sprE and sprT mutants. SprA, SprE, and SprT are needed for secretion of SprB and RemA but may not be needed for secretion of other proteins targeted to the T9SS. Genetic and molecular experiments demonstrate that gldK, gldL, gldM, and gldN form an operon and suggest that the proteins encoded by these genes may interact to form part of the F. johnsoniae T9SS.
INTRODUCTION
Cells of Flavobacterium johnsoniae move rapidly over surfaces by gliding motility. Flavobacterium gliding motility does not involve flagella or pili but instead relies on novel machinery that appears to be confined to members of the large and diverse phylum Bacteroidetes (1, 2). Genetic experiments identified Gld, Spr, and Rem proteins with roles in motility. A motor comprised of some of the Gld proteins is thought to propel cell surface adhesins such as SprB and RemA. These adhesins interact with the substratum or with polysaccharides that coat the substratum, and the action of the motor on the adhesins propels the cell (3, 4).
Some of the Gld and Spr proteins form a novel protein secretion system originally referred to as the Por protein secretion system (5–7). More recently, this has been called the type IX secretion system (T9SS) (2, 8), because the known components are not similar in sequence to proteins of the previously described type I to type VIII secretion systems (9–11). T9SSs are common in members of the phylum Bacteroidetes but are apparently not found outside this phylum (2). Proteins secreted by T9SSs have predicted N-terminal type 1 signal peptides and are thought to rely on the Sec system for export across the cytoplasmic membrane. They also have conserved C-terminal domains (CTDs) that appear to direct them to the T9SS for secretion across the outer membrane (4, 12–15).
T9SSs have been studied in the nonmotile periodontal pathogen Porphyromonas gingivalis and in F. johnsoniae. The P. gingivalis T9SS is involved in secretion of gingipain proteases and other virulence factors, and the F. johnsoniae T9SS is needed for secretion of the cell surface motility adhesins SprB and RemA and for secretion of a chitinase. In F. johnsoniae, GldN, SprE, SprF, and SprT (which are related to the P. gingivalis T9SS proteins PorN, PorW, PorP, and PorT, respectively) have been demonstrated to have roles in secretion (5–7, 16). F. johnsoniae GldK, GldL, GldM, and SprA were originally identified as proteins required for gliding motility (17, 18). Studies of P. gingivalis revealed that homologs of these proteins (PorK, PorL, PorM, and Sov, respectively) are required for gingipain secretion (7, 19). F. johnsoniae GldK, GldL, GldM, and SprA were assumed to also be involved in protein secretion (2), but the evidence supporting this had not been presented. Here we demonstrate that F. johnsoniae GldK, GldL, GldM, and SprA are each required for secretion of SprB and RemA and for utilization of chitin. gldK, gldL, gldM, and gldN form an operon, and the products of these genes each localize to the cell envelope, where they may interact to form a complex involved in protein secretion.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
F. johnsoniae ATCC 17061 strain UW101 was the wild-type strain used in this study. The streptomycin-resistant rpsL mutant of UW101 (CJ1827) was used to construct strains with unmarked deletions (20). F. johnsoniae strains were grown in Casitone-yeast extract (CYE) medium (21) at 25°C. To observe colony spreading, F. johnsoniae cells were grown at 25°C on PY2 medium supplemented with 10 g of agar per liter (22). Motility medium (MM) was used to observe movement of individual cells in wet mounts (23). Strains and plasmids used in this study are listed in Table 1, and primers used in this study are listed in Table S1 in the supplemental material. Sites of transposon insertions in gldK, gldL, gldM, and gldN are illustrated in Fig. 1, as are the regions carried on plasmids used for complementation experiments. The plasmids used for complementation were all derived from pCP1 and have copy numbers of approximately 10 in F. johnsoniae (21, 22, 24). Antibiotics were used at the following concentrations when needed: ampicillin at 100 μg/ml, cefoxitin at 100 μg/ml, erythromycin at 100 μg/ml, streptomycin at 100 μg/ml, and tetracycline at 20 μg/ml.
Table 1.
Strains and plasmids used in this study
| F. johnsoniae strain or plasmid | Genotype and/or descriptiona | Reference(s) or source |
|---|---|---|
| Strains | ||
| UW101 (ATCC 17061) | Wild type | 38, 39 |
| UW102-57 | Spontaneous gldK mutant | 17, 26 |
| UW102-141 | Nitrosoguanidine-induced gldK mutant | 17, 26 |
| CJ1372 | gldK::HimarEm1 (Emr) | 17 |
| CJ1373 | gldK::HimarEm1 (Emr) | 17 |
| CJ1827 | rpsL2 (Smr); wild-type strain used in construction of deletion mutants | 20 |
| CJ1922 | rpsL2 ΔsprB (Smr) | 20 |
| CJ1984 | rpsL2 ΔremA (Smr) | 4 |
| CJ1985 | rpsL2 ΔsprB ΔremA (Smr) | 4 |
| CJ2083 | rpsL2 remA::myc-tag-1 (Smr) | 4 |
| CJ2090 | rpsL2 Δ(gldN gldO) (Smr) | 4 |
| CJ2122 | rpsL2 ΔgldK (Smr) | This study |
| CJ2140 | rpsL2 ΔgldK in remA::myc-tag-1 background (Smr) | This study |
| CJ2157 | rpsL2 ΔgldL (Smr) | This study |
| CJ2262 | rpsL2 ΔgldM (Smr) | This study |
| CJ2263 | rpsL2 ΔgldM in remA::myc-tag-1 background (Smr) | This study |
| CJ2281 | rpsL2 ΔgldL in remA::myc-tag-1 background (Smr) | This study |
| CJ2089 | rpsL2 Δ(gldN gldO) remA::myc-tag-1 (Smr) | 4 |
| CJ2302 | rpsL2 ΔsprA (Smr) | This study |
| CJ2317 | rpsL2 ΔsprA in remA::myc-tag-1 background (Smr) | This study |
| CJ2327 | sprT disruption in remA::myc-tag-1 background (Emr Smr) | This study |
| KDF001 | sprT mutant (Emr) | 7 |
| FJ149 | sprE mutant (Emr) | 6 |
| Plasmids | ||
| pET30a | Protein expression vector; Kmr | Novagen |
| pCP23 | E. coli-F. johnsoniae shuttle plasmid; Apr (Tcr) | 22 |
| pCP29 | E. coli-F. johnsoniae shuttle plasmid; Apr (Cfr Emr) | 24 |
| pRR51 | rpsL-containing suicide vector; Apr (Emr) | 20 |
| pJVB4 | 1.3-kbp fragment of gldK amplified with primer pair 704/705 and ligated into EcoRI-, SalI-digested pET30a; Kmr | This study |
| pJVB2 | 0.6-kbp fragment of gldL amplified with primer pair 706/707 and ligated into EcoRI-, SalI-digested pET30a; Kmr | This study |
| pJVB6 | 1.3-kbp fragment of gldM amplified with primer pair 708/709 and ligated into EcoRI-, SalI-digested pET30a; Kmr | This study |
| pAB18 | 2.4-kbp region downstream of gldL amplified with primers 1199 and 1200 and ligated into XbaI-, SalI-digested pRR51; Apr (Emr) | This study |
| pAB19 | Construct used to delete gldL; 2.2-kbp region upstream of gldL amplified with primer pair 1197/1198 and ligated into BamHI-, XbaI-digested pAB18; Apr (Emr) | This study |
| pAB30 | Construct used to delete sprA; 2.5 kbp upstream and 2.5 kbp downstream of sprA amplified using primer pairs 1333/1334 and 1335/1336, respectively, and ligated into BamHI-, SalI-digested pRR51; Apr (Emr) | This study |
| pJJ01 | Construct used to delete gldK; 1.9-kbp region upstream and 1.9-kbp region downstream of gldK amplified using primer pairs 1209/1210 and 1211/1212, respectively, and ligated into BamHI-, SphI-digested pRR51; Apr (Emr) | This study |
| pJJ02 | Construct used to delete gldM; 2.6-kbp region upstream and 2.4-kbp region downstream of gldM amplified using primer pairs 1237/1214 and 1215/1238, respectively, and ligated into BamHI-, SphI-digested pRR51; Apr (Emr) | This study |
| pTB99 | pCP23 carrying gldK; Apr (Tcr) | 17 |
| pTB81a | pCP23 carrying gldL; Apr (Tcr) | 17 |
| pTB94a | pCP23 carrying gldM; Apr (Tcr) | 17 |
| pTB79 | pCP23 carrying gldN; Apr (Tcr) | 17 |
| pKF002 | pCP23 carrying sprT; Apr (Tcr) | 7 |
| pSN48 | pCP23 carrying sprA; Apr (Tcr) | 18 |
Antibiotic resistance phenotypes are as follows: ampicillin, Apr; cefoxitin, Cfr; erythromycin, Emr; streptomycin, Smr; tetracycline, Tcr. Unless indicated otherwise, the antibiotic resistance phenotypes are those expressed in E. coli. The antibiotic resistance phenotypes given in parentheses are those expressed in F. johnsoniae but not in E. coli.
Fig 1.

Map of the gldKLMN region. Numbers below the map refer to kilobase pairs of sequence. The sites of HimarEm insertions are indicated by triangles. Orientations of HimarEm insertions are indicated by the direction in which the triangles are pointing, with triangles pointing to the right having inverted repeat 2 (IR2) on the right side. The regions of DNA carried by plasmids used in this study are indicated beneath the map.
Deletion of gldK, gldL, gldM, and sprA.
Unmarked deletions were made as previously described (20). To make the gldL deletion, a 2.4-kbp fragment downstream of gldL was amplified by PCR using Phusion DNA polymerase (New England BioLabs, Ipswich, MA) and primers 1199 (introducing an XbaI site) and 1200 (introducing a SalI site). The fragment was digested with XbaI and SalI and ligated into pRR51 that had been digested with the same enzymes, to generate pAB18. A 2.2-kbp fragment spanning the upstream region of gldL was amplified by PCR with primers 1197 (introducing a BamHI site) and 1198 (introducing an XbaI site). The fragment was digested with BamHI and XbaI and fused to the region downstream of gldL by ligation with pAB18 that had been digested with the same enzymes, to generate the deletion construct pAB19. Plasmid pAB19 was introduced into streptomycin-resistant wild-type F. johnsoniae strain CJ1827 and remA::myc-tag-1 strain CJ2083 (4) by triparental conjugation, and gldL deletion mutants were isolated as previously described (20). Deletion of gldL was confirmed by PCR amplification using primers 690 and 707, which flank the gldL coding sequence. gldK, gldM, and sprA were deleted in wild-type CJ1827 and in remA::myc-tag-1 strain CJ2083 by the same approach, using the primers listed in Table S1 in the supplemental material and the plasmids listed in Table 1.
Transduction of the sprT mutation into CJ2083 (rpsL2 remA::myc-tag-1).
The sprT mutation in strain KDF001 was transduced into CJ2083 (rpsL2 remA::myc-tag-1) essentially as previously described (5), using phage ϕCj54 and selecting for erythromycin resistance.
RT-PCR to characterize the operon comprised of gldK, gldL, gldM, and gldN.
RNA was extracted from wild-type F. johnsoniae UW101 cells as described previously (16). Briefly, wild-type cells were grown in 25 ml of MM overnight at 25°C without shaking. Cells were harvested by centrifugation at 4,000 × g for 10 min, and the pellet was suspended in 450 μl of MM. RNAProtect (Qiagen, Valencia, CA) was added, and pellet was collected as described previously (16). RNA was extracted and purified by using the RNeasy minikit (Qiagen) according to the manufacturer's instructions, except that RNasin (Promega Corp., Madison, WI) and RQ1 RNase-free DNase (Promega) were used as described previously (16). RNA concentrations (optical density at 260 nm [OD260]) and purity (OD260/280) were determined by UV spectroscopy, and RNA integrity was verified by visualizing the intensity of the 16S and 23S rRNA bands on a 1% agarose gel.
Reverse transcriptase PCR (RT-PCR) was used to determine the transcriptional organization of gldK, gldL, gldM, and gldN. cDNA was generated by using the SuperScript first-strand synthesis kit for RT-PCR (Invitrogen Corp., Carlsbad, CA) according to the manufacturer's instruction. RNA was reverse transcribed by using gene-specific primer 695 essentially as described previously (16). For PCR, antisense primers used to generate the cDNA were used in various combinations with sense primers.
Expression of recombinant GldK, GldL, and GldM for antibody production.
A 1,308-bp fragment of gldK was amplified by using Phusion DNA polymerase and primers 705 (introducing a SalI site) and 704 (introducing an EcoRI site). The product was digested with EcoRI and SalI and inserted into pET30a (Novagen, Madison, WI) that had been digested with the same enzymes, to generate pJVB4. A 607-bp fragment of gldL was amplified by using primers 707 (introducing a SalI site) and 706 (introducing an EcoRI site), and the product was digested and inserted into pET30a as described above, generating pJVB2. A 1,311-bp fragment of gldM was amplified by using primer 709 (introducing a SalI site) and primer 708. The PCR product was digested with SalI and EcoRI (which cuts within gldM) and inserted into pET30a as described above, generating pJVB6. pJVB4, pJVB2, and pJVB6 encode proteins with amino-terminal His-tagged peptides fused to the C-terminal 434, 174, and 426 amino acids of GldK, GldL, and GldM, respectively.
pJVB4, pJVB2, and pJVB6 were introduced into Escherichia coli Rosetta2(DE3) (Novagen), which produces seven rare tRNAs required for efficient expression of F. johnsoniae proteins in E. coli (3, 18). Expression was induced by addition of 1.0 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and incubation for 3 h at 37°C. Cells were collected by centrifugation and disrupted by using a French pressure cell, and recombinant proteins were purified by Ni affinity chromatography. Polyclonal antibodies against recombinant GldK, GldL, and GldM were produced and affinity purified using the recombinant proteins by Proteintech Group, Inc. (Chicago, IL). Antibodies were also raised against recombinant GldN, as previously described (5).
Detection and localization of GldK, GldL, and GldM.
Wild-type and mutant cells of F. johnsoniae were grown to mid-log phase in CYE medium at 25°C. Cells were washed twice in sterile distilled water by centrifugation at 4,000 × g and were lysed by incubation for 5 min at 100°C in SDS-PAGE loading buffer. Proteins (15 μg per lane) were separated by SDS-PAGE, and Western blotting was performed as described previously (5). To determine protein localization, wild-type cells were grown to mid-log phase in MM at 25°C with shaking. The cells were washed twice with sterile distilled water, and EDTA-free Halt protease inhibitor cocktail was added (Thermo Fisher Scientific, Rockford, IL). The cells were disrupted by using a French pressure cell and fractionated into soluble inner membrane (Sarkosyl-soluble) and outer membrane (Sarkosyl-insoluble) fractions essentially as described previously (25). Proteins were separated by SDS-PAGE, equal amounts of each fraction based on the starting material were loaded per lane, and Western blotting was performed as described previously (5).
Analysis of wild-type and mutant cells for production and secretion of SprB and Myc-tagged RemA.
Cells were grown to mid-exponential phase in CYE medium at 25°C. Production of SprB and Myc-tagged RemA was determined by Western blot analyses using antisera against SprB and RemA, respectively, as previously described (4, 5). Secretion of SprB was examined essentially as previously described (6). Briefly, cells were grown overnight in MM at 25°C. Purified anti-SprB (1 μl of a 1:10 dilution of a 300-mg/liter stock), 0.5-μm-diameter protein G-coated polystyrene spheres (1 μl of a 0.1% stock preparation; Spherotech, Inc., Libertyville, IL), and bovine serum albumin (BSA) (1 μl of a 1% solution) were added to 7 μl of cells (approximately 5 × 108 cells per ml) in MM. The cells were introduced into a tunnel slide (4) and examined by phase-contrast microscopy at 25°C. Samples were examined 2 min after spotting, and images were recorded for 30 s to determine the percentage of cells that had anti-SprB-coated spheres attached to them. The major advantage of this method is that proteins are detected on the surface of live cells that have not been chemically or enzymatically altered. Surface-localized Myc-tagged RemA was detected similarly, except that antisera against the Myc tag (EQKLISEEDL; AbCam, Cambridge, MA) was used instead of anti-SprB (4).
Microscopic observations of cell movement.
Wild-type and mutant cells were examined for movement over glass by phase-contrast microscopy. Cells were grown overnight in MM at 25°C without shaking, as previously described (23). Simple tunnel slides were constructed by using double-stick tape, glass microscope slides, and glass coverslips, as previously described (4). Cells in MM were introduced into the tunnel slides, incubated for 5 min, and observed for motility by using an Olympus BH2 phase-contrast microscope with a heated stage at 25°C. Images were recorded with a Photometrics CoolSNAPcf2 camera and analyzed by using MetaMorph software (Molecular Devices, Downingtown, PA).
Measurements of bacteriophage sensitivity.
The bacteriophages active against F. johnsoniae that were used in this study were ϕCj1, ϕCj13, ϕCj23, ϕCj28, ϕCj29, ϕCj42, ϕCj48, and ϕCj54 (26–28). Sensitivity to bacteriophages was determined essentially as previously described, by spotting 5 μl of phage lysates (109 PFU/ml) onto lawns of cells in CYE overlay agar (5). The plates were incubated for 24 h at 25°C to observe lysis.
Measurement of chitin utilization.
The ability of F. johnsoniae to utilize chitin was assayed as described previously (5). F. johnsoniae cells were grown overnight in MM without shaking at 25°C. Two microliters of cells was spotted onto MYA-chitin containing appropriate antibiotics, and plates were incubated for 2.5 days.
Microscopic observations of cell attachment.
Wild-type and mutant cells of F. johnsoniae were examined for attachment to glass by using a Petroff-Hausser counting chamber as previously described, with slight modifications (5). Cells were grown overnight in MM without shaking at 25°C. The culture was diluted with an equal volume of fresh MM and incubated at 25°C without shaking until a cell density equivalent to an OD600 of 0.4 was reached. Cells (2.5 μl) were added to a Petroff-Hausser counting chamber, covered with a glass coverslip, and allowed to incubate for 2 min at 25°C. The number of cells attached to 9 randomly selected 0.03-mm2 regions of the glass coverslip was determined.
RESULTS
gldK, gldL, gldM, and gldN form an operon.
Previous results indicated that gldL, gldM, and gldN are cotranscribed (17). The situation regarding gldK, which lies upstream of gldL, was less clear. It was suggested that gldK was transcribed separately, but the gldK transcript was not detected by Northern blot analysis, leaving the operon structure uncertain. Complementation analyses used to determine operon structure were also inconclusive, because moderate overexpression of gldK and gldL together in wild-type cells resulted in dramatic decreases in growth and motility, complicating analysis of polar effects of mutations (17). To understand the importance of GldK, GldL, and GldM to gliding motility and protein secretion, we revisited the transcriptional organization of these genes.
The ability of pTB99, which carries gldK, to complement various gldK mutants was examined. Two of the mutants (UW102-57 and UW102-141) had single-base substitutions in gldK, whereas the other two (CJ1372 and CJ1373) had HimarEm transposon insertions in gldK. The sites of the mutations in UW102-57 and UW102-141 were determined by amplification and sequencing. UW102-57 had an “A”-to-“T” substitution at position 1054, numbered from the A of the gldK start codon, and UW102-141 had a G-to-A substitution at position 1158, as previously reported (17). The substitutions converted the codons for R352 and W386, respectively, to stop codons. pTB99 restored motility to UW102-57 and UW102-141 but failed to restore motility to either of the gldK HimarEm mutants (Fig. 2). These results suggested that the base substitution mutations resulted in nonpolar mutations, whereas the HimarEm insertions may have exhibited polar effects on downstream genes.
Fig 2.
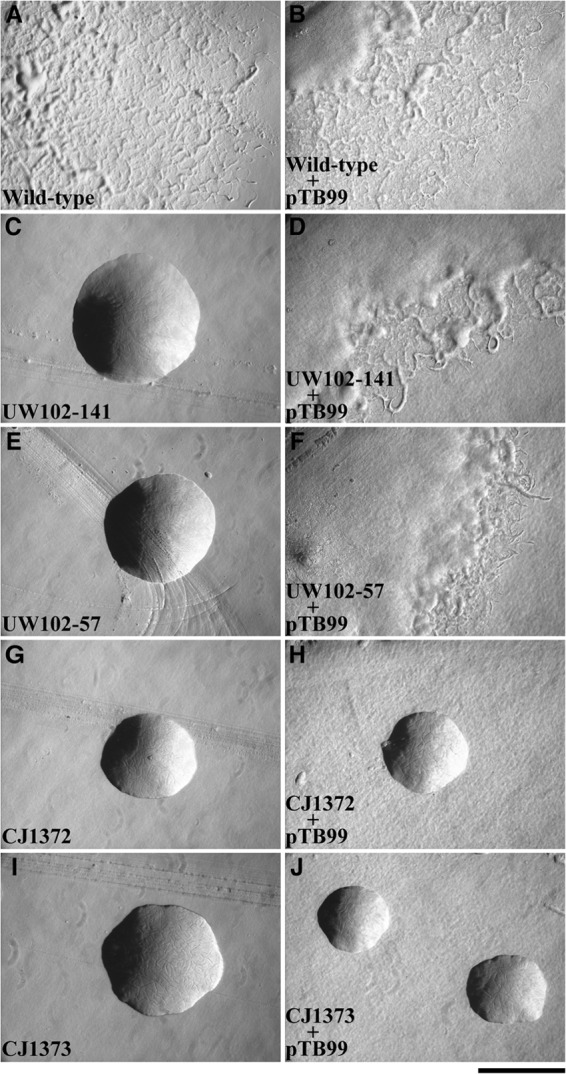
Photomicrographs of F. johnsoniae colonies. Colonies were incubated at 25°C on PY2 agar for 36 h. Photomicrographs were taken with a Photometrics Cool-SNAPcf2 camera mounted on an Olympus IMT-2 phase-contrast microscope. (A) Wild-type F. johnsoniae UW101. (B) F. johnsoniae UW101 with pTB99, which carries gldK. (C) gldK point mutant strain UW102-141. (D) UW102-141 complemented with pTB99. (E) gldK point mutant strain UW102-57. (F) UW102-57 complemented with pTB99. (G) gldK Himar mutant strain CJ1372. (H) CJ1372 with pTB99. (I) gldK Himar mutant strain CJ1373. (J) CJ1373 with pTB99. The bar indicates 1 mm and applies to all panels.
To explore the issue of polarity more directly, we examined the levels of GldK, GldL, GldM, and GldN proteins in HimarEm-induced GldK mutants. GldK, GldL, and GldM were overexpressed in E. coli, and antisera were raised against each protein. These proteins were each detected in cells of wild-type F. johnsoniae, but they were absent from appropriate gld deletion mutants (Fig. 3). Western blot analysis revealed that strains CJ1372 and CJ1373, which have transposon insertions in gldK, not only lacked GldK protein but also had little if any GldL, GldM, and GldN proteins (Fig. 3 and data not shown). Similarly, strains with transposon insertions in gldL produced little GldM or GldN, and strains with transposon insertions in gldM were deficient in GldN (data not shown). The results provide additional evidence of polarity of the gldK mutations and suggest that gldK, gldL, gldM, and gldN may be cotranscribed.
Fig 3.

Western blot immunodetection of GldK (A), GldL (B), GldM (C), and GldN (D) in whole-cell extracts of wild-type, mutant, and complemented strains of F. johnsoniae. The wild-type strain was CJ1827 (rpsL2), the gldK Himar mutant was CJ1372, and the strains with deletions in gldK, gldL, gldM, and gldNO were CJ2122, CJ2157, CJ2262, and CJ2090, respectively. pTB99, pTB81a, pTB94a, and pTB79 express GldK, GldL, GldM, and GldN, respectively, and thus complement the indicated mutants. Fifteen micrograms of protein was loaded into each lane.
We recently developed a method to generate in-frame deletions of F. johnsoniae genes (20). By using this method, strains with deletions of gldK, gldL, and gldM were generated. In contrast to the transposon mutants described above, CJ2122 (ΔgldK) produced GldL, GldM, and GldN, indicating that the mutation was nonpolar and that the GldK protein was not required for production of GldL, GldM, and GldN (Fig. 3). The gldK deletion mutant produced wild-type levels of GldL and GldM (Fig. 3B and C) but appeared to produce less GldN (Fig. 3D). Complementation of the gldK deletion mutant with pTB99, which carries gldK, restored GldK and GldN to wild-type levels. Restoration of GldN levels by expression of GldK argues against a polar effect of the gldK deletion mutation on expression of gldN. Analysis of CJ2157 (ΔgldL) and CJ2262 (ΔgldM) gave similar results. CJ2157 failed to produce GldL protein but produced GldM and GldN, and CJ2262 failed to produce GldM protein but produced GldN (Fig. 3C and D). The strains with deletions in gldL or gldM were similar to strains with gldK deletions in that they appeared to have low levels of GldN protein. One possible explanation for these results is that GldK, GldL, GldM, and GldN may form a complex, and GldN may be unstable in the absence of any of its partners. This may also explain the low levels of GldL observed in CJ2262 (ΔgldM) cells (Fig. 3B) and the reduced levels of GldK protein in strains with mutations in gldL, gldM, and gldN (Fig. 3A).
The results presented above suggest that gldK, gldL, gldM, and gldN form an operon. RT-PCR analysis confirmed the presence of a transcript spanning gldK, gldL, gldM, and gldN (Fig. 4). A predicted Flavobacterium consensus promoter sequence (TANNTTTG [29]) was identified 97 bp upstream of the gldK start codon, and 5′ rapid amplification of cDNA ends (RACE) analysis identified the start site of transcription 5 bp downstream of the final G of this promoter sequence (17). An inverted repeat (AAAAAACTCTTACTACCTTTGTAGTAAGAGTTTTTT), which may function as a transcription terminator, was present 22 bp downstream of the gldN stop codon. Genetic and molecular evidence previously demonstrated that gldO, which lies downstream of the gldKLMN operon and which is partially redundant with gldN, is transcribed independently. Transposon insertions in gldN were not polar on gldO, and the gldN mutants produced GldO protein (5).
Fig 4.

RT-PCR analysis of the gldKLMN operon. (A) Map of the gldKLMN operon with primers used for RT-PCR (numbered arrows). (B) RT-PCR of wild-type F. johnsoniae UW101 RNA. Reverse transcription was carried out with primer 695, which is complementary to a sequence internal to gldN. PCR was conducted by using the primer pairs shown. For each primer pair, three reaction mixtures were loaded onto the gel: a positive-control PCR mixture with F. johnsoniae chromosomal DNA as the template (lanes 2, 5, 8, and 11), a no-RT control reaction mixture (lanes 3, 6, 9, and 12), and an RT-PCR mixture (lanes 4, 7, 10, and 13). Lane 1, DNA ladder as a size standard; lanes 2 to 4, primer pair 685/705; lanes 5 to 7, primer pair 690/695; lanes 8 to 10, primer pair 616/695; lanes 11 to 13, primer pair 692/695.
GldK, GldL, and GldM are each required for gliding.
As previously reported, transposon insertions in gldK, gldL, and gldM result in a complete loss of motility (17). Given the concerns regarding polarity of these mutations, we analyzed the phenotypes of the in-frame deletions to determine the roles of the individual proteins. Cells with deletion mutations in gldK, gldL, or gldM were completely nonmotile. They formed nonspreading colonies on agar media (Fig. 5) and exhibited no cell movements in wet mounts on glass (see Movies S1 to S3 in the supplemental material). Cells of the deletion mutants were also examined by time-lapse microscopy for possible slow or intermittent movements on glass or agar using methods previously described (2), but no movements were observed. In each case, complementation with the appropriate gene restored motility. Complementation of the gldM deletion mutant was incomplete, perhaps because expression of gldM from a plasmid did not result in optimal GldM levels or did not allow optimal formation of a complex with the other Gld proteins. The results indicate that GldK, GldL, and GldM are each essential for gliding motility. Previous experiments demonstrated that the presence of GldN, or its paralog GldO, is also required for gliding (5).
Fig 5.
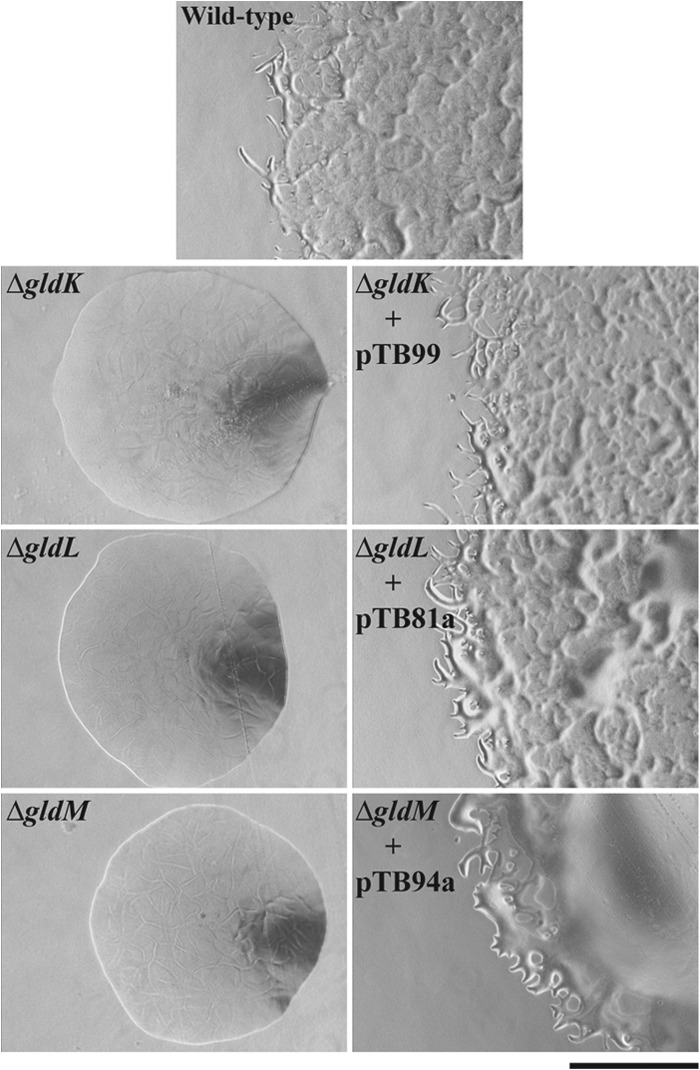
gldK, gldL, and gldM are each required for formation of spreading colonies. Colonies were grown for 36 h at 25°C on PY2 agar medium. Photomicrographs were taken with a Photometrics Cool-SNAPcf2 camera mounted on an Olympus IMT-2 phase-contrast microscope. The wild-type strain was CJ1827. CJ1827, CJ2122 (ΔgldK), CJ2157 (ΔgldL), and CJ2262 (ΔgldM) carried control vector pCP23 so that all strains were resistant to tetracycline and could be grown on identical media. pTB99, pTB81a, and pTB94a express GldK, GldL, and GldM, respectively. The bar indicates 0.5 mm and applies to all panels.
GldK, GldL, and GldM are each required for secretion of the cell surface motility adhesins SprB and RemA.
Components of the F. johnsoniae T9SS, such as GldN, SprE, SprF, and SprT, are required for surface localization of SprB (5–7, 16). GldN is also required for secretion of the mobile cell surface motility adhesin RemA (4, 5). Here we examined the roles of GldK, GldL, and GldM in secretion of SprB and of recombinant RemA that carried a Myc tag to allow easy detection. Cells with deletion mutations in gldK, gldL, or gldM produced SprB and Myc-tagged RemA proteins, as determined by Western blot analyses (Fig. 6), but they failed to secrete these proteins to the cell surface (Tables 2 and 3). Anti-SprB-coated polystyrene spheres attached readily to wild-type cells but failed to attach to cells of an sprB deletion mutant or to cells that had deletions in gldK, gldL, or gldM (Table 2). Complementation with a plasmid expressing the appropriate gene in each case restored SprB to the cell surface. Similarly, anti-Myc-coated polystyrene spheres attached readily to CJ2083 cells, which express Myc-tagged RemA, but they failed to attach to cells with mutations in gldK, gldL, or gldM (Table 3).
Fig 6.

Immunodetection of SprB and RemA in cells of wild-type and mutant F. johnsoniae strains. (A) Cell extracts (15 μg of protein) of wild-type (CJ1827), ΔsprB (CJ1922), ΔgldK (CJ2122), ΔgldL (CJ2157), and ΔgldM (CJ2262) strains were examined by Western blotting using antiserum against SprB. (B) Cell extracts (15 μg of protein) of wild-type (CJ2083), ΔremA (CJ1984), ΔgldK (CJ2140), ΔgldL (CJ2281), and ΔgldM (CJ2263) strains were examined by Western blotting using antiserum against RemA. All strains in panel B except the ΔremA strain (CJ1984) were derived from CJ2083 and thus expressed the Myc-tagged version of RemA.
Table 2.
Binding of protein G-coated polystyrene spheres carrying anti-SprB antibodies
| Strain | Description | Antibody added | Avg % of cells with spheres attached (SD)a |
|---|---|---|---|
| CJ1827/pCP23 | Wild type with control plasmid pCP23 | No antibody | 0.0 (0.0) |
| CJ1827/pCP23 | Wild type with control plasmid pCP23 | Anti-SprB | 53.7 (3.1) |
| CJ1922/pCP23 | ΔsprB with control plasmid pCP23 | Anti-SprB | 1.0 (1.0) |
| CJ2122/pCP23 | ΔgldK with control plasmid pCP23 | Anti-SprB | 0.3 (0.6) |
| CJ2122/pTB99 | ΔgldK with pTB99 carrying gldK | Anti-SprB | 54.0 (2.0) |
| CJ2157/pCP23 | ΔgldL with control plasmid pCP23 | Anti-SprB | 0.3 (0.6) |
| CJ2157/pTB81a | ΔgldL with pTB81a carrying gldL | Anti-SprB | 54.3 (3.5) |
| CJ2262/pCP23 | ΔgldM with control plasmid pCP23 | Anti-SprB | 0.0 (0.0) |
| CJ2262/pTB94a | ΔgldM with pTB94a carrying gldM | Anti-SprB | 56.0 (2.0) |
| CJ2302/pCP23 | ΔsprA with control plasmid pCP23 | Anti-SprB | 0.3 (0.6) |
| CJ2302/pSN48 | ΔsprA with pSN48 carrying sprA | Anti-SprB | 51.3 (4.0) |
Purified anti-SprB antiserum and 0.5-μm-diameter protein G-coated polystyrene spheres were added to cells as described in Materials and Methods. Samples were introduced into a tunnel slide, incubated for 2 min at 25°C, and examined by using a phase-contrast microscope. Images were recorded for 30 s, and 100 randomly selected cells were examined for the presence of spheres that remained attached to the cells during this time. Numbers in parentheses are standard deviations calculated from three measurements.
Table 3.
Binding of protein G-coated polystyrene spheres carrying antibodies against Myc-tagged peptide
| Strain | Description | Antibody added | Avg % of cells with spheres attached (SD)a |
|---|---|---|---|
| CJ1827 | Wild type | Anti-Myc | 0.0 (0.0) |
| CJ1984 | ΔremA | Anti-Myc | 0.0 (0.0) |
| CJ2083 | remA::myc-tag-1 | No antibody | 0.0 (0.0) |
| CJ2083 | remA::myc-tag-1 | Anti-Myc | 54.0 (4.0) |
| CJ2140/pCP23 | ΔgldK remA::myc-tag-1 with control plasmid pCP23 | Anti-Myc | 1.0 (0.0) |
| CJ2140/pTB99 | ΔgldK remA::myc-tag-1 with pTB99 carrying gldK | Anti-Myc | 57.0 (1.0) |
| CJ2281/pCP23 | ΔgldL remA::myc-tag-1 with control plasmid pCP23 | Anti-Myc | 1.0 (1.0) |
| CJ2281/pTB81a | ΔgldL remA::myc-tag-1 with pTB81a carrying gldL | Anti-Myc | 52.7 (2.1) |
| CJ2263/pCP23 | ΔgldM remA::myc-tag-1 with control plasmid pCP23 | Anti-Myc | 1.0 (1.0) |
| CJ2263/pTB94a | ΔgldM remA::myc-tag-1 with pTB94a carrying gldM | Anti-Myc | 53.0 (1.7) |
| CJ2317 | ΔsprA remA::myc-tag-1 | Anti-Myc | 0.6 (0.5) |
| CJ2317/pSN48 | ΔsprA remA::myc-tag-1 with pSN48 carrying sprA | Anti-Myc | 55.3 (2.5) |
| CJ2327 | sprT remA::myc-tag-1 | Anti-Myc | 0.0 (0.0) |
| CJ2327/pKF002 | sprT remA::myc-tag-1 with pKF002 carrying sprT | Anti-Myc | 64.6 (2.0) |
Purified anti-Myc tag antiserum and 0.5-μm-diameter protein G-coated polystyrene spheres were added to cells as described in Materials and Methods. Samples were introduced into a tunnel slide, incubated for 2 min at 25°C, and examined by using a phase-contrast microscope. Images were recorded for 30 s, and 100 randomly selected cells were examined for the presence of spheres that remained attached to the cells during this time. Numbers in parentheses are standard deviations calculated from three measurements.
Cells of gldK, gldL, and gldM mutants were also resistant to infection by bacteriophages that infect wild-type cells (Fig. 7). SprB is thought to be a receptor for phages ϕCj1, ϕCj13, ϕCj23, and ϕCj29 (3), and RemA appears to be a receptor for phages ϕCj42, ϕCj48, and ϕCj54 (4), so it is not surprising that mutants with defects in secretion of SprB, RemA, and perhaps additional cell surface motility proteins exhibit phage resistance.
Fig 7.
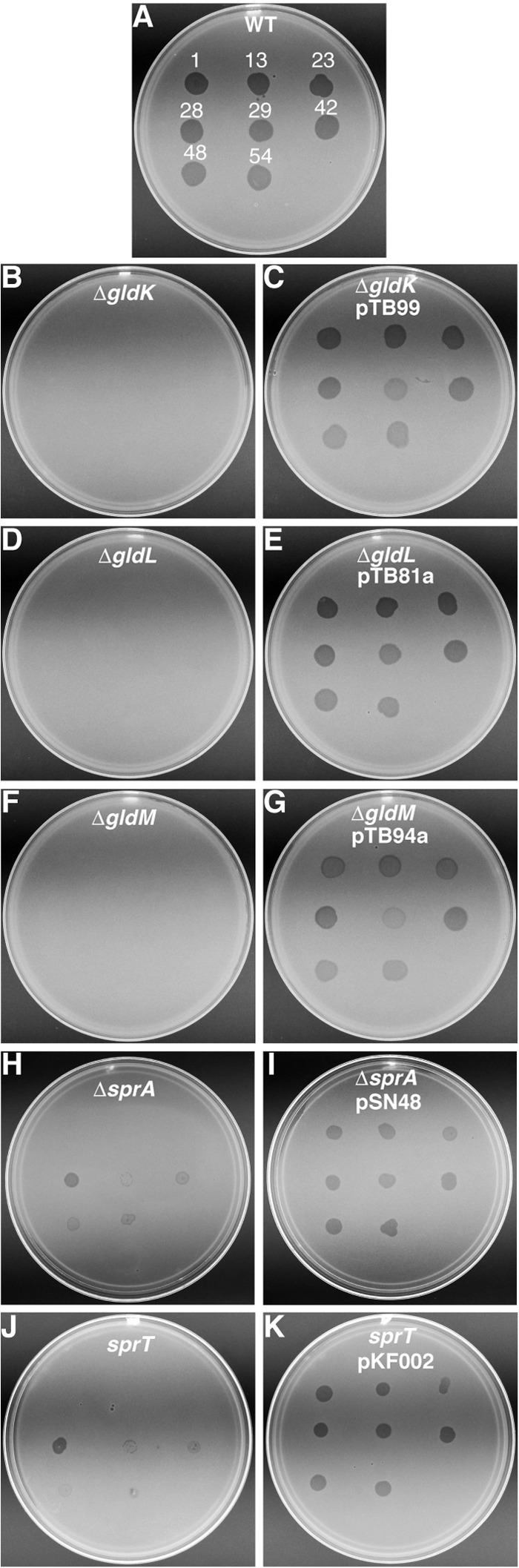
Effect of mutations on susceptibility to phages. Bacteriophages (5 μl of lysates containing approximately 109 PFU/ml) were spotted onto lawns of cells in CYE overlay agar. The plates were incubated at 25°C for 24 h to observe lysis. Bacteriophages were spotted in the following order from left to right, as also indicated by the numbers in panel A: top row, ϕCj1, ϕCj13, and ϕCj23; middle row, ϕCj28, ϕCj29, and ϕCj42; bottom row, ϕCj48 and ϕCj54. (A) Wild-type F. johnsoniae CJ1827. (B) CJ2122 (ΔgldK). (C) CJ2122 complemented with pTB99, which carries gldK. (D) CJ2157 (ΔgldL). (E) CJ2157 complemented with pTB81a, which carries gldL. (F) CJ2262 (ΔgldM). (G) CJ2262 complemented with pTB94a, which carries gldM. (H) CJ2302 (ΔsprA). (I) CJ2302 complemented with pSN48, which carries sprA. (J) KDF001 (sprT mutant). (K) KDF001 complemented with pKF002, which carries sprT.
Cells of the deletion mutants were also deficient in attachment to glass (Table 4), as has previously been observed for other motility mutants of F. johnsoniae (18). This may be the result of the absence of motility adhesins such as SprB and RemA on the cell surface. However, the lack of SprB and RemA on the cell surface cannot explain the entire attachment defect. Cells of an sprB mutant or of a remA mutant attached to glass almost as well as wild-type cells, and cells lacking both genes were only partially deficient in attachment to glass (Table 4). In contrast, cells with mutations in gldK, gldL, or gldM were completely deficient in attachment to glass. This suggests that cell surface adhesins in addition to SprB and RemA are secreted by the T9SS. Proteins secreted by T9SSs typically have conserved CTDs belonging to the TIGRFAMs TIGR04131 and TIGR04183. Analysis of the F. johnsoniae genome revealed 53 proteins with these T9SS CTDs (see Table S2 in the supplemental material). Some of these may be the additional cell surface adhesins that contribute to the ability of wild-type cells to attach to glass.
Table 4.
Effect of deletion of gldK, gldL, gldM, and sprA on attachment of cells to glass coverslips
| Strain | Description | Avg no. of cells attached to 0.03-mm2 region of glass coverslip (SD)a |
|---|---|---|
| CJ1827/pCP23 | Wild type with control vector pCP23 | 69.1 (4.9) |
| CJ2122/pCP23 | ΔgldK with control vector pCP23 | 0.3 (0.5) |
| CJ2122/pTB99 | ΔgldK with pTB99 carrying gldK | 67.7 (8.6) |
| CJ2157/pCP23 | ΔgldL with control vector pCP23 | 0.3 (0.5) |
| CJ2157/TB81a | ΔgldL with pTB81a carrying gldL | 60.7 (6.8) |
| CJ2262/pCP23 | ΔgldM with control vector pCP23 | 0.6 (1.0) |
| CJ2262/pTB94a | ΔgldM with pTB94a carrying gldM | 24.1 (2.5) |
| CJ2302/pCP23 | ΔsprA with control vector pCP23 | 0.1 (0.3) |
| CJ2302/pSN48 | ΔsprA with pSN48 carrying sprA | 64.2 (8.5) |
| KDF001 | sprT mutant | 1.4 (1.3) |
| KDF001/pKF002 | sprT mutant with pKF002 carrying sprT | 53.0 (5.4) |
| FJ149 | sprE mutant | 2.6 (1.2) |
| CJ1922/pCP23 | ΔsprB with control vector pCP23 | 66.6 (11.3) |
| CJ1984/pCP23 | ΔremA with control vector pCP23 | 54.8 (7.3) |
| CJ1985/pCP23 | ΔsprB ΔremA with control vector pCP23 | 22.6 (4.1) |
Approximately 106 cells in 2.5 μl of MM were introduced into a Petroff-Hausser counting chamber and incubated for 2 min at 25°C. Samples were observed by using an Olympus BH-2 phase-contrast microscope, and cells attached to a 0.03-mm2 region of the glass coverslip were counted. Numbers in parentheses are standard deviations calculated from 9 measurements.
GldK, GldL, and GldM are each required for chitin utilization.
F. johnsoniae was originally identified as a chitin-digesting bacterium (30). It produces a variety of proteins involved in chitin utilization, including at least one extracellular chitinase that we refer to as ChiA, encoded by Fjoh_4555. Mutations in the T9SS genes gldN, sprE, and sprT result in defects in chitin utilization and defects in secretion of ChiA (5–7). Cells with deletion mutations in gldK, gldL, and gldM were examined for chitin utilization (Fig. 8). Deletion of any of these genes resulted in an inability to digest colloidal chitin, indicating that GldK, GldL, and GldM are each required for chitin digestion and suggesting that they may be required for secretion of ChiA. In each case, complementation with plasmids carrying the appropriate genes restored the ability to digest chitin, although chitin utilization was only partially restored by complementation of the gldM mutant.
Fig 8.
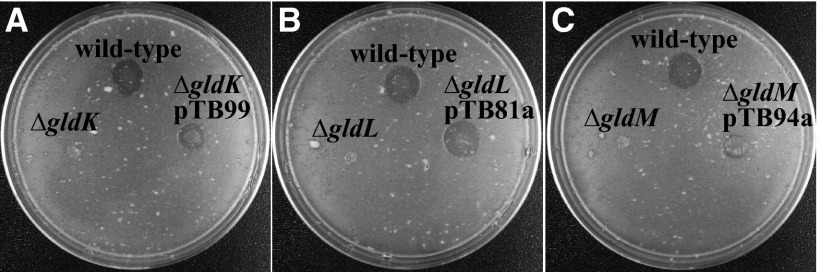
gldK, gldL, and gldM are each required for chitin utilization. Approximately 106 cells of F. johnsoniae were spotted onto MYA-chitin medium containing tetracycline and incubated at 25°C for 60 h. (A) F. johnsoniae CJ1827 (wild type), CJ2122 (ΔgldK), and CJ2122 complemented with pTB99, which carries gldK (ΔgldK pTB99). (B) F. johnsoniae CJ1827 (wild type), CJ2157 (ΔgldL), and CJ2157 complemented with pTB81a, which carries gldL (ΔgldL pTB81a). (C) F. johnsoniae CJ1827 (wild type), CJ2262 (ΔgldM), and CJ2262 complemented with pTB94a, which carries gldM (ΔgldM pTB94a). CJ1827, CJ2122, CJ2157, and CJ2262 carried control vector pCP23 so that all strains were resistant to tetracycline.
Localization of GldK, GldL, and GldM.
GldK, GldL, and GldM were each present primarily in the insoluble (membrane) fraction of cell extracts (Fig. 9). GldK is a predicted outer membrane lipoprotein, and GldL and GldM have hydrophobic alpha-helical segments that are predicted to anchor them to the cytoplasmic membrane (17). The mild detergent Sarkosyl solubilizes the F. johnsoniae cytoplasmic membrane while leaving the outer membrane intact (25). Sarkosyl solubilization was used to examine protein localization. GldL and GldM were enriched in the Sarkosyl-soluble (cytoplasmic membrane) fraction as predicted, whereas GldK was present in both the Sarkosyl-soluble (cytoplasmic membrane) and Sarkosyl-insoluble (outer membrane) fractions. While integral outer membrane proteins of F. johnsoniae are generally not solubilized by Sarkosyl, lipoproteins such as GldK might have a more tenuous connection to the membrane and thus might be partially solubilized by this treatment. The absence of GldL and GldM in the outer membrane fraction suggests that the cytoplasmic membrane was completely solubilized by the detergent, and the enrichment of GldK in the outer membrane fraction suggests that GldK may be associated primarily with the outer membrane.
Fig 9.

Cell fractionation and immunodetection of GldK, GldL, and GldM. Cell fractions of wild-type F. johnsoniae CJ1827 cells (lanes 1 to 4) and extracts of gld mutants (lanes 5) were examined for GldK (A), GldL (B), and GldM (C) by Western blot analyses. Lanes 1, soluble fraction of cell extract (cytoplasmic and periplasmic proteins); lanes 2, insoluble fraction of cell extract (primarily membrane proteins); lanes 3, Sarkosyl-soluble fraction of membranes (primarily cytoplasmic membrane proteins [CM]); lanes 4, Sarkosyl-insoluble fraction of membranes (primarily outer membrane proteins [OM]); lanes 5, cell extracts of gldK, gldL, or gldM mutants to demonstrate specificity of the antisera used in each panel.
SprA is required for secretion of SprB and RemA and for utilization of chitin.
SprA is an outer membrane protein that is involved in gliding motility (18). SprA is similar in sequence to the P. gingivalis T9SS protein Sov (18, 19). The similarity between SprA and Sov suggested that SprA might have a role in secretion. sprA deletion mutants were constructed and examined for secretion of SprB and RemA. The mutants produced SprB and RemA proteins (see Fig. S1 in the supplemental material) but failed to secrete them to the cell surface (Tables 2 and 3), suggesting that SprA has a role in secretion. Previous results using transposon-induced sprA mutants suggested that SprA was not completely essential for chitin utilization (18). The sprA mutants were severely but incompletely deficient in chitin utilization. Because the mutants analyzed would have produced truncated SprA protein that might have been partially functional, we revisited this phenotype with the deletion mutant. sprA deletion mutant strain CJ2302 failed to utilize chitin, suggesting a more severe defect in chitinase secretion than was observed for the transposon-induced mutant (see Fig. S2 in the supplemental material).
SprT and SprE have previously been shown to be required for secretion of SprB and for chitin utilization (6, 7). Like SprA, SprT was also required for secretion of RemA (Table 3, and see Fig. S1 in the supplemental material). The involvement of SprE in RemA secretion is less certain, but the resistance of sprE mutants to phages ϕCj42, ϕCj48, and ϕCj54, which are thought to use RemA as a receptor, suggests a role for SprE in RemA secretion also (4, 6). Cells with mutations in sprA, sprE, and sprT were each deficient in attachment to glass (Table 4), presumably as a result of a failure to secrete SprB, RemA, and other adhesins to the cell surface.
SprA, SprE, and SprT may be less central than GldK, GldL, GldM, and GldN to protein secretion and to gliding motility.
Cells with mutations in sprA, sprE, and sprT differ from cells of the gldK, gldL, gldM, and gldNO deletion mutants in two respects. First, cells of the gld mutants are completely nonmotile, whereas cells of sprA, sprE, and sprT mutants are severely deficient in motility but exhibit weak gliding movements on wet glass surfaces (6, 7, 18). This phenotype was verified for the newly constructed sprA deletion mutant (see Movie S4 in the supplemental material). Second, cells of the gld mutants were completely resistant to infection by all bacteriophages tested, whereas cells of sprA, sprE, and sprT mutants exhibited resistance to some but not all bacteriophages. They exhibited increased resistance to ϕCj1, ϕCj13, ϕCj23, and ϕCj29, as do sprB mutants (3, 16), and they were partially resistant to ϕCj42, ϕCj48, and ϕCj54, as are remA mutants (4), but they remained susceptible to ϕCj28 (Fig. 7). Resistance of gldK, gldL, gldM, and gldNO mutants to phages such as ϕCj28, which infect cells of CJ1985 (ΔsprB ΔremA) (4), supports the idea suggested above that the T9SS secretes cell surface proteins in addition to SprB and RemA. The sensitivity of sprA, sprE, and sprT mutants to phages such as ϕCj28 suggests that although SprA, SprE, and SprT are needed for secretion of some proteins by the T9SS, they may be less central to the function of this secretion system than are GldK, GldL, GldM, and GldN/O. The similar phenotypes of sprA, sprE, and sprT mutants also suggest the possibility that the proteins encoded by these genes may function together to support T9SS function.
DISCUSSION
gldK, gldL, gldM, and gldN were originally discovered as genes required for F. johnsoniae gliding motility (17). Later, orthologs of these genes (porK, porL, porM, and porN, respectively) were demonstrated to be required for secretion of P. gingivalis gingipains (7). The P. gingivalis secretion system was referred to as the Por secretion system (7) or, more recently, as the T9SS (2, 8). In F. johnsoniae, gldN and/or its paralog gldO was shown to be essential for secretion of the motility adhesins SprB and RemA (4, 5). The results presented here demonstrate that F. johnsoniae gldK, gldL, and gldM are cotranscribed with gldN and that the products of each of these genes are required for secretion of SprB and RemA. The outer membrane motility protein SprA, an ortholog of the P. gingivalis T9SS protein Sov, is also shown to be required for secretion of SprB and RemA. The results suggest that GldK, GldL, GldM, GldN, and SprA are each components of the F. johnsoniae T9SS. Other components of this system, previously shown to be involved in secretion in F. johnsoniae, include SprE, SprF, and SprT (6, 7, 16), which are similar in sequence to the P. gingivalis T9SS proteins PorW, PorP, and PorT, respectively (7, 31). The demonstration that the F. johnsoniae orthologs of the P. gingivalis T9SS proteins have roles in protein secretion provides independent confirmation of the function of these proteins. Similar proteins are found in many members of the large and diverse phylum Bacteroidetes, suggesting that T9SSs are common in this phylum (2).
The phenotypes of the F. johnsoniae mutants suggest that GldK, GldL, GldM, and GldN are essential for T9SS function and that SprA, SprE, and SprT are essential for secretion of many, but perhaps not all, proteins targeted to this system. Cells lacking GldK, GldL, GldM, or GldN and its paralog GldO failed to secrete SprB and RemA and were completely deficient in gliding motility and attachment to glass. They were also deficient in chitin utilization. The defect in chitin utilization is likely the result of a failure to secrete the chitinase encoded by Fjoh_4555, as previously reported for an sprT mutant (7). SprB and RemA appear to function as receptors for some but not all F. johnsoniae phages. Cells lacking GldK, GldL, GldM, or GldN and GldO were resistant to all phages tested, suggesting that some other proteins secreted by the T9SS also serve as phage receptors. In contrast, cells with mutations in sprA, sprE, and sprT exhibited partial phage resistance. They were resistant to phages ϕCj1, ϕCj13, ϕCj23, and ϕCj29 (indicating the absence of SprB on the cell surface) and were partially resistant to ϕCj42, ϕCj48, and ϕCj54 (suggesting the absence of RemA on the cell surface). They remained fully susceptible to ϕCj28. These results support the suggestion that GldK, GldL, GldM, and GldN (or GldO) are essential components of the T9SS, whereas SprA, SprE, and SprT are required for secretion of SprB and RemA but not for secretion of some other cell surface phage receptors secreted by the T9SS.
GldK, GldL, GldM, and GldN may form a complex.
Orthologs of gldK, gldL, gldM, and gldN are present in many members of the phylum Bacteroidetes, and they appear to be invariably clustered together on the genomes in this order, suggesting that, as in F. johnsoniae, they may be transcribed as a unit. Of 27 sequenced genomes analyzed that had each of these genes (2), 26 had gldK, gldL, gldM, and gldN organized as in F. johnsoniae, and the 27th (Chitinophaga pinensis) had the same organization except that a 201-bp open reading frame of unknown function was present between gldK and gldL. Unlike the highly conserved gene order, the sequences of the individual proteins have diverged substantially (see Fig. S3 to S6 in the supplemental material). This argues against recent horizontal gene transfer as an explanation for the conserved gene order. Phylogenetic trees based on GldK, GldL, GldM, and GldN sequences are similar to those based on 16S rRNAs (see Fig. S7 to S11 in the supplemental material), suggesting that the T9SS genes have primarily been passed vertically. The genetic organization of gldK, gldL, gldM, and gldN has probably been conserved because it is important for function. For example, the products of conserved gene clusters often interact (32). GldK, GldL, GldM, and GldN may interact to form a T9SS complex, and coordinated expression may be required for efficient assembly of this complex.
Several experimental observations support the suggestion that GldK, GldL, GldM, and GldN interact. First, as indicated above, cells with nonpolar mutations in gldL, gldM, and gldN had decreased levels of GldK protein. Similarly, cells with mutations in gldM had decreased levels of GldL protein, and cells with nonpolar mutations in gldK, gldL, and gldM appeared to have decreased levels of GldN protein (Fig. 3). These results suggest the possibility of instability of the proteins in the absence of their partners. Second, moderate overexpression (less than 10-fold) of gldK and gldL together, but not individually, in wild-type cells resulted in adverse effects on growth and motility, suggesting interaction between the encoded proteins (17). Third, a gldK mutant was identified that could be partially suppressed by moderate overexpression of gldL (17). Finally, the P. gingivalis T9SS proteins PorK, PorL, PorM, and PorN (orthologs of F. johnsoniae GldK, GldL, GldM, and GldN, respectively) migrate as a large complex by blue native PAGE (7).
Comparative analysis of GldK, GldL, GldM, and GldN proteins.
GldK, GldL, GldM, and GldN are novel proteins, and their exact functions in secretion and motility are not known. Analysis of genome sequences revealed orthologs of the genes encoding each of these proteins in many members of the phylum Bacteroidetes but not in bacteria belonging to other phyla (2). GldK is the most highly conserved of the four proteins. Fifty-six residues are completely conserved among the 26 sequences examined (see Fig. S3 in the supplemental material). Each of the proteins has an N-terminal type 2 signal peptide followed immediately by a cysteine, characteristic of bacterial lipoproteins. GldK is similar to sulfatase-modifying enzymes such as human formylglycine-generating enzyme (FGE). FGE activates sulfatases by catalyzing the conversion of the active-site cysteine to formylglycine. GldK is not thought to have this activity, however, because it lacks the active-site cysteine residues corresponding to C336 and C341 of FGE (33). The conserved regions of GldK may be important for protein folding and/or for protein-protein interactions, but further studies are needed to determine the exact functions of these regions. GldL and GldM localize to the cytoplasmic membrane, and sequence analyses suggest that they each have hydrophobic alpha-helical membrane-spanning regions. The N-terminal region of GldL is highly conserved and has two predicted membrane-spanning helices (see Fig. S4 in the supplemental material). The second transmembrane helix has a glutamate residue that is conserved in all 26 sequences. The high level of conservation of this charged residue within a hydrophobic stretch is unusual and suggests the possibility that it may have an important function. For example, it may be involved in harvesting cellular energy to power secretion, although other explanations are possible. The remainder of the GldL protein is predicted to be soluble. Several regions of strong conservation are scattered across this soluble region of the protein, including a pocket of high identity near the C terminus. GldM has a single predicted transmembrane helix. This region lies near the N terminus and is very highly conserved (see Fig. S5 in the supplemental material). The high levels of conservation in the transmembrane regions of GldL and GldM suggest that these have functions in secretion and/or motility beyond simple membrane anchoring. GldN is the least conserved of the four proteins (see Fig. S6 in the supplemental material), with only two residues (N77 and N265; numbered based on F. johnsoniae GldN) present in each sequence. GldN appears to have a cleavable type 1 signal peptide, but there is little else to assist in prediction of function. The presence of GldN, or the nearly identical protein GldO, is essential for motility and for secretion of SprB, RemA, and chitinase (4, 5). Future studies will be needed to elucidate their roles in these processes.
The results presented indicate that GldK, GldL, GldM, and SprA are each required for secretion of SprB, RemA, and chitinase. These proteins presumably function with other previously known components of the F. johnsoniae T9SS, including GldN, SprE, and SprT, that are also required for secretion of the same proteins. The predicted outer membrane protein SprF, which exhibits similarity to the P. gingivalis T9SS protein PorP, is also required for secretion of SprB but is not needed for secretion of RemA or for chitin utilization (4, 16). Unlike P. gingivalis, F. johnsoniae has many sprF-like (porP-like) genes that may encode semiredundant proteins involved in secretion of different proteins. A speculative model depicting our current understanding of the F. johnsoniae T9SS is illustrated in Fig. 10. As shown in this diagram, GldL and GldM are the only known cytoplasmic membrane components of the F. johnsoniae T9SS. As such, they have potential roles in harvesting cellular energy to power secretion, as suggested above, but further experiments will be needed to determine their exact functions.
Fig 10.

Speculative model of the F. johnsoniae T9SS. GldK, GldL, GldM, and GldN form the core components that are required for secretion. GldO (not shown) is nearly identical to GldN and is partially redundant with it. The outer membrane proteins SprA and SprT and the lipoprotein SprE are required for secretion of SprB, RemA, and ChiA, but they may not be required for secretion of all proteins targeted to the T9SS. SprF is needed for secretion of SprB but not for secretion of RemA and ChiA. F. johnsoniae has many paralogs of sprF that may encode proteins involved in secretion of proteins other than SprB. Proteins secreted by the T9SS all have predicted type 1 signal peptides and are predicted to be exported across the cytoplasmic membrane by the Sec system before being secreted across the outer membrane by the T9SS. Black lines indicate lipid tails on lipoproteins. Proteins are not drawn to scale, and stoichiometry of components is not known.
GldK, GldL, and GldM are required not only for secretion but also for gliding motility. The motility defects of cells lacking these proteins may be explained by their inability to deliver motility adhesins, such as SprB and RemA, to the cell surface. However, GldK, GldL, and GldM may also have more direct roles in cell movement. Although the cell surface adhesins involved in motility are known, the motor for cell movement has not yet been identified. Gliding of F. johnsoniae and related bacteria requires the proton gradient across the cytoplasmic membrane (34–37), and proteins that span this membrane must be involved in converting this into cell movement. Twelve Gld proteins required for gliding of F. johnsoniae have been identified (17), and only four of these (GldF, GldG, GldL, and GldM) are thought to span the cytoplasmic membrane. GldF and GldG do not appear to be central components of the gliding machinery because some relatives of F. johnsoniae lack these proteins and yet exhibit rapid gliding motility (2). This leaves GldL and GldM as candidates for the motor proteins that harvest cellular energy and propel SprB and RemA, resulting in cell movement. Alternatively, some other unidentified cytoplasmic membrane proteins may perform these motor functions. Further study is needed to determine whether GldL and GldM have roles in gliding beyond secretion of SprB and RemA and to determine the exact functions of each of the T9SS proteins in protein secretion.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by grant MCB-1021721 from the National Science Foundation.
Footnotes
Published ahead of print 10 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00333-13.
REFERENCES
- 1. Jarrell KF, McBride MJ. 2008. The surprisingly diverse ways that prokaryotes move. Nat. Rev. Microbiol. 6:466–476 [DOI] [PubMed] [Google Scholar]
- 2. McBride MJ, Zhu Y. 2013. Gliding motility and Por secretion system genes are widespread among members of the phylum Bacteroidetes. J. Bacteriol. 195:270–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nelson SS, Bollampalli S, McBride MJ. 2008. SprB is a cell surface component of the Flavobacterium johnsoniae gliding motility machinery. J. Bacteriol. 190:2851–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shrivastava A, Rhodes RG, Pochiraju S, Nakane D, McBride MJ. 2012. Flavobacterium johnsoniae RemA is a mobile cell surface lectin involved in gliding. J. Bacteriol. 194:3678–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rhodes RG, Samarasam MN, Shrivastava A, van Baaren JM, Pochiraju S, Bollampalli S, McBride MJ. 2010. Flavobacterium johnsoniae gldN and gldO are partially redundant genes required for gliding motility and surface localization of SprB. J. Bacteriol. 192:1201–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rhodes RG, Samarasam MN, Van Groll EJ, McBride MJ. 2011. Mutations in Flavobacterium johnsoniae sprE result in defects in gliding motility and protein secretion. J. Bacteriol. 193:5322–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sato K, Naito M, Yukitake H, Hirakawa H, Shoji M, McBride MJ, Rhodes RG, Nakayama K. 2010. A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 107:276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sato K, Yukitake H, Narita Y, Shoji M, Naito M, Nakayama K. 2013. Identification of Porphyromonas gingivalis proteins secreted by the Por secretion system. FEMS Microbiol. Lett. 338:68–76 [DOI] [PubMed] [Google Scholar]
- 9. Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. 2007. Type VII secretion—mycobacteria show the way. Nat. Rev. Microbiol. 5:883–891 [DOI] [PubMed] [Google Scholar]
- 10. Desvaux M, Hebraud M, Talon R, Henderson IR. 2009. Secretion and subcellular localizations of bacterial proteins: a semantic awareness issue. Trends Microbiol. 17:139–145 [DOI] [PubMed] [Google Scholar]
- 11. Economou A, Christie PJ, Fernandez RC, Palmer T, Plano GV, Pugsley AP. 2006. Secretion by numbers: protein traffic in prokaryotes. Mol. Microbiol. 62:308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen KA, Travis J, Potempa J. 2007. Does the importance of the C-terminal residues in the maturation of RgpB from Porphyromonas gingivalis reveal a novel mechanism for protein export in a subgroup of Gram-negative bacteria? J. Bacteriol. 189:833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seers CA, Slakeski N, Veith PD, Nikolof T, Chen YY, Dashper SG, Reynolds EC. 2006. The RgpB C-terminal domain has a role in attachment of RgpB to the outer membrane and belongs to a novel C-terminal-domain family found in Porphyromonas gingivalis. J. Bacteriol. 188:6376–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shoji M, Sato K, Yukitake H, Kondo Y, Narita Y, Kadowaki T, Naito M, Nakayama K. 2011. Por secretion system-dependent secretion and glycosylation of Porphyromonas gingivalis hemin-binding protein 35. PLoS One 6:e21372. 10.1371/journal.pone.0021372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Slakeski N, Seers CA, Ng K, Moore C, Cleal SM, Veith PD, Lo AW, Reynolds EC. 2011. C-terminal domain residues important for secretion and attachment of RgpB in Porphyromonas gingivalis. J. Bacteriol. 193:132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rhodes RG, Nelson SS, Pochiraju S, McBride MJ. 2011. Flavobacterium johnsoniae sprB is part of an operon spanning the additional gliding motility genes sprC, sprD, and sprF. J. Bacteriol. 193:599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Braun TF, Khubbar MK, Saffarini DA, McBride MJ. 2005. Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J. Bacteriol. 187:6943–6952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nelson SS, Glocka PP, Agarwal S, Grimm DP, McBride MJ. 2007. Flavobacterium johnsoniae SprA is a cell surface protein involved in gliding motility. J. Bacteriol. 189:7145–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saiki K, Konishi K. 2007. Identification of a Porphyromonas gingivalis novel protein Sov required for the secretion of gingipains. Microbiol. Immunol. 51:483–491 [DOI] [PubMed] [Google Scholar]
- 20. Rhodes RG, Pucker HG, McBride MJ. 2011. Development and use of a gene deletion strategy for Flavobacterium johnsoniae to identify the redundant motility genes remF, remG, remH, and remI. J. Bacteriol. 193:2418–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McBride MJ, Kempf MJ. 1996. Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 178:583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Agarwal S, Hunnicutt DW, McBride MJ. 1997. Cloning and characterization of the Flavobacterium johnsoniae (Cytophaga johnsonae) gliding motility gene, gldA. Proc. Natl. Acad. Sci. U. S. A. 94:12139–12144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu J, McBride MJ, Subramaniam S. 2007. Cell surface filaments of the gliding bacterium Flavobacterium johnsoniae revealed by cryo-electron tomography. J. Bacteriol. 189:7503–7506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kempf MJ, McBride MJ. 2000. Transposon insertions in the Flavobacterium johnsoniae ftsX gene disrupt gliding motility and cell division. J. Bacteriol. 182:1671–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hunnicutt DW, McBride MJ. 2000. Cloning and characterization of the Flavobacterium johnsoniae gliding motility genes gldB and gldC. J. Bacteriol. 182:911–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang LYE, Pate JL, Betzig RJ. 1984. Isolation and characterization of nonspreading mutants of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 159:26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pate JL, Petzold SJ, Chang L-YE. 1979. Phages for the gliding bacterium Cytophaga johnsonae that infect only motile cells. Curr. Microbiol. 2:257–262 [Google Scholar]
- 28. Wolkin RH, Pate JL. 1985. Selection for nonadherent or nonhydrophobic mutants co-selects for nonspreading mutants of Cytophaga johnsonae and other gliding bacteria. J. Gen. Microbiol. 131:737–750 [Google Scholar]
- 29. Chen S, Bagdasarian M, Kaufman MG, Bates AK, Walker ED. 2007. Mutational analysis of the ompA promoter from Flavobacterium johnsoniae. J. Bacteriol. 189:5108–5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stanier RY. 1947. Studies on non-fruiting myxobacteria. I. Cytophaga johnsonae, n. sp., a chitin-decomposing myxobacterium. J. Bacteriol. 53:297–315 [PMC free article] [PubMed] [Google Scholar]
- 31. Sato K, Sakai E, Veith PD, Shoji M, Kikuchi Y, Yukitake H, Ohara N, Naito M, Okamoto K, Reynolds EC, Nakayama K. 2005. Identification of a new membrane-associated protein that influences transport/maturation of gingipains and adhesins of Porphyromonas gingivalis. J. Biol. Chem. 280:8668–8677 [DOI] [PubMed] [Google Scholar]
- 32. Dandekar T, Snel B, Huynen M, Bork P. 1998. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem. Sci. 23:324–328 [DOI] [PubMed] [Google Scholar]
- 33. Dierks T, Dickmanns A, Preusser-Kunze A, Schmidt B, Mariappan M, von Figura K, Ficner R, Rudolph MG. 2005. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell 121:541–552 [DOI] [PubMed] [Google Scholar]
- 34. Duxbury T, Humphrey BA, Marshall KC. 1980. Continuous observations of bacterial gliding motility in a dialysis microchamber: the effects of inhibitors. Arch. Microbiol. 124:169–175 [Google Scholar]
- 35. Dzink-Fox JL, Leadbetter ER, Godchaux W., III 1997. Acetate acts as a protonophore and differentially affects bead movement and cell migration of the gliding bacterium Cytophaga johnsonae (Flavobacterium johnsoniae). Microbiology 143:3693–3701 [DOI] [PubMed] [Google Scholar]
- 36. Pate JL, Chang L-YE. 1979. Evidence that gliding motility in prokaryotic cells is driven by rotary assemblies in the cell envelopes. Curr. Microbiol. 2:59–64 [Google Scholar]
- 37. Ridgway HF. 1977. Source of energy for gliding motility in Flexibacter polymorphus: effects of metabolic and respiratory inhibitors on gliding movement. J. Bacteriol. 131:544–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McBride MJ, Braun TF. 2004. GldI is a lipoprotein that is required for Flavobacterium johnsoniae gliding motility and chitin utilization. J. Bacteriol. 186:2295–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McBride MJ, Xie G, Martens EC, Lapidus A, Henrissat B, Rhodes RG, Goltsman E, Wang W, Xu J, Hunnicutt DW, Staroscik AM, Hoover TR, Cheng YQ, Stein JL. 2009. Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75:6864–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.