

Abstract
Intramolecular hydrogen bonding is an important determinant of enzyme structure, catalysis, and inhibitor action. Monoacylglycerol lipase (MGL) modulates cannabinergic signaling as the main enzyme responsible for deactivating 2-arachidonoylglycerol (2-AG), a primary endocannabinoid lipid messenger. By enhancing tissue-protective 2-AG tone, targeted MGL inhibitors hold therapeutic promise for managing pain and treating inflammatory and neurodegenerative diseases. We report study of purified, solubilized human MGL (hMGL) to explore the details of hMGL catalysis by using two known covalent hMGL inhibitors, the carbamoyl tetrazole AM6701 and N-arachidonoylmaleimide (NAM), that act through distinct mechanisms. Using proton nuclear magnetic resonance spectroscopy (NMR) with purified wild-type and mutant hMGLs, we have directly observed a strong hydrogen-bond network involving Asp239 and His269 of the catalytic triad and neighboring Leu241 and Cys242 residues. hMGL inhibition by AM6701 alters this hydrogen-bonding pattern through subtle active-site structural rearrangements without influencing hydrogen-bond occupancies. Rapid carbamoylation of hMGL Ser122 by AM6701 and elimination of the leaving group is followed by a slow hydrolysis of the carbamate group, ultimately regenerating catalytically competent hMGL. In contrast, hMGL titration with NAM, which leads to cysteine alkylation, stoichiometrically decreases the population of the active-site hydrogen bonds. NAM prevents reformation of this network, and in this manner inhibits hMGL irreversibly. These data provide detailed molecular insight into the distinctive mechanisms of two covalent hMGL inhibitors and implicate a hydrogen-bond network as a structural feature of hMGL catalytic function.
Introduction
The endocannabinoid system is a ubiquitous cellular information-transducing element whose components are present to varying extents across mammalian tissue types. This intrinsic signaling pathway includes two cloned and expressed cannabinoid G protein-coupled receptors (GPCRs), designated CB1 and CB2; endogenous lipid messengers (endocannabinoids) that engage and activate these cannabinoid GPCRs; and transporters and enzymes that modulate endocannabinoid tone and signaling intensity.1,2 The most extensively studied endocannabinoids, the cannabinoid-receptor agonists N-arachidonoylethanolamine (anandamide) (AEA) and 2-arachidonoylglyerol (2-AG), are synthesized “on demand” and influence diverse physiological and homeostatic processes. In the central nervous system, for example, 2-AG modulates retrograde synaptic efficiency and neuronal activity by regulating the production/release of various neurochemicals.3 Endocannabinoid transmission through CB1 impacts cardiometabolic risk by influencing lipid and glucose metabolism and peripheral fat deposition.4 Various disease states activate endocannabinoid biosynthesis in a temporally and spatially defined manner to augment local endocannabinoid levels, which are kept in check by rapid enzymatic deactivation. Resultant potentiation of cannabinergic signaling may be regarded as a cytoprotective physiological response to limit the adverse sequellae of pathological stressors and help restore homeostasis.2,5,6 The tissue-protective nature of this response has placed great interest on targeted inhibitors of enzymatic endocannabinoid deactivation as potential pharmacotherapeutics that would elicit and/or enhance the salutary effect of endocannabnoid-system activation under disease conditions.2,7,8 As compared to exogeneously administered cannabinoid-receptor agonists, inhibitors of endocannabinoid deactivation appear less likely to incite adverse cannabinergic hyperactivity and unwanted psychotrophic responses, inviting a therapeutically attractive dissociation between beneficial and undesirable effects of pharmacological cannabinoid-receptor agonism.2,9,10
In this regard, monoacylglycerol lipase (MGL) (EC 3.1.1.23) is the focus of increasing scrutiny as a drug target. Particularly abundant in brain presynaptic terminals, MGL is a 33 kDa serine hydrolase that cleaves the ester bond of monoacylglycerols to generate free fatty acid and glycerol, a key step in lipolytic fatty acid mobilization from hepatic and adipose triacylglycerol stores.11–13 In the nervous system, MGL’s main function is to catalyze the hydrolytic biotransformation of the endocannabinoid 2-AG, its best-characterized substrate and the predominant brain endocannabinoid.14 A recent activity-based proteomic analysis has quantified that MGL mediates some 85% of the 2-AG hydrolysis in the brain.15 Since 2-AG is a very efficacious, CB1 and CB2 full agonist influencing a number of physiological processes including pain sensation, neurotransmission, synaptic plasticity, inflammation, and immune-cell function, rapid termination of 2-AG bioactivity by MGL constitutes a primary physiological control-point for quenching endocannabinoid drive.16 MGL’s propensity to co-localize with CB1 in the brain suggests that MGL serves in the CNS to limit 2-AG action through CB1.17 This suggestion is supported by findings that pharmacological MGL inhibition and RNA interference-mediated silencing of MGL expression increase tissue 2-AG levels and amplify 2-AG-mediated cannabinergic signaling in a CB1-sensitive, tissue-specific manner with antinociceptive and anti-inflammatory effects of likely clinical benefit.18–20 In human cancer cells, MGL hydrolysis of cellular lipid stores controls the production of free fatty acid-derived oncogenic lipid messengers that promote cancer-cell migration and invasiveness.21 The key roles that MGL plays in tuning homeostatic endocannabinoid signaling and supporting aggressive tumorigenesis make MGL inhibition a promising therapeutic modality for managing pain and treating inflammatory, neurodegenerative, and immunological disorders as well as cancer.
Thorough understanding of MGL’s (patho)physiological relevance and its optimal exploitation as a pharmacotherapeutic target have been hampered by at least two factors: (1) the limited availability of MGL inhibitors that are both potent and selective; (2) gaps in our understanding of the details associated with MGL’s catalytic and inactivation properties. Many MGL inhibitors yet identified inhibit multiple serine hydrolases and/or other enzymes, while others exhibit striking species selectivity.1–2,20 Off-target inhibition of fatty acid amide hydrolase (FAAH), the principal inactivating enzyme for the endocannabinoid AEA, by a nonselective MGL inhibitor is of particular concern in light of evidence that each of these two endocannabinoid-inactivating enzymes serves as a key point of control over specific downstream signaling events in vivo and regulates thereby a particular subset of bioresponses.10 Two novel compounds, the piperazine carbamate JZL184 and the tetrahydrolipstatin analog OMDM169, have recently been characterized as MGL inhibitors with >300-fold selectivity for inhibition of MGL vs. FAAH and, in the case of JZL184, no detectable interaction with other serine hydrolases, other protein components of the endocannabinoid system, or phospholipases involved in arachidonic acid mobilization.20,22,23 Although the absolute potencies and species-selectivities of JZL184 or OMDM169 as MGL inhibitors vary with assay conditions and MGL source,20,22–24 the selectivity of these two compounds for MGL and their efficacy at enhancing tissue 2-AG levels in vivo have invited their use as pharmacological probes to block catalytic 2-AG inactivation. Publications in the patent and biomedical literatures within the last year have disclosed a refined hMGL homology model25 and the first three-dimensional crystal structures of preparations of hMGL with N-terminal His-6 and C-terminal Strep tags26,27 and monomeric, His-tag modified hMGL in apo and liganded states.28 These reports offer the initial computational and experimental insight into the structural aspects of hMGL catalysis and its inhibition. Yet this structural information is subject to the extrapolations inherent to homology modeling and the possibility that the conformations of the hMGL crystals studied could reflect the influence of diverse experimental factors (e.g., crystal packing, detergents used in enzyme purification, co-crystalized ligand). Thorough multidisciplinary evaluation will be required to validate these newly-advanced MGL inhibitors and structural templates.
Two covalent hMGL inhibitors are recognized as valuable tools for interrogating the amino acids critical to hMGL catalytic activity and pharmacological modulation by active site-directed small molecules. Recognition that hMGL is highly sensitive to sulfhydryl-modifying reagents led to the identification of the potent hMGL inhibitor N-arachidonoylmaleimide (NAM), which acts as a Michael acceptor toward free thiols.29 NAM binding to at least one of three candidate cysteine residues (i.e., Cys201, Cys208, Cys242) in the vicinity of the hMGL active site has been implicated in the mechanism of hMGL inhibition by NAM,27–33 although direct, mass spectrometry-based proteomic demonstration of this covalent interaction has been made only for NAM binding at hMGL Cys208 and Cys242.30 A representative from another class of potent, covalent MGL inhibitors, AM6701, is a 2,5-substituted carbamoyl tetrazole that has been demonstrated by mass spectrometry to carbamoylate Ser122 in the hMGL catalytic triad,30 as does JZL184.20 Although neither NAM nor AM6701 is truly selective for MGL, newer-generation N-substituted maleimide and carbamoyl-tetrazole derivatives have been advanced as hMGL inhibitors with improved pharmacological profiles.32,33 Therefore, thorough understanding of the molecular basis of hMGL inhibition by AM6701 and NAM and any structural impact these inhibitors may have upon the hMGL active site would serve as a valuable aid to the design and targeting of future drug candidates that effectively modulate hMGL activity for therapeutic gain.
These considerations prompted us to apply nuclear magnetic resonance spectroscopy (NMR) to study the interaction of NAM and AM6701 with purified hMGL and investigate potential structural effects of these inhibitors on hMGL. NMR is an ideal analytical method for probing the nature and dynamic consequences of ligand-binding to protein therapeutic targets.34,35 In this initial NMR study of hMGL, we have identified and observed directly resonances in a low-field region of the hMGL 1D proton (1H) NMR spectrum indicative of an extensive hydrogen-bond network involving two amino acid residues in the hMGL catalytic triad and others in its close proximity. NAM–hMGL adduct formation induces a structural perturbation of this hydrogen-bond network that may contribute to NAM’s irreversible inhibitor effect. In contrast, these short-distance amino-acid interactions are only subtly influenced by covalent occupation of the enzyme’s nucleophilic Ser122 residue by AM6701 resulting from a carbamoylation reaction that prevents hMGL catalytic activity. However, AM6701 inhibition of hMGL occurs without apparent disruption of active-site architecture in the catalytic pocket, for hMGL carbamoylation by AM6701 can be fully reversed under physiological conditions to regenerate catalytically-competent enzyme. The present study offers new experimental insight into the hMGL catalytic domain and provides guidance for the design of active site-targeted small molecules as potential hMGL-inhibitor drugs.
Results and discussion
hMGL preparation and quantification of catalytic activity
Through a novel isolation and purification protocol to be detailed elsewhere, we were able to obtain purified, monomeric recombinant hMGL (WT and C242A and D239T mutants) as well as the uniformly-labeled protein (u-15N hMGL) in solution (Fig. 1). Although results from a very recent crystallization study of WT hMGL modified with N-terminal His-6 and C-terminal Strep tags invited speculation that hMGL is a biological dimer,27 the present results and those of another laboratory’s hMGL crystallization study28 do not support this speculation. Our hMGL preparation not only enabled the initial 1H NMR analysis of hMGL, but was also readily amenable to kinetic assay for hydrolytic activity using the fluorogenic substrate arachidonoyl, 7-hydroxy-6-methoxy-4-methylcoumarin ester (AHMMCE).30 Similar fluorescence-based assays with reporter substrates have found utility in quantifying hMGL catalytic activity and its inhibition by small molecules.24,36,37 Since the absolute MGL activity profile may vary with assay conditions (including substrate),31,36,37 quantitative activity results are most reliably compared internally within a given assay paradigm.
Fig. 1.
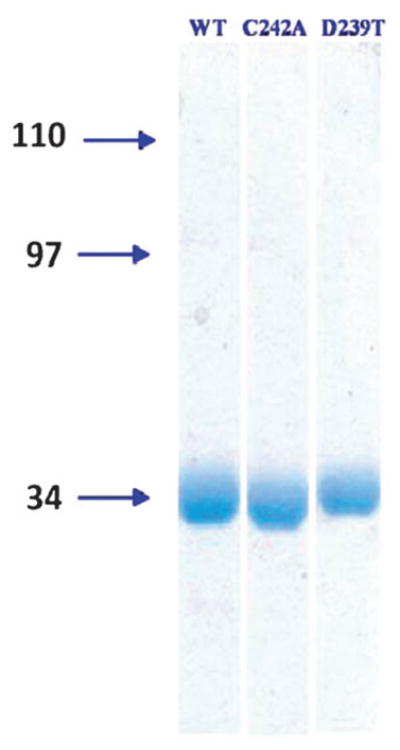
Coomassie-stained 10% SDS-PAGE gel analysis of purified, recombinant, His6-tagged WT hMGL and C242A and D239T hMGL mutants. Molecular masses (kDa) are designated to the left of the gels.
1D 1H NMR spectrum of WT hMGL
The 1D 1H NMR spectrum of WT hMGL obtained in the absence of detergent and with water suppression using excitation sculpting with gradients38 is shown in Fig. 2. We observe a large chemical-shift dispersion, especially in the methyl-group region between +1.0 ppm and −1.0 ppm and in the amide region downfield of 8.5 ppm. This chemical shift dispersion of proton signals is indicative of the enzyme’s proper 3D folding. Evaluation of line widths for individual peaks from these regions (Δν1/2 ~25 Hz) supports the conclusion that the enzyme is monomeric under these NMR experimental conditions, even in the absence of detergent. Addition of detergent does, however, stabilize the monomeric hMGL and prevent enzyme aggregation (data not shown).
Fig. 2.

1D proton NMR spectrum of WT hMGL (30 μM, final protein concentration) in 20 mM phosphate buffer, pH 7.4, containing 100 mM NaCl.
Detection and assignment of low-field 1H NMR resonances
Notable within the WT hMGL 1D 1H NMR spectrum is the presence of four distinct proton resonances in the low-field region (12–16 ppm) component observed at 37 °C with 1331 pulse sequence for minimal water excitation39 (Fig. 3A). Each of these four resonances is split into a doublet of 1JNH = 90 Hz in the u-15N hMGL spectrum (Fig. 3A), indicating that all signals belong to –NH groups. We attribute these four low-field peaks to the presence of a strong hydrogen-bond network on the following basis: enzyme active-site amide and imidazole −NH protons usually exchange rapidly with bulk water and thus may not be observable by NMR. Nevertheless, factors including low temperature, active-site inaccessibility to solvent, and formation of strong hydrogen bonds that markedly attenuate the exchange rate may allow successful detection of such proton resonances.40 X-Ray structural data indicate that the hMGL catalytic triad is indeed buried at the bottom of the substrate binding pocket with limited water accessibility.26–28 Low-field (12–18 ppm) resonances corresponding to strong hydrogen bonds in the active site of enzymes are usually not observable with the use of standard water-suppression schemes. Our application of pulse sequences oriented for observing rapidly-exchanging protons can enable successful detection of hydrogen bond-related, low-field proton NMR resonances.39,41 These considerations offer initial support to our attributing the low-field NMR resonances from hMGL we have detected to hydrogen bonds in the hMGL active site. Recently reported hMGL crystal structures are suggestive of such a strong hydrogen-bond network involving not only residues of the hMGL catalytic triad (i.e., Ser122, Asp239, His269), but also Cys242, which is buried in the active site, close to Ser122.26,27 X-Ray data also suggest that Asp239 might serve as the acceptor of three strong hydrogen bonds in such a putative short-distance interaction network.27 Accordingly, the most likely contributors of protons sufficiently de-shielded to participate in the hydrogen-bond pattern in the hMGL low-field spectral region are the amide –NH group of Ser122 and the Nδ1H of His269, two residues of the hMGL catalytic triad, a triad that is also a hallmark of the catalytic triad of other “classical” serine hydrolases (lipases, proteases).42
Fig. 3.
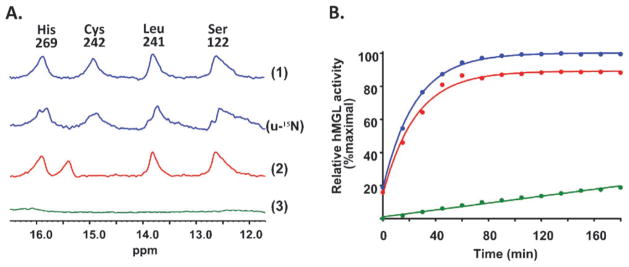
Panel A: Low-field region of the 1D 1H hMGL NMR spectrum. The quartet of peaks is indicative of four hydrogen bonds involving the catalytic triad within the enzyme active site. (1) WT hMGL; (u-15N) u-15N labeled WT hMGL; (2) hMGL C242A mutant; (3) hMGL D239T mutant. The basis for the peak assignments indicated is detailed in the text. Panel B: Best-fit plots of the time-course of WT (blue), C242A mutant (red), and D239T mutant (green) hMGL hydrolytic activity.
Actual peak assignment in the downfield region of the 1D 1H NMR spectrum of hMGL was based upon several different criteria. The resonance at 15.9 ppm evidenced a pH-dependent chemical shift (data not shown) and on this basis was assigned to the Nδ1H proton of His269 in the hMGL catalytic triad.43 Site-directed mutation of Cys242 to alanine abrogated the WT hMGL spectral peak at 14.9 ppm, identifying this signal as originating from the Cys242 –NH group (Fig. 3A). Mutation of Asp239 to threonine abolished all four downfield resonances (Fig. 3A), a finding supporting the postulation that Asp239 accepts three hydrogen bonds as a key residue of this hMGL amino-acid interaction network. From this result with the D239T hMGL mutant and the N–O distances derived from the high-resolution (1.3Å ) hMGL X-ray structures reported,26,28 the resonances at 13.8 ppm and 12.7 ppm were provisionally assigned to backbone Leu241 and Ser122 amides, respectively. These complementary lines of evidence suggest that optimal positioning of the catalytic-triad residues in the hMGL active site is supported by a network of relatively strong, short-distance amino-acid interactions reflected in the downfield hMGL NMR pattern shown in Fig. 3A. Along with the high structural precision (1.3Å ) of the high-resolution hMGL X-ray data26,28 we have used to assign the low-field NMR resonances in Fig. 3 (vide supra), the loss of the hydrogen-bond network observed upon mutation of Asp239 (the acceptor of three hydrogen bonds and a residue in the hMGL catalytic triad42) and the perturbation of the network by mutation of Cys242, which is buried in the active site close to the Ser122 of the catalytic triad26,27 all support the conclusion that the hMGL hydrogen-bond network we have identified is present within the enzyme’s active site.
Enzymatic activity was measured over time for WT hMGL and the C242A and D239T mutants that had been analyzed by NMR. As summarized in Fig. 3B, WT hMGL and the C242A mutant displayed comparably high hydrolytic activity, whereas mutation of Asp239 to threonine substantially compromised hMGL activity. These data confirm our prior activity analysis of the hMGL Cys242 mutant,30 although mutating Cys242 of recombinant rat MGL to a nonpolar amino acid other than alanine (i.e., glycine) has been reported to reduce its ability to hydrolyze 2-oleoylglycerol.31 Given the appreciable (at least 84%) sequence identity among rat, mouse, and human MGL orthologs,30,42,43 this apparent inconsistency regarding the effect of mutating Cys242 on MGL catalysis may reflect intrinsic structural features of MGL that are species-related.
Effect of AM6701 binding on the hMGL active-site hydrogen-bond network and enzymatic activity
The active-site hydrogen-bond network of hMGL may be involved in fundamental aspects of hMGL catalysis, such as substrate-induced hMGL conformational mobility supporting transition-state stabilization as well as molecular dynamics during hMGL-inhibitor interaction.44,45 On this basis, we next used NMR to explore the potential effect of known, potent, covalent hMGL inhibitors on the enzyme’s catalytic-site hydrogen-bond network, as deduced from changes in the low-field NMR pattern. We have previously offered direct demonstration by mass spectrometry that hMGL inhibition by AM6701 involves a covalent interaction resulting in the enzyme’s rapid, selective carbamoylation at its catalytic serine nucleophile (Ser122) and release of the AM6701 leaving group.30 As shown in Fig. 4A, addition of equimolar AM6701 induced perturbations in the low-field region of the hMGL NMR spectrum, as evidenced by the shift of an amide resonance assigned to Cys242 from 14.9 to 13.7 ppm. The shift suggests that covalent modification of hMGL Ser122 by AM6701 induces subtle structural rearrangements within the enzyme’s catalytic pocket which are reflected in the changes observed in the down-field region of the hMGL NMR spectrum. These NMR changes indicate that hMGL carbamoylation by AM6701 alters the active-site hydrogen-bond pattern of the enzyme without affecting the hydrogen-bond occupancies. Interestingly, the chemical shift of the peak provisionally assigned to an amide of Ser122 remained unchanged. Over time, the peak at 14.9 ppm that had shifted to 13.7 ppm upon incubating AM6701 with WT hMGL gradually reappeared such that the original downfield hydrogen-bond NMR pattern of WT hMGL was completely reestablished by 17 h.
Fig. 4.
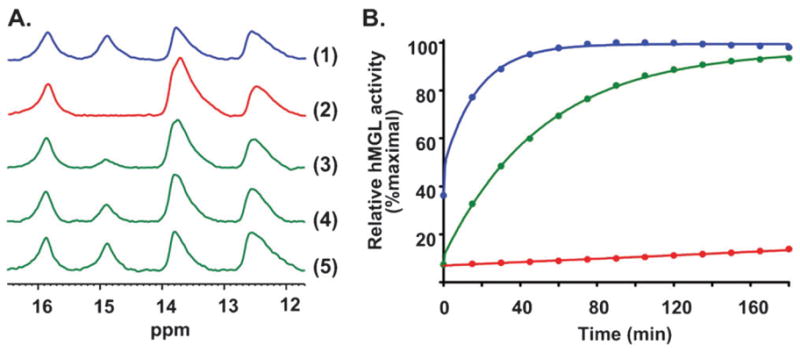
Panel AEffect of the active site-directed inhibitor, AM6701, on the low-field region of the 1D 1H NMR spectrum of WT hMGL. (1) hMGL, no AM6701; (2) AM6701–hMGL, 1 : 1 molar ratio, immediately after mixing; (3) AM6701–hMGL, 1 : 1 molar ratio, after 4.5 h; (4) AM6701–hMGL, 1 : 1 molar ratio, after 9 h; (5) AM6701–hMGL, 1 : 1 molar ratio, after 17 h. Peak assignments are given in Fig. 3A. Panel B: Best-fit plots of the time-course of the hydrolytic activity of WT hMGL under the following conditions: no AM6701 (blue); immediately after mixing with AM6701 at a 1 : 1 molar ratio with hMGL protein (red); 20 h after start of hMGL incubation with AM6701 (green).
Equimolar AM6701 elicited a dramatic reduction of hMGL hydrolytic activity (Fig. 4B), confirming our prior results.30 Samples of hMGL taken 3 h after introduction of AM6701 still showed appreciable hMGL inhibition (data not shown). By 20 h, however, the enzyme recovered full hydrolytic activity (Fig. 4B). These aggregate NMR and biochemical assay data support the conclusion that the carbamoylated enzyme undergoes slow, spontaneous hydrolysis, making AM6701 a reversible, covalent hMGL inhibitor. The reported28 high-resolution structure of hMGL in complex with a covalent triazolocarboxamide inhibitor (SAR629) suggested to those authors the possibility of coordinating a water molecule between the catalytic Ser122 and a carbamate functionality to which it may be liganded, a coordination scheme that offers indirect support for our observation that the covalent bond between AM6701 and hMGL Ser122 is hydrolytically reversible. Yet the carbamoylation product formed between the hMGL inhibitor JZL184 and the enzyme’s Ser122 was reported to be stable for at least 30 h at room temperature.20 Local geometry dependent, at least in part, upon the chemical structure of the carbamoylating inhibitor may be a decisive determinant of the reversibility of hMGL inhibition by AM6701 we have documented.
Effect of NAM binding on the hMGL active-site hydrogen-bond network and enzymatic activity
MGL is sensitive to chemically-selective, sulfhydryl-modifying alkylating reagents.29 As demonstrated directly by mass spectrometry-based proteomic analysis supplemented with mutational studies,30 NAM inhibits hMGL through partial enzyme alkylation at Cys208 and/or Cys242 (Cys242 being favored). The remaining two hMGL cysteine residues, Cys32 and Cys201, may not be targets for NAM alkylation.30 These results are consonant with hMGL crystallographic data for apo and SAR629-liganded hMGL,28 which suggests the possibility that NAM binding to Cys242, a residue localized close to active-site Ser122,26,27 might sterically hinder substrate access to the catalytic pocket. According to the hMGL X-ray structures reported,28 hMGL Cys208 is further from the catalytic triad than Cys242 and oriented away from the putative substrate-entrance tunnel, although Cys208 solvent exposure could facilitate its reaction with NAM. In contrast to the response of the hMGL active-site hydrogen-bond pattern to AM6701, hMGL titration with NAM markedly affected the low-field region of the 1D 1H NMR spectrum in a different manner. Increasing NAM:hMGL molar ratios appeared to progressively decrease the occupancy of all hydrogen bonds within the hMGL catalytic triad (Fig. 5A). These NMR data support conclusion that covalent NAM binding at equimolar hMGL–NAM was sufficient to disrupt completely the hMGL active-site hydrogen-bond network, which remained disrupted during the subsequent 24 h.
Fig. 5.
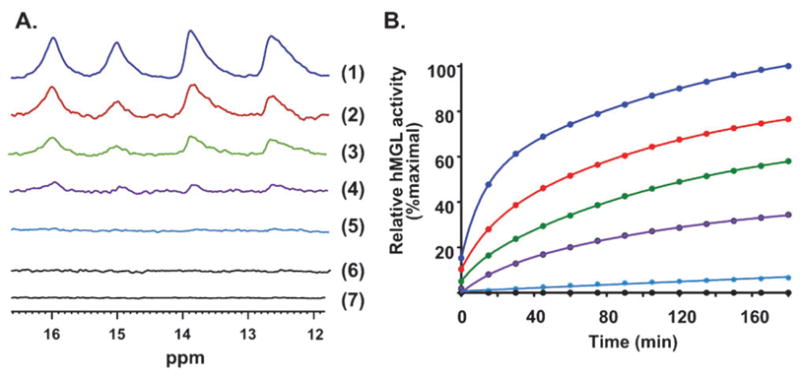
Panel AEffect of the active site-directed covalent inhibitor, NAM, on the low-field NMR spectral region of WT hMGL. Spectra denote the effect of NAM at the following NAM–hMGL molar ratios: 0 : 1 (spectrum 1); 0.25 : 1 (spectrum 2); 0.5 : 1 (spectrum 3); 0.75 : 1 (spectrum 4); 1 : 1 (spectrum 5); 2 : 1 (spectrum 6). Spectrum (7) denotes hMGL with NAM at a 2 : 1 NAM–hMGL molar ratio after a 24 h incubation. Peak assignments are as given in Fig. 3A. Panel B: Best-fit plots of the time-course of WT hMGL hydrolytic activity in the absence (blue) or presence of increasing molar ratios of NAM, as designated by the color-code in Panel A.
The activity correlation to the NMR hMGL titration experiment with NAM demonstrates a progressive hMGL inactivation with increasing NAM:hMGL molar ratio (Fig. 5B). Substantial hMGL inhibition by NAM was evident at a NAM–hMGL molar ratio of 1 : 1, and hydrolytic activity of the enzyme did not recover over a subsequent 24 h period (Fig. 5B). These combined data suggest that NAM disrupts functionally important hydrogen-bond interactions involving the hMGL catalytic triad, thereby inactivating the enzyme as an irreversible covalent inhibitor. Thus, hMGL inhibition by NAM likely reflects durable changes in the enzyme’s conformational properties critical to catalysis. Such NAM-induced conformational changes might prevent amino-acid residues in the active site hydrogen-bond network from adequately (re)forming these critical hydrogen-bond interactions over time. Along with prior demonstration that mutation of Cys208 and/or Cys242 to alanine did not affect hMGL catalytic activity, whereas partial NAM alkylation of either cysteine residue inhibited the enzyme by ~80%,30 the data herein underscore the critical role of hMGL cysteine residues neighboring the active-site in hMGL inhibition by NAM, but not in hMGL catalytic function.
Experimental
Materials
Standard laboratory chemicals, culture media, and buffers were purchased from Sigma Chemical (St. Louis, MO, USA) and Fisher Chemical (Pittsburg, PA, USA), unless otherwise specified.30 SDS-PAGE supplies where from Bio-Rad (Hercules, CA, USA). NAM was purchased from Cayman Chemical (Ann Arbor, MI, USA). AM6701 and AHMMCE were synthesized at the Center for Drug Discovery, Northeastern University, by standard routes.
Expression and isolation of recombinant WT and mutant hMGLs
The hMGL C242A and D239T mutants were generated using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). The DNA primary structures of the mutants were confirmed by sequencing. Recombinant WT and mutant hMGLs were expressed essentially as previously detailed.30 In brief, a single E. coli BL21 (DE3) colony containing the appropriate pET45His6hMGL plasmid was inoculated into 10 ml of Luria broth supplemented with ampicillin (100 μg ml−1) and grown overnight at 30 °C with shaking (250 rpm). The next morning, these 10 ml were used to inoculate 500 ml of fresh Luria broth-ampicillin medium and allowed to grow under the specified conditions until the culture reached an OD600 of 0.6–0.8, at which time expression was induced by adding 0.4 mM (final concentration) isopropyl-β-D-thiogalactopyranoside. After 4 h induction, the cells were harvested by centrifugation, washed with phosphate-buffered saline, and held at −80 °C. Three grams (wet-weight) of cells were resuspended in 20 ml lysis buffer [10 mM Na-phosphate, pH 7.45, containing 100 mM NaCl and up to 1.0% (w/v) N-dodecyl-β-D-maltoside detergent] and disrupted on ice by six, 30 s sonication cycles, each cycle consisting of 1 s bursts at 50 W separated by a 5 s interval. The resulting lysate, after centrifugation (10 000g, 20 min, 4 °C), was used as the basis for hMGL purification through an immobilized metal-affinity chromatography-based procedure to be detailed elsewhere. Functional, monomeric hMGL (WT and mutants) was obtained, the purity of which was checked under denaturing conditions on 10% SDS-PAGE gels. Prior to enzyme assay or NMR analyses, hMGL samples in elution buffer (10 mM Na-phosphate, pH 7.45, containing 100 mM NaCl and 300 mM imidazole) were dialyzed for 12 h to ensure thorough imidazole removal using a membrane with a molecular-weight cutoff of 12 000–14 000 Da. Enzyme protein concentration was determined spectrophotometrically using the molar extinction coefficient, ε280 = 24 910 M−1 cm−1. In this report, the wild-type hMGL sequence (without His6-tag) is used as the basis for the numerical descriptor of the enzyme’s amino-acid residues.
MGL assay
hMGL activity was measured as the hydrolysis of the reporter fluorogenic substrate AHMMCE to coumarin fluorophore, essentially as developed and detailed previously by us.30 AHMMCE was stored at −20 °C as a 10 mM DMSO stock solution, which was thawed and diluted 1 : 1 (v/v) with assay buffer (50 mM Tris-HCl, pH 7.4) such that the final DMSO concentration in each assay reaction was well below 8%. This procedure ensures that AHMMCE remains in solution during the assay. To start the enzyme reaction, AHMMCE from the diluted stock was added to known amounts of (WT or mutant) hMGL protein to achieve a final substrate concentration of 50 or 100 μM in a total assay volume of 200 μl. Reactions were incubated at 22 °C, and fluorescence readings at 360 nm/460 nm (λexcitation/λemission) were taken every 15 min over a 3 h period using a Synergy HT Plate Reader (BioTek Instruments, Winooski, VT, USA) in kinetic mode. Relative fluorescence units were converted to the amount of coumarin formed based on a standard coumarin fluorescence curve. Under these conditions, nonenzymatic AHMMCE hydrolysis to fluorescent product (coumarin) was negligible.
NMR spectroscopy
Enzyme samples for NMR were prepared in a solution of 93% H2O–7% D2O (v/v) containing 20 mM Na-phosphate, 100 mM NaCl, 0.02% (w/v) NaN3, and a low content of residual N-dodecyl-β-D-maltoside, at pH 7.4. hMGL protein concentration was 80–100 μM. AM6701 and NAM (50 mM) were dissolved in DMSO-d6 and added to the NMR tube to give a desired, final inhibitor–hMGL molar ratio.
All 1D 1H NMR spectra were obtained at 37 °C on a 700-MHz Bruker AVANCE II NMR spectrometer equipped with a 5 mm triple resonance probe. NMR spectra for low-field exchangeable protons were recorded using the 1331 pulse sequence38,39,41 with the excitation maximum at 16 ppm to minimize water excitation at 4.7 ppm. Application of binominal 1331 pulse sequence provided the best results for water suppression (higher receiver gain), but introduced significant offset-dependent phase distortions. These distortions cannot be compensated for by data processing. Acquisition was performed with a 90° flip angle, recycle delay 0.6 s, and 32 K data points for 4 K scans. Data were processed and analyzed using the Topspin 2.1 software package (Bruker). Exponential weighting resulting in a 30 Hz line broadening was applied. The proton chemical shift values were directly referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid at 0 ppm.
Conclusion
Indirect potentiation of cannabinergic signaling by inhibiting MGL-catalyzed 2-AG inactivation is an attractive modality for exploiting therapeutically the tissue-protective function of the endocannabinoid system. The present work constitutes an NMR-based approach toward elucidating the molecular details involved in the inhibition/recovery of hMGL enzymatic activity. The results offer detailed structural insights into small-molecule inhibition of hMGL and mechanistic aspects of the enzyme’s catalysis useful for the development of novel, pharmacologically attractive hMGL inhibitors as potential drugs. More generally, this study demonstrates the utility of solution 1H NMR for direct mechanistic exploration of enzyme-inhibitor interaction in drug design.
Acknowledgments
This work has been supported by the United States National Institutes of Health (NIH)/National Institute on Drug Abuse (NIDA) through grants DA09158, DA00493, DA03801, and DA07312 to AM.
Footnotes
This article is part of a Molecular BioSystems themed issue on Chemical Genomics.
References
- 1.Di Marzo V. Trends Pharmacol Sci. 2006;27:134–140. doi: 10.1016/j.tips.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Hwang J, Adamson C, Butler D, Janero DR, Makriyannis A, Bahr BA. Life Sci. 2010;86:615–623. doi: 10.1016/j.lfs.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- 4.Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong WI, Bátaki S, Marsicano G, Lutz B, Buetther C, Kunos G. J Clin Invest. 2008;118:3160–3169. doi: 10.1172/JCI34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Argenio G, Valenti M, Scaglione G, Cosenza V, Sorrentini I, Di Marzo V. FASEB J. 2006;20:2544–570. doi: 10.1096/fj.05-4943fje. [DOI] [PubMed] [Google Scholar]
- 6.Degn M, Lambertsen KL, Petersen G, Meldgaard M, Artmann A, Clausen BH, Hansen SH, Finsen B, Hansen HS, Lund TM. J Neurochem. 2007;103:1907–1916. doi: 10.1111/j.1471-4159.2007.04892.x. [DOI] [PubMed] [Google Scholar]
- 7.Magrioti V, Naxakis G, Hadjipavlou-Litina D, Makriyannis A, Kokotos G. Bioorg Med Chem Lett. 2008;18:5424–5427. doi: 10.1016/j.bmcl.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vandevoorde S. Curr Top Med Chem. 2008;8:247–267. doi: 10.2174/156802608783498005. [DOI] [PubMed] [Google Scholar]
- 9.Janero DR, Vadivel SK, Makriyannis A. Int Rev Psychiatry. 2009;21:122–133. doi: 10.1080/09540260902782778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long JZ, Nomura DK, Vann RE, Walentiny M, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Proc Natl Acad Sci U S A. 2009;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 12.Zvonok N, Williams J, Johnston M, Pandarinathan L, Janero DR, Li J, Krishnan SC, Makriyannis A. J Proteome Res. 2008;7:2158–2164. doi: 10.1021/pr700839z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. J Lipid Res. 2008;50:3–21. doi: 10.1194/jlr.R800031-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Proc Natl Acad Sci U S A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blankman JL, Simon GM, Cravatt BF. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugiura T. BioFactors. 2009;35:88–97. doi: 10.1002/biof.18. [DOI] [PubMed] [Google Scholar]
- 17.Dinh TP, Freund TF, Piomelli D. Chem Phys Lipids. 2002;121:149–158. doi: 10.1016/s0009-3084(02)00150-0. [DOI] [PubMed] [Google Scholar]
- 18.Dinh TP, Kathuria S, Piomelli D. Mol Pharmacol. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- 19.Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long JZ, Nomura DK, Cravatt BF. Chem Biol. 2009;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bisogno T, Ortar G, Petrosino S, Morera E, Palazzo E, Nalli M, Maione S, Di Marzo V and the Endocanabinoid Research Group. Biochim Biophys Acta, Mol Cell Biol Lipids. 2009;1791:53–60. doi: 10.1016/j.bbalip.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Holtfrerich A, Makharadze T, Lehr M. Anal Biochem. 2010;399:218–224. doi: 10.1016/j.ab.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 25.Bowman AL, Makriyannis A. J Comput-Aided Mol Des. 2009;23:799–806. doi: 10.1007/s10822-009-9289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janssen Pharmaceutica NV, Schubert C, Grasberger BL, Schalk-Hihi C, Maguire DM, Lewandowski FA, Milligan CM, Alexander RS. PCT Application WO/2009/132267. 2009
- 27.Labar G, Bauvois C, Borel F, Ferrer JL, Wouters J, Lambert DM. ChemBioChem. 2010;11:218–227. doi: 10.1002/cbic.200900621. [DOI] [PubMed] [Google Scholar]
- 28.Bertrand T, Augé F, Houtmann J, Rak A, Vallée F, Mikol V, Berne PF, Michot N, Cheuret D, Hoornaert C, Mathieu M. J Mol Biol. 2010;396:663–673. doi: 10.1016/j.jmb.2009.11.060. [DOI] [PubMed] [Google Scholar]
- 29.Saario SM, Salo OMH, Nevalainen T, Poso A, Laitinen JT, Järvinen T, Niemi R. Chem Biol. 2005;12:649–656. doi: 10.1016/j.chembiol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Zvonok N, Pandarinathan L, Williams J, Johnston M, Karageorgos I, Janero DR, Krishnan SC, Makriyannis A. Chem Biol. 2008;15:854–862. doi: 10.1016/j.chembiol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King AR, Lodola A, Carmi C, Fu J, Mor M, Piomelli D. Br J Pharmacol. 2009;157:974–983. doi: 10.1111/j.1476-5381.2009.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matuszak N, Muccioli GG, Labar G, Lambert DM. J Med Chem. 2009;52:7410–7420. doi: 10.1021/jm900461w. [DOI] [PubMed] [Google Scholar]
- 33.Ortar G, Cascio MG, Moriello AS, Camalli M, Morera E, Nalli M, Di Marzo V. Eur J Med Chem. 2008;43:62–72. doi: 10.1016/j.ejmech.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 34.Boehr DD, Dyson HJ, Wright PE. Chem Rev. 2006;106:3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 35.Baldwin AJ, Kay LE. Nat Chem Biol. 2009;5:808–814. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Chandra P, Jones PG, Kennedy JD. Assay Drug Dev Technol. 2008;6:387–393. doi: 10.1089/adt.2007.122. [DOI] [PubMed] [Google Scholar]
- 37.Savinainen JR, Yoshno M, Minkkilä A, Nevalainen T, Laitinen JT. Anal Biochem. 2010;399:132–134. doi: 10.1016/j.ab.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 38.Hwang TL, Shaka AJ. J Magn Reson, Ser A. 1995;112:275–279. [Google Scholar]
- 39.Hore PJ. J Magn Reson. 1983;54:283–300. [Google Scholar]
- 40.Wuthrich K. NMR of Proteins and Nucleic Acids. Wiley; New York: 1986. [Google Scholar]
- 41.Plateau P, Gueron M. J Am Chem Soc. 1982;104:7310–7311. [Google Scholar]
- 42.Ekici OD, Paetzel M, Dalbey RE. Protein Sci. 2008;17:2023–2037. doi: 10.1110/ps.035436.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tyukhtenko SI, Litvinchuk AV, Chang C-F, Leu RJ, Shaw J-F, Huang T-H. FEBS Lett. 2002;528:203–206. doi: 10.1016/s0014-5793(02)03308-2. [DOI] [PubMed] [Google Scholar]
- 44.Berlow RB, Igumenova TI, Loria JP. Biochemistry. 2007;46:6001–6010. doi: 10.1021/bi700344v. [DOI] [PubMed] [Google Scholar]
- 45.Loria JP, Berlow RB, Watt ED. Acc Chem Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]