

Abstract
Heart failure is a major cause of disability and death worldwide, and approximately half of heart failure-related deaths are sudden and presumably due to ventricular arrhythmias. Patients with heart failure have been shown to be at 6- to 9-fold increased risk of sudden cardiac death compared to the general population. (AHA. Heart Disease and Stroke Statistics—2003 Update. Heart and Stroke Facts. Dallas, TX: American Heart Association; 2002) Thus, electrophysiological remodelling associated with heart failure is a leading cause of disease mortality and has been a major investigational focus examined using many animal models of heart failure. While these studies have provided an important foundation for understanding the arrhythmogenic pathophysiology of heart failure, the need for corroborating studies conducted on human heart tissue has been increasingly recognized. Many human heart studies of conduction and repolarization remodelling have now been published and shed some light on important, potentially arrhythmogenic, changes in human heart failure. These studies are being conducted at multiple experimental scales from isolated cells to whole-tissue preparations and have provided insight into regulatory mechanisms such as decreased protein expression, alternative mRNA splicing of ion channel genes, and defective cellular trafficking. Further investigations of heart failure in the human myocardium will be essential for determining possible therapeutic targets to prevent arrhythmia in heart failure and for facilitating the translation of basic research findings to the clinical realm.
Keywords: Heart failure, Remodelling, Arrhythmias, Human heart, Conduction, Repolarization, Ion channels, Action potential
Introduction
Heart failure is a major problem in the western world, with prevalence rates estimated at 1–2% and incidence approaching 5–10 per 1000 persons annually.1 Heart failure patients generally have a poor prognosis, as 5-year survival rates have been reported to range from 35 to 60%,1 and approximately half of these heart failure-associated deaths are sudden and presumably due to ventricular tachyarrhythmias.1,2 Continuous, 24-hour electrocardiogram (ECG) monitoring of patients with New York Heart Association stage II–III heart failure revealed that 50% demonstrate episodes of ventricular tachycardia.3 Despite the burden of arrhythmias in patients with heart failure, the current therapeutic options for the prevention of sudden cardiac death in heart failure are rather limited. Antiarrhythmic drugs can actually produce worse outcomes in terms of mortality for heart failure patients,4 and due to the failure of pharmacotherapy, primary prevention with implantable cardioverter defibrillators has become the recommended practice for heart failure patients with an ejection fraction <30%.5–7 Thus, the translation of basic research findings into improved therapies for arrhythmia in heart failure is desperately needed.
Animal model investigations have provided many mechanistic insights into electrophysiological changes during heart failure. Dog models of heart failure have demonstrated evidence for the disruption of proper gap junction localization and decreased fast sodium currents, potentially explaining arrhythmogenic slowing down of conduction in heart failure.8–10 Rabbit and canine models have also revealed downregulations of various repolarizing potassium currents, increases in late sodium current, or alterations in sodium–calcium exchanger activity, which could explain characteristic action potential prolongation in heart failure.11–15 Indeed, animal model studies have lead to various hypotheses for the mechanisms of arrhythmogenesis in heart failure. However, the heart failure-like syndrome that can be rapidly induced in rabbits and dogs with chronic fast pacing has several important differences from human heart failure. In these tachypacing-induced models of heart failure, neither species develops left ventricular hypertrophy, and the heart failure phenotype is largely reversible with the cessation of pacing.16 Also, the complex nature of the progression and pathogenesis of human heart failure make it difficult to faithfully replicate in an animal model. These limitations make the translation of results from heart failure models to humans a difficult challenge. As has been pointed out in a recent perspective article by Robbins, the translation of basic cardiovascular research into the improvement of human health has been less significant than was hoped.17
There is now a growing literature of detailed functional, structural, cellular, and molecular studies comparing normal and failing human hearts that provide evidence of the proarrhythmic mechanisms, originally uncovered in animal models, that are most likely to be important in humans. We focus our review on studies of arrhythmogenic remodelling of activation and repolarization in human heart failure, highlighting research areas with concordance and potential discrepancies between animal models and humans. We review findings from human heart failure investigations of activation remodelling, concentrating on gap junctional and sodium channel alterations, and we also discuss repolarization remodelling, including studies on transmural repolarization gradients, potassium channels, late sodium current, and the sodium-calcium exchanger. For more thorough reviews of electrical remodelling in animal models of heart failure, we refer the reader to several detailed reviews on the subject.2,18–21
Activation remodelling
Conduction velocity slowing down is a characteristic change of the failing myocardium, and it leads to QRS complex widening on the surface ECGs of patients with heart failure.22 Slowed conduction in the failing myocardium could potentially be arrhythmogenic by promoting unidirectional conduction block and re-entrant arrhythmias.23 The rapid propagation of action potentials in the myocardium is dependent upon proper gap junctional coupling between adjacent cardiomyocytes and swift activation of the cardiac sodium current; thus, gap junctional proteins and sodium channels are important potential targets for arrhythmogenic remodelling of activation in the human heart.24,25
Sodium current
The cardiac sodium current, INa, produced by voltage-dependent sodium channels, is responsible for the upstroke, or phase 0, of the cardiac action potential.21 These rapidly activating and inactivating cardiac sodium channels are comprised of α and β protein subunits, which are encoded by the SCN5A and SCN1B genes, respectively.26
Many groups have examined cardiac myocytes that have been dissociated from non-failing and failing human ventricular tissue. These studies have been particularly informative for the investigation of sodium currents in the human heart. Valdivia et al.9 demonstrated that peak density of transient sodium currents was decreased by 57% in cardiomyocytes isolated from failing hearts as compared with non-failing hearts. However, mRNA expression for the both the α or β sodium channel subunits was not different between groups, suggesting that the sodium channel may not be transcriptionally regulated in human heart failure. Thus, they postulated that the reduction in cardiac sodium current in heart failure could be due to non-functional channels or decreased functional expression of the sodium channels at the cell surface.
The molecular mechanism for sodium channel regulation in human heart failure has been further examined, and alternative splicing of sodium channel mRNA has been demonstrated. Non-functional splice variants of SCN5A are increased in human heart failure and the full-length functional mRNA only represents approximately half of the total SCN5A mRNA in the failing myocardium.27 Although the non-failing and failing human heart tissues used in this study were obtained under different conditions and the physiological role of non-functional sodium channel proteins is unclear, this study strengthens the argument that non-functional sodium channels may lead to reduced peak sodium current in heart failure. In a more recent study, the same group investigated the role of splicing factor upregulation in generating the increased splicing of SCN5A in human heart failure.28 They identified two splicing factors, RBM25 and LUC7L3, that regulate the E28C and E28D splice variants of SCN5A and are upregulated in human heart failure. Interestingly, in complementary model studies, they also demonstrate that these splicing factors may be regulated by hypoxic stimuli and angiotensin II, suggesting signaling pathways that could link the syndrome of heart failure to increased splicing factor activity. Taken together, these results suggest that reduction of sodium current could be involved in arrhythmogenic conduction slowing and that altered sodium channel mRNA splicing is a potential contributing mechanism.
Connexin43
Gap junctions serve as electrical conduits between adjacent myocardial cells, and are necessary for the fast and efficient spread of electrical excitation through the heart.24 Gap junctions are comprised of connexin channels, which are formed by joining two connexon hemichannels from adjacent cells.29 Each hemichannel is formed by the oligomerization of six connexin subunits, and the most prevalent connexin subunit in the human ventricle is connexin43.30 In the normal heart, gap junctions are concentrated at the intercalated disc to allow for rapid longitudinal spread of myocardial activation.24
Gap junctional protein expression and localization are important for the normal spread of conduction.24 Connexin43 expression is decreased at both the mRNA and protein levels in end-stage failing human hearts due to both ischaemic and dilated cardiomyopathy compared with non-failing hearts.31 In the failing human heart, connexin43 is also improperly localized. Work by Smyth et al.29 demonstrated that one potential mechanism for improper targeting of connexin43 to the intercalated disc is the disruption of proper microtubule trafficking. They showed reduced levels of connexin43 in the ventricle of end-stage ischaemic failing human hearts, with both immunostaining of tissue sections and biochemical isolation of junctional membrane fractions. They also demonstrated that the microtubule plus end protein, EB1, which functions to target the delivery of connexin43 to the intercalated disc, also has decreased intercalated disc expression in the failing human heart. Supporting experiments, performed in other model systems, suggest that oxidative stress disrupts proper microtubular trafficking.29
Optical mapping of coronary-perfused left ventricular wedge preparations has also been used to examine conduction changes in human hearts with end-stage, non-ischaemic cardiomyopathy and non-failing controls (Figure 1). Glukhov et al.32 examined transmural conduction velocities as a function of distance between the epicardium and endocardium in both non-failing and non-ischaemic human hearts, finding that conduction velocities were lowest at the epicardium and increased towards the endocardium. However, conduction velocities were reduced in failing hearts compared with non-failing hearts in each transmural region. Interestingly, they also studied transmural connexin43 expression as a function of distance from epicardium to endocardium and demonstrated a trend for connexin43 quantity that paralleled the conduction velocity changes. Connexin43 levels increased from epicardium to endocardium and were uniformly reduced in failing hearts compared with non-failing hearts. In addition to overall connexin43 levels, they also examined the spatial distribution of connexin43 in failing hearts by co-immunostaining with N-cadherin, which is a structural protein that localizes to the intercalated discs of cardiomyocytes. They found that less connexin43 was localized with N-cadherin in failing compared with non-failing hearts and showed that the most pronounced differences were in the subepicardium. While such investigations are limited to the correlation of molecular and electrophysiological results, this study serves as an excellent example of the type of multiscale approach to human heart investigation—from molecular and cellular techniques to functional electrophysiology—that is needed to elucidate the arrhythmogenic changes in human heart failure and the molecular targets underlying these alterations. Taken together, disruption of normal connexin43 targeting and localization at the intercalated disc could lead to delayed activation and arrhythmogenesis in human heart failure.
Figure 1.
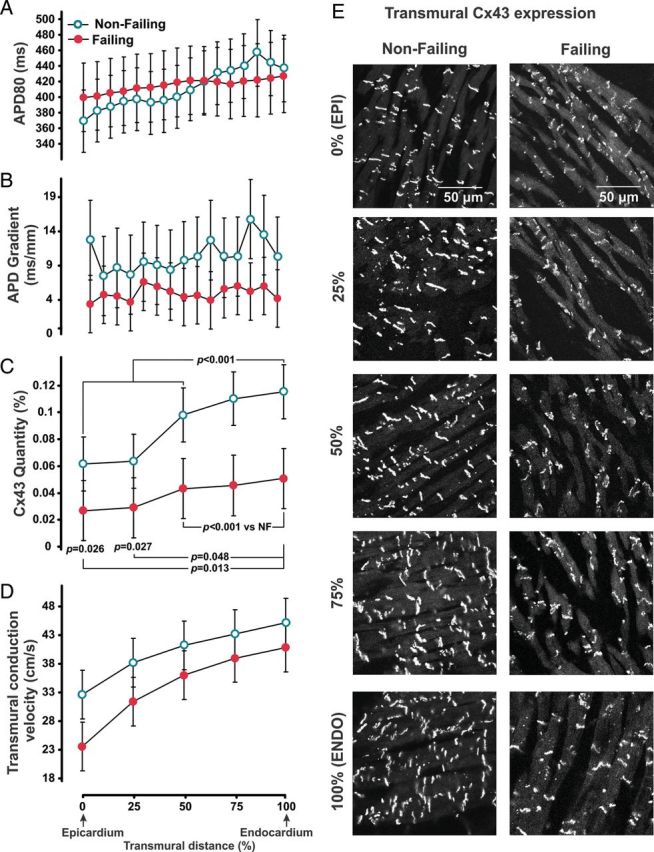
Summary transmural electrophysiological data and representative connexin43 immunolabelling measured from left ventricular preparations isolated from non-failing and dilated cardiomyopathic failing human hearts. Conduction velocity in non-failing and failing human hearts parallels connexin43 quantity, suggesting that connexin43 is an important determinant of conduction velocity in the human myocardium. (A) Average action potential duration at 80% repolarization measured at a cycle length of 2000 ms plotted as a function of transmural distance between the epicardium and endocardium. Transmural distance expressed as a percentage of transmural thickness. (B) Spatial derivative of the action potential duration as a function of transmural depth. (C) Average connexin43 protein quantity measured from transmural layers between the epicardium and endocardium. (D) Conduction velocity determined as a function of transmural distance. (E) Immunolabelling images from non-failing and failing hearts showing connexin43 expression at different transmural layers. Reprinted with permission from Glukhov et al.32
Repolarization remodelling
Action potential prolongation is a hallmark change of the failing myocardium and has been reported in isolated cardiac preparations and cardiac myocytes from failing human hearts as compared with non-failing hearts.33–35 Prolongation of the action potential underlies QT prolongation on the surface ECG of heart failure patients22 and could be proarrhythmic by promoting triggered activity or, if heterogeneous, by causing enhanced dispersion of repolarization. Triggered activity in myocardial cells occurs when action potential prolongation leads to early or delayed after depolarizations, in which calcium can re-enter the cardiomyocyte and stimulate a new action potential.23 Enhanced dispersion of repolarization could lead to functional re-entry and arrhythmias.23 Thus, there are several targets for which altered regulation could cause action potential prolongation, and many ionic currents and exchangers have been shown to be remodelled in human heart failure.
Dispersion of repolarization
Action potential duration prolongation and dispersion of repolarization were examined in coronary-perfused left ventricular wedge preparations from the human heart using optical mapping.36 Transmural action potential durations were analysed at 80 percent repolarization, and Glukhov et al. found that action potentials were prolonged in heart failure in a transmurally heterogeneous manner, with the greatest prolongation occurring in the subepicardial layer. Counter to findings from animal models of heart failure, they also found that the gradients of action potential duration from subendocardium to subepicardium were significantly decreased in failing compared with non-failing hearts (29 ± 6 ms versus 111 ± 13 ms, P < 0.005), demonstrating greater action potential duration homogeneity. They also went on to examine whether gap junctional coupling correlated with this reduced transmural dispersion of repolarization in the failing human myocardium. Immunostaining of connexin43 showed a similar pattern to the other connexin43 results described above,32 with lower connexin43 in the failing heart. Connexin43 downregulation in heart failure suggests that reduced gap junctional cell-to-cell coupling does not necessarily lead to increased action potential heterogeneity, as would have been predicted. Therefore, remodelling of other ion channels or membrane transporters must be considered to explain this observation.
Late sodium current
Under normal conditions, voltage-dependent cardiac sodium channels rapidly inactivate, thus producing minimal current during the plateau phase of the cardiac action potential.26 However, during pathological conditions, these sodium channels can reopen in the later phases of the action potential, resulting in late inward sodium current. Due to relatively low ionic conductance during the action potential plateau, the delicate balance of inward and outward currents could be disrupted by relatively small perturbations in current densities, leading to prolongation of the action potential.37–39
The role of late sodium current activity in action potential prolongation has been investigated in human heart failure. Maltsev et al.40 recorded late sodium current from left ventricular cardiomyocytes isolated from donor and failing human hearts. This late INa was demonstrated to have similar sensitivities to tetrodotoxin and saxitoxin to the cardiac sodium channel, suggesting that the cardiac sodium channel isoform underlies this current. However, they also observed that the sizes of the late current and the transient sodium current were independent. While late INa densities and biophysical properties were not different between non-failing and failing heart groups in this study, early after depolarizations from failing ventricular myocytes were observed and could be suppressed by the application of tetrodotoxin. Conversely, Valdivia et al.9 demonstrated increased late sodium current in cardiomyocytes from failing left ventricular tissue when compared with normal hearts. They measured late sodium current as a percentage of the transient current, and showed that late sodium current was 2.4 ± 0.5% of the peak sodium current in cells from failing hearts, while only 0.2 ± 0.1% in cardiomyocytes from normal hearts. However, in both of these investigations, the human heart sample sizes were small, particularly in the non-failing heart category, and thus the role of late INa in prolongation of the action potential during heart failure may require further investigation.
The biophysical mechanism for late sodium current in the human heart was investigated by Undrovinas et al.41 using cell-attached patch preparations from isolated normal and failing human ventricular myocytes. They observed two types of late sodium channel openings—scattered late and burst—that happen within the range of membrane potentials where early after depolarizations are also observed. They further showed that failing ventricular cells demonstrated sodium channel openings with slower inactivation and longer burst durations than those in normal ventricular cells, suggesting greater late INa in heart failure.
Repolarizing potassium currents
Outward myocardial potassium currents are responsible for the repolarization phase of the cardiac action potential.26 While there are many potassium currents that may be expressed in the human heart, the most prominent are the transient outward current, the rapid and slow delayed rectifier currents, and the inward rectifier current.39 The transient outward potassium current, Ito, is responsible for phase 1 repolarization, while the delayed rectifier potassium currents, IKr and IKs, contribute to repolarization during phases 2 and 3 of the action potential. The inward rectifier current, IK1, maintains the resting membrane potential of cardiac myocytes, but also contributes to terminal repolarization.26
Transient outward current
Of the repolarizing potassium currents, alterations of the transient outward current have been the most thoroughly investigated to date. Several studies have found Ito to be downregulated in human heart failure. Beuckelmann et al.34 compared transient outward current density from cardiomyocytes isolated from control non-failing and end-stage failing hearts with dilated cardiomyopathy and found that Ito was reduced in failing hearts. Differences in transmural heterogeneity of transient outward current density have also been examined in non-failing and failing human hearts.42 In the non-failing heart, Ito density was shown to be four-fold lower in subendocardial compared with subepicardial cells. In cells from failing hearts, Ito was reduced relative to cells from non-failing hearts only in the subepicardial region. Interestingly, the biophysical properties of Ito also varied transmurally, with the potential at half-maximal activation being more positive in subendocardial cardiomyocytes; however, transient outward current biophysical properties were not different between non-failing and failing hearts. Thus, regionally specific regulation of the transient outward current may be important in both health and disease.
Kaab et al.43 investigated the potential molecular mechanism for transient outward current downregulation in human heart failure. This study demonstrated that Kv4.3 mRNA, which encodes the Ito α subunit, is decreased by 30% in human tissues isolated from failing hearts compared with non-failing hearts. To correlate molecular findings with electrophysiological data, they also isolated cardiac myocytes from adjacent regions of the same hearts and confirmed that peak transient outward current density was reduced in heart failure. Despite the consistency of Ito downregulation in human heart failure, the role of Ito in the remodelling of action potential duration remains somewhat unclear due to its primary role during phase 1 of the action potential. Thus, decreased expression of the transient outward current may not be directly related to action potential prolongation. Transient outward current density may actually be involved in regulating the degree of calcium influx during the action potential plateau, such that reduced Ito may lead to decreased calcium influx and abbreviated action potentials.44,45 Other groups have suggested that Ito would have minimal impact on the action potential duration in large mammals.21,46 Thus, the role of transient outward current density in the determination of action potential durations in the human heart requires further investigation.
Delayed rectifier currents
Delayed rectifier potassium currents have been particularly difficult to evaluate in human isolated cardiomyocytes. In two different studies, current amplitudes recorded in isolated cells from both non-failing and failing hearts were small, precluding comparison.34,47 In addition, previous gene expression studies did not reveal any differences in KCNH2 or KCNQ1 mRNA expression, which encode the α subunits of IKr and IKs, respectively.43,48 However, mRNA expression for the delayed rectifier accessory subunit, KCNE1, was upregulated, which could potentially alter the activation kinetics of IKs. Because of the difficulty in investigating IKr in isolated cells, we recently examined its expression pharmacologically in non-failing and failing human hearts using optical mapping of coronary-perfused left ventricular wedge preparations.49 IKr was blocked using the high-affinity drug, E-4031, and transmural action potential durations were prolonged with IKr blockade in non-failing hearts, as anticipated. However, the degree of action potential prolongation was significantly less with rapid delayed rectifier current blockade in failing hearts, suggesting that functional expression of IKr is reduced in failing hearts. Western blot analysis was also performed on protein samples prepared from the same hearts used for functional electrophysiological examination, and the human ether-a-go-go related gene 1a α subunit protein, which underlies IKr, was found to be reduced in failing hearts compared with non-failing hearts. Due to their role during repolarization phases 2 and 3, the delayed rectifier potassium currents may represent important targets that are downregulated in human heart failure, leading to arrhythmogenic action potential prolongation. However, more detailed investigations, particularly of the slow delayed rectifier current, IKs, are needed in the human heart.
Inward rectifier current
Densities of the inward rectifier potassium current have been found to be decreased in human heart failure. One study demonstrated reduced IK1 at negative voltages in isolated cells from failing compared with non-failing hearts.34 Additionally, Koumi et al.50 examined IK1 whole-cell current density and channel properties from human isolated cardiomyocytes and found reduction in inward rectifier current in cells from hearts with dilated cardiomyopathy relative to controls. They went on to investigate differences in IK1 with regard to aetiology of cardiomyopathy and found that single-channel properties were different between IK1 channels isolated from hearts with ischaemic versus dilated cardiomyopathy. Thus, aetiology-specific regulation of the inward rectifier current may result in different effects on action potential duration. The same group also investigated the role of β-adrenergic modulation on the inward rectifier current in myocytes isolated from non-failing and failing human hearts.51 In the normal heart, they found that application of isoproterenol could suppress IK1 by reducing the channel open probability. Conversely, in cells from failing hearts, the modulation of IK1 by isoproterenol was significantly reduced. Because this and other repolarizing potassium currents are regulated by β-adrenergic stimulation, detailed analysis of action potential duration regulated in human heart failure in response to β-adrenergic activation is needed.
Despite these changes in IK1 current, the molecular mechanism for inward rectifier current downregulation in the human heart during heart failure is not yet understood. IK1 does not appear to be transcriptionally regulated, as Kir2.1 mRNA, which encodes the IK1 α subunit, does not to appear to downregulated in human heart failure.43,52
Sodium–calcium exchanger
In addition to being important for calcium handling, the sodium–calcium exchanger, NCX, may also regulate cardiac action potential duration.53 The normal forward activity of NCX allows three sodium ions to be transported into the cell in exchange for pumping one calcium ion out of the cell. Thus, the sodium–calcium exchanger serves as an electrogenic pump, increased activity of which could potentiate the plateau phase of the action potential.
Sodium–calcium exchanger expression and current density have been inconsistently altered in human heart failure.54 Studer et al.55 quantified mRNA gene expression of the sodium–calcium exchanger in large samples from normal donors and heart failure patients, and found that NCX mRNA was increased by 55% in patients with dilated cardiomyopathy and 41% in patients with coronary artery disease. Sodium–calcium exchanger protein levels are either increased or unchanged in the failing human myocardium.56–58 However, functional studies of NCX current density have shown that NCX current, as a function of calcium concentration, is unchanged in human heart failure, although the sodium–calcium exchanger did have a greater contribution to intracellular calcium relaxation due to the depressed sarcoplasmic reticulum calcium uptake.59 Weber et al.60 also demonstrated that NCX current direction during the action potential plateau shifted from forward mode (calcium efflux and sodium influx) to reverse mode (calcium influx and sodium efflux) due to an increased intracellular sodium and reduced submembrane intracellular calcium concentration in the failing human heart. Reverse mode sodium–calcium exchanger activity would serve to hyperpolarize the cell membrane, which could abbreviate instead of prolong the action potential. Thus, findings regarding altered NCX expression and regulation in the human heart have not clearly indicated that NCX plays an important role in action potential prolongation in human heart failure.
Validation of and contradiction with animal model findings
Several of the major targets that are found to be altered in animal models of heart failure are similarly remodelled in human heart failure (Table 1). Altered regulation and localization of connexin43 has been demonstrated in canine heart failure models,8 and has been validated at multiple scales in human heart studies as an important cause of conduction slowing in human heart failure. Changes in late sodium current from canine models also parallel the increase in late sodium current seen in human heart failure,9,61 suggesting an important role for late INa in repolarization abnormalities. Despite its unclear function in action potential duration, downregulation of the transient outward current appears to be a very consistent finding in both human heart failure and animal heart failure models.11,12,62–66 These results implicate Ito as an important target for repolarization remodelling in humans.
Table 1.
Summary of human heart failure investigations of activation and repolarization compared with findings from rabbit and dog models of heart failure
| Activation/repolarization remodelling | Target current/protein | Human investigations | Rabbit and dog investigations |
|---|---|---|---|
| Activation | Connexin43 | ↓29,31,32,36 | ↓8 |
| Activation | Peak INa | ↓9,27,28 | ↓9,10 |
| =62 | |||
| Repolarization | Late INa | ↑9,41,72 | ↑9,61 |
| =40 | |||
| Repolarization | Ito | ↓34,42,43 | ↓11,12,62–66 |
| Repolarization | IKr | ↓49 | ↓12 |
| =43,48 | =11,66 | ||
| ↑65 | |||
| Repolarization | IKs | ? | ↓11,12 |
| =65 | |||
| Repolarization | IK1 | ↓34,50 | ↓11,62,69,70 |
| =43,52 | =12,63,65,66 | ||
| Repolarization | NCX | =56,58,59 | ↑15,68–70 |
| ↑55–57 | ↓73 |
↓, decreased expression; =, no change; ↑, increased expression; ?, currently unknown
Although there are many similar findings between animal and human studies, there are notable differences as well. While dispersion of repolarization has been suggested to be a major arrhythmogenic substrate in heart failure because it has been demonstrated in dog models,67 results from left ventricular wedge preparations in the human heart suggest that the action potential duration becomes more transmurally homogeneous in the failing human myocardium.36 Dispersion of repolarization may still be present or increased between other anatomic regions in the human heart, such as between the apex and base; however, currently the role for enhanced dispersion of repolarization as an arrhythmogenic mechanism in human heart failure has not been substantiated. Additionally, the sodium–calcium exchanger has consistently been shown to be upregulated in canine and rabbit heart failure studies, which could lead to action potential prolongation.15,68–70 However, results from human heart failure investigations are much less clear as to whether NCX is significantly remodelled in heart failure and, if so, whether the changes would lead to prolongation or shortening of the action potential duration.
Finally, some activation and repolarization targets appear to be more consistently altered in human heart failure than in studies conducted in animal models. This could be the result of variability in animal model species or heart failure-induction methods or could indicate the need for larger sample sizes in validating human heart investigations. Peak sodium current in animal models has been demonstrated to be either decreased or unchanged.9,61,62 While functional examinations of IK1 in human heart failure have shown decreased current density,34,50,62,65,66,69,70 animal investigations have shown the inward rectifier current to be unchanged or reduced.11,12 Also, the delayed rectifier currents have been demonstrated to be either reduced, unchanged, or even increased in rabbit and dog models of heart failure.12,65,66,71
Future considerations
The expression of many cardiac ion channels and transporters is heterogeneous throughout the ventricular myocardium. Differences have been demonstrated from epicardium to endocardium and from apex to base in cardiac potassium channels,34,39,42 and connexin43 has been shown to vary transmurally.8,36 While some studies highlighted above have taken myocardial heterogeneity into consideration, future investigations of arrhythmogenic remodelling in human heart failure should better account for this heterogeneity in anatomical tissue sampling strategies. Such studies will be necessary to thoroughly understand the normal gradients of activation and repolarization in the human heart and how they may be modified in heart failure. Also, because heart failure is a heterogeneous condition, resulting from congenital cardiomyopathy, ischaemic heart disease, and hypertension, among other causes, aetiologic differences will need to be examined in greater detail. Arrhythmogenic mechanisms may vary depending on aetiology, and therefore, appropriate therapeutic options in heart failure could be aetiology-specific. In addition, most heart failure studies to date have investigated end-stage heart failure from transplant recipients compared with normal controls. While it is important to understand the difference between these two extremes, the changes in late-stage heart disease may not accurately reflect the early remodelling during heart failure development. It will also be important to understand early electrophysiological changes that occur during hypertrophy and the earlier stages of heart failure in humans, as these changes may be more amenable to targeted intervention before significant irreversible structural damage has occurred.
Final remarks
Human heart investigations will not replace well-executed, controlled studies conducted in animal models for understanding the pathophysiological mechanisms of heart failure. Rather, studies on human heart tissue will continue to provide insight into what might be the most important electrophysiological alterations to pursue for potential therapeutic intervention. Parallel lines of animal model and human heart investigation, often with animal models providing the first clues toward the most fundamental experiments to pursue in human hearts, will be necessary for continued translational progress. Additionally, human studies coupled with investigations in other model systems may help better elucidate the molecular regulatory mechanisms of arrhythmogenic remodelling. Without such studies, most human heart investigations are limited to the correlation of molecular and cellular findings with electrophysiological results. Ultimately, we anticipate that increased understanding of activation and repolarization remodelling in human heart failure will promote better integration between basic science and clinical cardiology.
Conflict of interest: none declared.
References
- 1.Mosterd A, Hoes AW. Clinical epidemiology of heart failure. Heart. 2007;93:1137–46. doi: 10.1136/hrt.2003.025270. doi:10.1136/hrt.2003.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomaselli GF, Zipes DP. What causes sudden death in heart failure. Circ Res. 2004;95:754–63. doi: 10.1161/01.RES.0000145047.14691.db. doi:10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 3.Ponikowski P, Anker SD, Amadi A, Chua TP, Cerquetani E, Ondusova D, et al. Heart rhythms, ventricular arrhythmias, and death in chronic heart failure. J Cardiac Fail. 1996;2:177–83. doi: 10.1016/s1071-9164(96)80039-x. doi:10.1016/S1071-9164(96)80039-X. [DOI] [PubMed] [Google Scholar]
- 4.Buxton AE, Lee KL, Fisher JD, Josephson ME, Prystowsky EN, Hafley G. A randomized study of the prevention of sudden death in patients with coronary artery disease.: Multicenter Unsustained Tachycardia Trial Investigators. N Engl J Med. 1999;341:1882–90. doi: 10.1056/NEJM199912163412503. doi:10.1056/NEJM199912163412503. [DOI] [PubMed] [Google Scholar]
- 5.Glatter KA, Young JN, McElvany MD. Implantable cardioverter-defibrillators: a new preventive medical option. Prev Cardiol. 2006;9:49–53. doi: 10.1111/j.1520-037x.2006.3647.x. quiz 4–5 doi:10.1111/j.1520-037X.2006.3647.x. [DOI] [PubMed] [Google Scholar]
- 6.Moss AJ, Hall WJ, Cannom DS, Daubert JP, Higgins SL, Klein H, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. Multicenter Automatic Defibrillator Implantation Trial Investigators. N Engl J Med. 1996;335:1933–40. doi: 10.1056/NEJM199612263352601. doi:10.1056/NEJM199612263352601. [DOI] [PubMed] [Google Scholar]
- 7.Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877–83. doi: 10.1056/NEJMoa013474. doi:10.1056/NEJMoa013474. [DOI] [PubMed] [Google Scholar]
- 8.Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1762–70. doi: 10.1152/ajpheart.00346.2004. doi:10.1152/ajpheart.00346.2004. [DOI] [PubMed] [Google Scholar]
- 9.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–83. doi: 10.1016/j.yjmcc.2004.12.012. doi:10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Maltsev VA, Sabbab HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effect of long-term therapy with carvedilol. Cell Mol Life Sci. 2002;59:1561–8. doi: 10.1007/s00018-002-8529-0. doi:10.1007/s00018-002-8529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1031–41. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- 12.Tsuji Y, Opthof T, Kamiya K, Yasui K, Liu W, Lu Z, et al. Pacing-induced heart failure causes a reduction of delayed rectifier potassium currents along with decreases in calcium and transient outward currents in rabbit ventricle. Cardiovasc Res. 2000;48:300–9. doi: 10.1016/s0008-6363(00)00180-2. doi:10.1016/S0008-6363(00)00180-2. [DOI] [PubMed] [Google Scholar]
- 13.Tsuji Y, Zicha S, Qi XY, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. 2006;113:345–55. doi: 10.1161/CIRCULATIONAHA.105.552968. doi:10.1161/CIRCULATIONAHA.105.552968. [DOI] [PubMed] [Google Scholar]
- 14.Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, et al. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol. 2005;288:H2077–87. doi: 10.1152/ajpheart.00526.2003. doi:10.1152/ajpheart.00526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hobai IA, O'Rourke B. Enhanced Ca(2+)-activated Na(+)-Ca(2+) exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–8. doi: 10.1161/01.res.87.8.690. doi:10.1161/01.RES.87.8.690. [DOI] [PubMed] [Google Scholar]
- 16.Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res. 1998;39:60–76. doi: 10.1016/s0008-6363(98)00110-2. doi:10.1016/S0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- 17.Robbins J. Twenty years of gene targeting: what we don't know. Circ Res. 2011;109:722–3. doi: 10.1161/CIRCRESAHA.111.249912. doi:10.1161/CIRCRESAHA.111.249912. [DOI] [PubMed] [Google Scholar]
- 18.Nabauer M, Kaab S. Potassium channel down-regulation in heart failure. Cardiovasc Res. 1998;37:324–34. doi: 10.1016/s0008-6363(97)00274-5. doi:10.1016/S0008-6363(97)00274-5. [DOI] [PubMed] [Google Scholar]
- 19.Aiba T, Tomaselli GF. Electrical remodeling in the failing heart. Current opinion in cardiology. 2010;25:29–36. doi: 10.1097/HCO.0b013e328333d3d6. doi:10.1097/HCO.0b013e328333d3d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michael G, Xiao L, Qi XY, Dobrev D, Nattel S. Remodelling of cardiac repolarization: how homeostatic responses can lead to arrhythmogenesis. Cardiovasc Res. 2009;81:491–9. doi: 10.1093/cvr/cvn266. doi:10.1093/cvr/cvn266. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Hill JA. Electrophysiological remodeling in heart failure. J Mol Cell Cardiol. 2010;48:619–32. doi: 10.1016/j.yjmcc.2010.01.009. doi:10.1016/j.yjmcc.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin H, Lyon AR, Akar FG. Arrhythmia mechanisms in the failing heart. Pacing Clin Electrophysiol. 2008;31:1048–56. doi: 10.1111/j.1540-8159.2008.01134.x. doi:10.1111/j.1540-8159.2008.01134.x. [DOI] [PubMed] [Google Scholar]
- 23.Wit AL, Rosen MR. Pathophysiologic mechanisms of cardiac arrhythmias. Am Heart J. 1983;106(4 Part 2):798–811. doi: 10.1016/0002-8703(83)90003-0. doi:10.1016/0002-8703(83)90003-0. [DOI] [PubMed] [Google Scholar]
- 24.Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res. 2004;62:309–22. doi: 10.1016/j.cardiores.2003.11.035. doi:10.1016/j.cardiores.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 25.Rohr S, Kucera JP, Kleber AG. Slow conduction in cardiac tissue, I: effects of a reduction of excitability versus a reduction of electrical coupling on microconduction. Circ Res. 1998;83:781–94. doi: 10.1161/01.res.83.8.781. doi:10.1161/01.RES.83.8.781. [DOI] [PubMed] [Google Scholar]
- 26.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–53. doi: 10.1152/physrev.00002.2005. doi:10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 27.Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, et al. Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ Res. 2007;101:1146–54. doi: 10.1161/CIRCRESAHA.107.152918. doi:10.1161/CIRCRESAHA.107.152918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao G, Xie A, Huang SC, Zhou A, Zhang J, Herman AM, et al. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure. Circulation. 2011;124:1124–31. doi: 10.1161/CIRCULATIONAHA.111.044495. doi:10.1161/CIRCULATIONAHA.111.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, et al. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest. 2010;120:266–79. doi: 10.1172/JCI39740. doi:10.1172/JCI39740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko YS, Matsushita T. Gap junction alterations in human cardiac disease. Cardiovasc Res. 2004;62:368–77. doi: 10.1016/j.cardiores.2003.12.007. doi:10.1016/j.cardiores.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 31.Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, et al. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–71. doi: 10.1006/jmcc.2000.1308. doi:10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- 32.Glukhov AV, Fedorov VV, Kalish PW, Ravikumar VK, Lou Q, Janks D, et al. Conduction remodeling in human end-stage non-ischemic left ventricular cardiomyopathy. Circulation. 2012;125:1835–47. doi: 10.1161/CIRCULATIONAHA.111.047274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beuckelmann DJ, Erdmann E. Ca(2+)-currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Res Cardiol. 1992;87:235–43. doi: 10.1007/978-3-642-72474-9_19. [DOI] [PubMed] [Google Scholar]
- 34.Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379–85. doi: 10.1161/01.res.73.2.379. doi:10.1161/01.RES.73.2.379. [DOI] [PubMed] [Google Scholar]
- 35.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, et al. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–6. doi: 10.1161/01.res.61.1.70. doi:10.1161/01.RES.61.1.70. [DOI] [PubMed] [Google Scholar]
- 36.Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, et al. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res. 2010;106:981–91. doi: 10.1161/CIRCRESAHA.109.204891. doi:10.1161/CIRCRESAHA.109.204891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, et al. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90:2534–9. doi: 10.1161/01.cir.90.5.2534. doi:10.1161/01.CIR.90.5.2534. [DOI] [PubMed] [Google Scholar]
- 38.Weidmann S. Effect of current flow on the membrane potential of cardiac muscle. J Physiol. 1951;115:227–36. doi: 10.1113/jphysiol.1951.sp004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nabauer M, Beuckelmann DJ, Uberfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 1996;93:168–77. doi: 10.1161/01.cir.93.1.168. doi:10.1161/01.CIR.93.1.168. [DOI] [PubMed] [Google Scholar]
- 40.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–52. doi: 10.1161/01.cir.98.23.2545. doi:10.1161/01.CIR.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 41.Undrovinas AI, Maltsev VA, Kyle JW, Silverman N, Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–89. doi: 10.1006/jmcc.2002.2100. doi:10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- 42.Wettwer E, Amos GJ, Posival H, Ravens U. Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ Res. 1994;75:473–82. doi: 10.1161/01.res.75.3.473. doi:10.1161/01.RES.75.3.473. [DOI] [PubMed] [Google Scholar]
- 43.Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, et al. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–93. doi: 10.1161/01.cir.98.14.1383. doi:10.1161/01.CIR.98.14.1383. [DOI] [PubMed] [Google Scholar]
- 44.Zygmunt AC, Robitelle DC, Eddlestone GT. Ito1 dictates behavior of ICl(Ca) during early repolarization of canine ventricle. Am J Physiol. 1997;273(3 Part 2):H1096–106. doi: 10.1152/ajpheart.1997.273.3.H1096. [DOI] [PubMed] [Google Scholar]
- 45.Coraboeuf E, Coulombe A, Deroubaix E, Hatem S, Mercadier JJ. Transient outward potassium current and repolarization of cardiac cells. Bull Acad Natl Med. 1998;182:325–33. discussion 33–5. [PubMed] [Google Scholar]
- 46.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–83. doi: 10.1016/s0008-6363(99)00017-6. doi:10.1016/S0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 47.Veldkamp MW, van Ginneken AC, Opthof T, Bouman LN. Delayed rectifier channels in human ventricular myocytes. Circulation. 1995;92:3497–504. doi: 10.1161/01.cir.92.12.3497. doi:10.1161/01.CIR.92.12.3497. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe E, Yasui K, Kamiya K, Yamaguchi T, Sakuma I, Honjo H, et al. Upregulation of KCNE1 induces QT interval prolongation in patients with chronic heart failure. Circ J. 2007;71:471–8. doi: 10.1253/circj.71.471. doi:10.1253/circj.71.471. [DOI] [PubMed] [Google Scholar]
- 49.Holzem KM, Glukhov AV, Efimov IR. The role of IKr in transmural repolarization abnormalities in human heart failure [Abstract] Circulation. 2011;124(A16014) [Google Scholar]
- 50.Koumi S, Backer CL, Arentzen CE. Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation. 1995;92:164–74. doi: 10.1161/01.cir.92.2.164. doi:10.1161/01.CIR.92.2.164. [DOI] [PubMed] [Google Scholar]
- 51.Koumi S, Backer CL, Arentzen CE, Sato R. beta-Adrenergic modulation of the inwardly rectifying potassium channel in isolated human ventricular myocytes. Alteration in channel response to beta-adrenergic stimulation in failing human hearts. J Clin Invest. 1995;96:2870–81. doi: 10.1172/JCI118358. doi:10.1172/JCI118358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Yue L, White M, Pelletier G, Nattel S. Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation. 1998;98:2422–8. doi: 10.1161/01.cir.98.22.2422. doi:10.1161/01.CIR.98.22.2422. [DOI] [PubMed] [Google Scholar]
- 53.Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O'Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. doi: 10.1161/01.RES.0000080932.98903.D8. doi:10.1161/01.RES.0000080932.98903.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bers DM. Excitation-contraction Coupling and Cardiac Contractile Force. Netherlands: Springer; 2001. [Google Scholar]
- 55.Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, et al. Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–53. doi: 10.1161/01.res.75.3.443. doi:10.1161/01.RES.75.3.443. [DOI] [PubMed] [Google Scholar]
- 56.Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34:951–69. doi: 10.1006/jmcc.2002.2037. doi:10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- 57.Reinecke H, Studer R, Vetter R, Holtz J, Drexler H. Cardiac Na+/Ca2+ exchange activity in patients with end-stage heart failure. Cardiovasc Res. 1996;31:48–54. [PubMed] [Google Scholar]
- 58.Schwinger RH, Wang J, Frank K, Muller-Ehmsen J, Brixius K, McDonough AA, et al. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation. 1999;99:2105–12. doi: 10.1161/01.cir.99.16.2105. doi:10.1161/01.CIR.99.16.2105. [DOI] [PubMed] [Google Scholar]
- 59.Piacentino V, 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, et al. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–8. doi: 10.1161/01.RES.0000062469.83985.9B. doi:10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 60.Weber CR, Piacentino V, 3rd, Houser SR, Bers DM. Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation. 2003;108:2224–9. doi: 10.1161/01.CIR.0000095274.72486.94. doi:10.1161/01.CIR.0000095274.72486.94. [DOI] [PubMed] [Google Scholar]
- 61.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. doi:10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaab S, Nuss HB, Chiamvimonvat N, O'Rourke B, Pak PH, Kass DA, et al. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–73. doi: 10.1161/01.res.78.2.262. doi:10.1161/01.RES.78.2.262. [DOI] [PubMed] [Google Scholar]
- 63.Rozanski GJ, Xu Z, Whitney RT, Murakami H, Zucker IH. Electrophysiology of rabbit ventricular myocytes following sustained rapid ventricular pacing. J Mol Cell Cardiol. 1997;29:721–32. doi: 10.1006/jmcc.1996.0314. doi:10.1006/jmcc.1996.0314. [DOI] [PubMed] [Google Scholar]
- 64.Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, et al. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004;561(Part 3):735–48. doi: 10.1113/jphysiol.2004.075861. doi:10.1113/jphysiol.2004.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akar FG, Wu RC, Juang GJ, Tian Y, Burysek M, Disilvestre D, et al. Molecular mechanisms underlying K+ current downregulation in canine tachycardia-induced heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H2887–96. doi: 10.1152/ajpheart.00320.2004. doi:10.1152/ajpheart.00320.2004. [DOI] [PubMed] [Google Scholar]
- 66.Verkerk AO, Baartscheer A, de Groot JR, Wilders R, Coronel R. Etiology-dependency of ionic remodeling in cardiomyopathic rabbits. Int J Cardiol. 2011;148:154–60. doi: 10.1016/j.ijcard.2009.10.047. doi:10.1016/j.ijcard.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 67.Akar FG, Laurita KR, Rosenbaum DS. Cellular basis for dispersion of repolarization underlying reentrant arrhythmias. J Electrocardiol. 2000;33(Suppl):23–31. doi: 10.1054/jelc.2000.20313. doi:10.1054/jelc.2000.20313. [DOI] [PubMed] [Google Scholar]
- 68.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–19. doi: 10.1161/01.res.85.11.1009. doi:10.1161/01.RES.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 69.Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97(Suppl 1):I36–42. doi: 10.1007/s003950200027. [DOI] [PubMed] [Google Scholar]
- 70.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–67. doi: 10.1161/hh1101.091193. doi:10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 71.Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, et al. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101:2631–8. doi: 10.1161/01.cir.101.22.2631. doi:10.1161/01.CIR.101.22.2631. [DOI] [PubMed] [Google Scholar]
- 72.Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116–27. doi: 10.1016/j.cardiores.2005.08.015. doi:10.1016/j.cardiores.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yao A, Su Z, Nonaka A, Zubair I, Spitzer KW, Bridge JH, et al. Abnormal myocyte Ca2+ homeostasis in rabbits with pacing-induced heart failure. Am J Physiol. 1998;275(4 Part 2):H1441–8. doi: 10.1152/ajpheart.1998.275.4.H1441. [DOI] [PubMed] [Google Scholar]