

Recently, the U.S. Food and Drug Administration (FDA), the U.S. National Institutes of Health, and the stem cell research community have collaborated on a series of workshops that address moving pluripotent stem cell therapies into the clinic. The first two workshops in the series focused on preclinical science, and a third, future workshop will focus on clinical trials. This summary addresses major points from both of the recent preclinically focused meetings.
Keywords: Pluripotent stem cells, Stem cells, Clinical translation, Clinical trials
Abstract
Recently, the U.S. Food and Drug Administration (FDA), the U.S. National Institutes of Health, and the stem cell research community have collaborated on a series of workshops that address moving pluripotent stem cell therapies into the clinic. The first two workshops in the series focused on preclinical science, and a third, future workshop will focus on clinical trials. This summary addresses major points from both of the recent preclinically focused meetings. When entering into a therapeutics developmental program based on pluripotent cells, investigators must make decisions at the very early stages that will have major ramifications during later phases of development. Presentations and discussions from both invited participants and FDA staff described the need to characterize and document the quality, variability, and suitability of the cells and commercial reagents used at every translational stage. This requires consideration of future regulatory requirements, ranging from donor eligibility of the original source material to the late-stage manufacturing protocols. Federal, industrial, and academic participants agreed that planning backward is the best way to anticipate what evidence will be needed to justify human testing of novel therapeutics and to eliminate wasted efforts.
Introduction
The need to develop new and more effective drug-based treatments is well recognized; however, recent advances in molecular, cellular, genetic, and material sciences have provided additional opportunities as novel interventions that may address unmet medical needs. Pluripotent stem cell-based therapeutics is arguably one promising approach. Although a great deal has been written about the promise of stem cells in the treatment of a host of diseases and injuries, the field is at a relatively early stage [1]. Much work spanning basic, translational, and clinical science is needed for these cells to meet their anticipated potential. This research will require collaborations from both public and private institutions. One such interaction has been a recent collaboration between the U.S. Food and Drug Administration (FDA), the U.S. National Institutes of Health (NIH), and the stem cell research community in a series of workshops that address moving pluripotent stem cell therapies into the clinic. The first two workshops in the series focused on preclinical science, whereas a third, future workshop will focus on clinical trials. Our goal in this brief report is to summarize several salient themes that emerged from the first two workshops.
To enable more effective translation of scientific progress to clinical testing and use, the NIH provides support to researchers developing novel therapies, and the FDA is charged with regulation of manufacturing, clinical testing, and marketing of therapeutics. The NIH is essentially the nation's medical research agency. Its mission is to “seek fundamental knowledge about the nature and behavior of living systems and the application of that knowledge to enhance health, lengthen life, and reduce the burdens of illness and disability” (http://www.nih.gov). The FDA is primarily a regulatory body, with a responsibility to protect public health “by assuring the safety, efficacy and security of human and veterinary drugs, biological products, medical devices,” among other things (http://www.fda.gov). Although the respective missions and activities of NIH and FDA differ, there is common ground between them, and increased coordination will advance the goals of both agencies.
With the establishment of the FDA-NIH Joint Leadership Council and the 2010 launch of the NIH-FDA Regulatory Science Initiative (see [2, 3]), interactions between the NIH and FDA continue to grow. Recently, the NIH and FDA began collaborating on a series of workshops that address moving pluripotent stem cell therapies into the clinic (Fig. 1). The first in the series, “Pluripotent Stem Cells in Translation: Early Decisions,” held March 21 and 22, 2011, focused on the considerations necessary to making pluripotent cells into a product suitable for clinical use. Topics included the choice, characterization, and biology of pluripotent cells; regulatory requirements and challenges; and emerging technologies that may facilitate the translational trajectory for pluripotent cells. The second workshop, held on July 10 and 11, 2012, focused on animal models, preclinical safety, and proof-of-concept testing.
Figure 1.
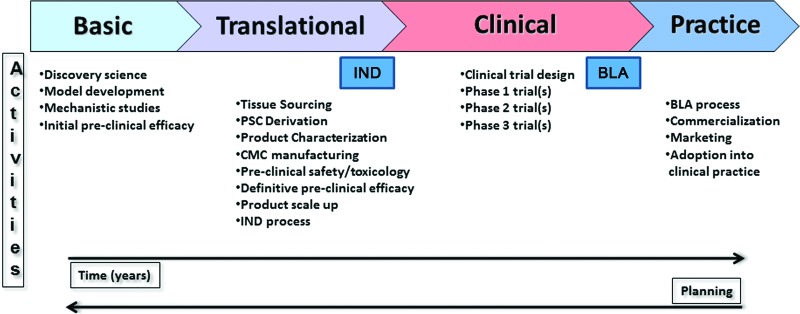
An outline of the steps involved in generating novel PSC-based medicines. From left to right, the figure illustrates the transition from research stages to medical practice. Major activities in each step are listed. Two key regulatory processes, the IND application and the BLA, are highlighted. Planning backward from clinical trial design and/or the envisioned marketed use of the cellular product, as recommended in both workshops, is also depicted. Abbreviations: BLA, Biologic License Application; CMC, chemistry, manufacturing, and controls; IND, Investigational New Drug; PSC, pluripotent stem cell.
Planning and Design
The concept of “planning backward” was a primary theme that emerged from the first workshop and was reemphasized in the second. When entering into a therapeutics developmental program based on pluripotent cells, investigators must make decisions at the very early stages that will have major ramifications during later phases of development. Federal, industrial, and academic participants agreed that planning backward is the best way to anticipate what evidence will be needed to justify human testing of novel therapeutics and to eliminate wasted efforts. For example, Dr. Malcolm Moos (FDA, Bethesda, MD) stressed that attention should be paid during early decision-making to three key controls to help ensure a safe and potent product: (a) source control (i.e., control over where the cells and reagents used in manufacture come from, and how they are tested to ensure suitability for manufacture); (b) product testing (e.g., potency assays); and (c) process control (e.g., design of a reproducible, well-controlled process for cellular differentiation/enrichment/purification, appropriate testing of manufacturing intermediates, validation of the process, and adherence to current good manufacturing practices).
It is particularly important and relatively common to give careful thought to these variables as one initiates translational phases of research; however, Dr. Gordon Keller (McEwen Centre for Regenerative Medicine, Toronto, ON, Canada) stressed that care should be taken even at the most basic stages of research. Successful manipulations and reproducible results require high-quality reagents, and it is incumbent on the investigator to characterize and document the quality, variability, and suitability of the commercial reagents that they use. If one intends for one's work to be relevant to products ultimately used for clinical testing, it is never too soon to envision future regulatory requirements, including the late-stage manufacturing challenges that will exist. As covered by Dr. Jane Lebkowski (Geron Corporation, Menlo Park, CA), attention needs to be paid at an early point to the effective dose (number of cells and/or repeated dosing), materials required to produce the cells, long-term storage of the final product, and even how the cellular product will need to be shipped, since all of these parameters will impact the manufacturing process. In essence, even at the earliest stages of testing a cell preparation intended for use as a clinical product, one should be considering how that product will be scaled through to market: cell source, cell bank size, scalable manufacturing processes, improved raw materials, and infrastructure to support development and manufacturing. The theme of advanced planning, careful characterization, and justification for the proposed approach reemerged in the second workshop, which is discussed later.
Design decisions that will determine the success of the translational effort begin, in fact, with the initial creation of pluripotent cell lines themselves. During the first workshop, it became evident that NIH and FDA requirements can differ in important ways. Meeting the NIH funding eligibility requirements for use of human embryonic stem (hES) cells (http://stemcells.nih.gov/policy/Pages/2009guidelines.aspx) does not necessarily guarantee that these cells will also meet FDA regulations for clinical use. The FDA donor eligibility rule, 21 CFR 1271 (subpart C), effective on May 25, 2005, “requires human cell, tissue, and cellular and tissue-based product (HCT/Ps) establishments to screen and test cell and tissue donors for risk factors for, and clinical evidence of, relevant communicable disease agents or diseases” (http://www.fda.gov/BiologicsBloodVaccines/TissueTissueProducts/QuestionsaboutTissues/ucm102842.htm). It is not sufficient that the cell, tissue, and cellular or tissue-based product itself is tested, but it is required that the donor of the product must be screened and tested at the time of tissue recovery, using methods specified by the FDA. The documentation of these tests must be available when the product is being evaluated by the FDA. To re-emphasize this key point, the fact that a human embryonic stem cell line meets the requirements of the NIH Human Embryonic Stem Cell Registry does not necessarily ensure that the eligibility rule has been met. It is also important to note, however, that this rule does not apply to tissue recovered before the eligibility rule was finalized, that is, May 25, 2005. For tissue recovered after May 25, 2005, applicants may pursue an exemption to the rule. Regardless, the best course is to become familiar with the FDA donor eligibility rule and to consider its ramifications during the earliest planning stages of producing a cellular product destined for human use.
Since the FDA and the NIH are both part of the U.S. Department of Health and Human Services, one could reasonably ask why such a disparity exists in the first place. Each agency has a congressionally mandated mission that specifies its activities. NIH funds research at all stages, and much of the work it funds is not directed at the clinical use of reagents created for research; it is not always necessary, or financially prudent, to meet all regulatory requirements for early-stage research. Additionally, the NIH is prohibited from funding the creation of hES cell lines; therefore it cannot establish rules regarding the embryo donation and cell line creation process. For now, what the NIH can do is to work together with the FDA to ensure that the community is informed of the distinctions between the agencies' activities.
Animal Models
The second meeting focusing on preclinical considerations, held July 10–11, 2012, explored the use of in vivo and in vitro animal models that could be used to justify the potential benefits of a pluripotent cell-derived therapeutic and establish potential risks in consideration for approval of clinical testing. Drs. Walter Koroshetz (National Institute of Neurological Disorders and Stroke [NINDS]/NIH) and Mahendra Rao (Center for Regenerative Medicine/NIH) again emphasized the importance of planning preclinical studies based on the intended design of clinical studies, that is, planning backward by incorporating the clinical model into the design of the preclinical studies. Whether investigating mechanism of action or safety of a novel cell product, sponsors should consider current FDA regulations (under 21CFR312.22) and published guidances (see: http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM329861.pdf). Dr. Patrick Au (Center for Biologics Evaluation and Research [CBER]/FDA) explained that the diversity of cell products and the unique biological properties of specific cell products necessitate that CBER maintain a flexible, case-by-case approach to reviewing cellular products. The best strategy for gaining clarity on the regulatory expectations for a specific preclinical development program is to schedule an early interaction with the CBER through a pre-pre-Investigational New Drug (pre-pre-IND) dialogue. The intent of the pre-pre-IND dialogue is to provide a forum for discussing the nature of the planned clinical indication and the preclinical development program that would allow for an adequate assessment of both the risks and potential benefits of the therapeutic candidate.
Animal modeling plays an important role in justifying potential efficacy but also in assessing potential risks presented by a candidate therapeutic. The meeting participants discussed the choices of models that are used, both in general and with specific examples that clarified the rationale behind these choices. Examples included cellular interventions for macular degeneration, cardiovascular disease, diabetes, stroke, and neurological disorders. The structure, physiology, and immunological state of these systems differ, as does the homology of specific organs in humans compared with different species (rodent, rabbit, dog, pig, and nonhuman primates). In addition, relatively few large animal models of specific disorders are available, often necessitating the use of sequential systems to assess mechanism of action, pivotal efficacy, pharmacokinetics, and toxicology. Speakers emphasized that there is no single recipe for success, nor are there shortcuts. There is no substitute for rigorous evaluation of oncological risks, effect of the cell product on host tissues, and effect of host tissues on the cells at every stage of study.
Before discussing particular types of animal models used for testing pluripotent cells, it is important to note that many of the concerns relating to these cells are similar to those relating to differentiated cell products, whether they persist after implantation or can be expected to have paracrine or immunological effects independent of their continued survival. As such, established precedents are relevant to evaluating the merit of experimental designs (e.g., in review of grant applications) and in regulatory decisions. These common regulatory concerns include identification of cell distribution and cell persistence, toxicity, dosing, safety of delivery procedure, and development of potency assays and available marker antibodies, and so forth. There are important differences with pluripotent cells, however, that must be addressed. Dr. Robert Deans (Athersys, Cleveland, OH) emphasized that pluripotent cells themselves are not the actual product of interest—they can divide, differentiate, and mature after transplantation to exert the desired effects. Therefore, the risk of ectopic differentiation, tumorigenicity, migration to nontarget sites, and immunological properties pose serious and sometimes unique concerns. Induced pluripotent cells (iPSCs) in particular raise unique questions for autologous therapeutic strategies. Despite the autologous source, in vitro manipulations may induce changes that could cause immunological rejection. It is also unclear at this point how individualized cell preparations will be regulated. One unresolved questioned identified by Dr. Peter Coffey (Institute of Ophthalmology, London, U.K.) is whether equally extensive characterization protocols will have to be repeated for every iPSC line developed for autologous use. These and other product pluripotent product issues will require continued evaluation and discussion.
The key issues that can be addressed in testing with animal models include mechanism of action in disease-specific models; cell fate, migration, and continued differentiation; effect of the disease processes on implanted cells; delivery and dosing paradigms; optimization of trial design and patient selection criteria; outcome assessment, including surrogate outcomes for potency assays and lot release criteria; and risk of ectopic differentiation or tumor development. Like other cellular therapies, pluripotent cells may be ascribed to have pleiotropic (also known as uncertain) mechanism(s) of action. The question of establishing mechanism of action was a recurring theme throughout the workshop.
Whereas mechanism of action (MOA) studies are a hallmark of NIH research, Dr. Au pointed out that, to the FDA, understanding of MOA is helpful in the development of a particular cell product (nice to know), but it is not required (need to know). Having an understanding of the MOA can help guide product characterization and manufacturing, as well as designing appropriate preclinical studies and clinical trials. From a regulatory perspective, safety data to support the proposed dose range and anatomic site of delivery in humans is critical information that needs to be ascertained. Dr. Rajesh Ranganathan (NINDS/NIH) discussed the differing agency roles in regulation of product-in-human testing (FDA) versus prioritization of resources at every stage in the development of novel therapeutics that may relieve the burden of disease (NIH). Experiences in the pharmaceutical industry show that potency observed in model systems does not ensure efficacy in humans. Therefore, proof of concept (POC) may be achievable only in human studies. MOA studies are critical, however, for de-risking the translational process, even in the absence of full understanding of the disease. Animal models and the associated endpoints designed to test the effect of the cell product on a particular target can be used to justify surrogate markers or potency assays, to assess toxicities, and to justify the types of in vitro and animal studies that should be conducted before POC human studies should begin.
One important question posed during the panel discussion is whether there is valid evidence that any observed effects are attributable to the cells themselves versus the host reactions they elicit. That is, how can it be demonstrated that the cells are actually needed for therapeutic benefit? During POC testing, selecting appropriate control conditions can provide important information about the role of the specific pluripotent cell-derived product on host responses, compared with other foreign cells. The question of selecting the appropriate “control” conditions (live or killed cells vs. media vs. needle puncture) also deserves careful consideration. Speakers cautioned that, again, there is no one-size fits all control cell: the nature of the cell product and the chosen control should be justified by information about both—Dr. Clive Svendsen (Cedars-Sinai Regenerative Medicine Institute, Los Angeles, CA) cautioned that if the control cell injections themselves cause damage, the comparison might unrealistically amplify the apparent benefit of the cell product being tested.
One of the inevitable difficulties of testing of human cell products in animals is immune rejection of xenografts. This difficulty was cited as one place where standards in the field are not sufficient, which can adversely affect the evaluation of both funding applications and papers submitted for publication if reviewers do not value non-hypothesis-driven studies required for INDs. Preclinical studies using homologous cells (cells derived from donors of the same species as the host) might be used to address early POC, host responses, and migration, if there is sufficient justification that testing of the human cell product in the disease model is not feasible. In fact, it was pointed out that xenografting has distinct disadvantages for early POC and safety testing in some disease models. Dr. Arnold Kriegstein (University of California, San Francisco, San Francisco, CA) described the differentiation of human cells, which is significantly slower than that of rodent cells, yet their function may depend on progressing through their full maturation process to adequately determine their effect on hosts. As Dr. Svendsen described, this can be particularly problematic in rodent models of progressive lethal disorders, suggesting that following homologous species studies it would be reasonable to use nondisorder large animals for later safety studies of the human cell product itself. Dr. Emerson Perin (Texas Heart Institute, Galveston, TX) compared small and large species models used to directly test human cell delivery, safety, retention, and immune issues. If developmental models are used, it is important to consider the developmental milestones of host species, as the graft-host responses can change during maturation of the host, which could affect migration of grafted cells, for example.
Conclusion
Overall, the discussion of different types of animal models illustrated many relevant preclinical issues, but few issues unique to testing of pluripotent cells emerged, compared with testing of other cell products. Several excellent summaries of testing and translation of cells for diabetes and retinal, cardiac, and neurological disorders were presented. Perhaps the greatest advantage of testing in animals is the potential to understand the fate and function of the cells in complex and relevant disease conditions. Strategies to address immunological responses, immune-privileged sites, humanized hosts (mice or pigs), antirejection drugs, in vitro testing, and homologous species testing were described; each model has advantages and limitations to be considered. In fact, the speakers agreed that every model system has limitations and that the best strategy is to understand and address the various strengths and limitations as directly as possible. These issues include size, life span, understanding and fidelity of disease models, delivery devices, and structural/physiological differences between species. Dosing choice for cellular therapies remains a challenge, with characterization of the maximum feasible dose being more relevant than the maximum tolerable dose used in drug testing. The endpoints chosen to assess both positive and negative effects of the cells are also an emerging areas where standardization of assays, tracking to evaluate cell survival and fate, justification of parameters, sensitivity, and reliability all need to be better developed.
The ultimate resolution of these questions will depend on continuing and informed discussion between the agencies and the scientific community. We refer readers to the videocast of these meetings for a more detailed account of both workshops (http://videocast.nih.gov/summary.asp?Live=10013&bhcp=1; http://videocast.nih.gov/summary.asp?Live=10081&bhcp=1; http://videocast.nih.gov/summary.asp?live=11344&bhcp=1; http://videocast.nih.gov/summary.asp?Live=11345&bhcp=1). We also encourage investigators interested in this area to consult published literature where many similar topics have been discussed [4–7]. The third meeting in this series, which is in development, will address moving pluripotent cellular products out of the laboratory into human testing and issues of clinical trial design. Overall, these meetings illustrate federal agencies working together in collaboration with the research community to address this nascent field in a rational fashion. Claims of stem cell treatments and cures have captured the attention of both the public and the media, but the field of pluripotent cell therapy, although promising, is at an early stage. Significant technical hurdles remain that will be overcome only through years of concomitant research at the basic, translational, and clinical levels.
Acknowledgments
We thank the participants of both workshops for providing invaluable insights into this emerging area, our FDA colleagues for comments on a draft of this report, and Drs. Story Landis (NINDS/NIH) and Celia Witten (CBER/FDA) for providing overall leadership. We also thank members of the planning committees from both the FDA and the NIH for making these workshops a reality. FDA: Drs. Rachael F. Anatol, Patrick Au, Kimberly A. Benton, Theresa T. Chen, Donald W. Fink, Thomas P. Finn, Ying Huang, Deborah A. Hursh, Wei Liang, Malcolm C. Moos, Stephanie L. Simek, and Steven O. Winitsky. NIH: Drs. Kristin Abraham (National Institute of Diabetes and Digestive and Kidney Diseases [NIDDK]), Rosemarie Hunziker (National Institute of Biomedical Imaging and Bioengineering [NIBIB]), Christine Kelley (NIBIB), Naomi Kleitman (NINDS), Susan Marino (NINDS), David Owens (NINDS), Mahendra Rao (National Institute of Arthritis and Musculoskeletal and Skin Diseases [NIAMS]), Pamela Robey (National Institute of Dental and Craniofacial Research), Sheryl Sato (NIDDK), Sonia Skarlatos (National Heart, Lung, and Blood Institute [NHLBI]), and John W. Thomas (NHLBI). We would like to also acknowledge the contributions made by Dr. Laura Cole (National Institute on Deafness and Other Communication Disorders [NIDCD]), Buck Wong (NIDCD), Dr. Scott Lipnick (Office of the Director), and Megan Laycock (NIAMS). Funding for both workshops was provided by the NIH.
Author Contributions
N.K., M.S.R., and D.F.O.: manuscript writing.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Ho P-J, Yen M-L, Yet S-F, et al. Current applications of human pluripotent stem cells: Possibilities and challenges. Cell Transplantation. 2012;21:801–814. doi: 10.3727/096368911X627507. [DOI] [PubMed] [Google Scholar]
- 2.Hamburg MA. Advancing regulatory science. Science. 2011;331:987. doi: 10.1126/science.1204432. [DOI] [PubMed] [Google Scholar]
- 3.Collins FS. Reengineering translational science: The time is right. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3002747. 90cm17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter MK, Frey-Vasconcells J, Rao MS. Developing safe therapies from human pluripotent stem cells. Nat Biotechnol. 2009;27:606–613. doi: 10.1038/nbt0709-606. [DOI] [PubMed] [Google Scholar]
- 5.Fink DW., Jr FDA regulation of stem cell-based products. Science. 2009;324:1662–1663. doi: 10.1126/science.1173712. [DOI] [PubMed] [Google Scholar]
- 6.Aboody K, Capela A, Niazi N, et al. Translating stem cell studies to the clinic for CNS repair: current state of the art and the need for a Rosetta Stone. Neuron. 2011;70:597–613. doi: 10.1016/j.neuron.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Au P, Hursh DA, Lim A, et al. FDA oversight of cell therapy clinical trials. Sci Transl Med. 2012;4 doi: 10.1126/scitranslmed.3004131. 149fs31. [DOI] [PubMed] [Google Scholar]