

This study showed that an alternative cell detachment of mesenchymal stromal/stem cells (MSCs) with pronase instead of trypsin significantly accelerated the lung clearance of the cells and, importantly, increased their targeting to an area of injury. Pronase detachment could be used as a method to improve the MSC lung clearance and targeting in vivo. This may have a major impact on the bioavailability of MSCs in future therapeutic regimes.
Keywords: Mesenchymal stem cells, Bone marrow stromal cells, Cell adhesion molecules, Cell transplantation, Experimental models
Abstract
Systemic infusion of therapeutic cells would be the most practical and least invasive method of administration in many cellular therapies. One of the main obstacles especially in intravenous delivery of cells is a massive cell retention in the lungs, which impairs homing to the target tissue and may decrease the therapeutic outcome. In this study we showed that an alternative cell detachment of mesenchymal stromal/stem cells (MSCs) with pronase instead of trypsin significantly accelerated the lung clearance of the cells and, importantly, increased their targeting to an area of injury. Cell detachment with pronase transiently altered the MSC surface protein profile without compromising cell viability, multipotent cell characteristics, or immunomodulative and angiogenic potential. The transient modification of the cell surface protein profile was sufficient to produce effective changes in cell rolling behavior in vitro and, importantly, in the in vivo biodistribution of the cells in mouse, rat, and porcine models. In conclusion, pronase detachment could be used as a method to improve the MSC lung clearance and targeting in vivo. This may have a major impact on the bioavailability of MSCs in future therapeutic regimes.
Introduction
Mesenchymal stromal/stem cells (MSCs) are a particularly promising option for cell therapy. MSCs are easily accessible from several sources, can be highly expanded in culture, and have shown low immunogenic activity [1–3]. This is a major advantage, allowing them to be potentially used as allogeneic “off-the-shelf” products. In fact, MSCs have already been explored in many clinical trials for treating, for example, cardiovascular disease, stroke, Crohn's disease, and graft-versus-host disease [4, 5]. In all cell therapies, efficient and controllable cell delivery is crucial for successful clinical outcome. Cell delivery is indication-dependent and often local transplantation is considered as the primary choice. Systemic infusion, however, would be more practical, enabling rapid and less invasive off-the-shelf therapy. However, this means that cells must be able to home to the target tissue where they could exert their beneficial effects.
The mechanism of MSC homing is currently being studied vigorously and is likely to have similar features with the homing of leukocytes to sites of inflammation. The vascular endothelium and leukocytes undergo a series of coordinated adhesion steps mediated by selectins and their ligands. Subsequently, cells start to roll and encounter local chemokines, which activate integrins resulting in firm adhesion, invasion, and extravasation [6]. Similar mechanisms have been suggested for MSCs engaging P-selectin and vascular cell adhesion molecule-1/very late antigen (VLA)-4 to mediate firm adhesion on endothelial cells and transendothelial migration [7–9]. MSCs can be found in many tissues after systemic infusion in healthy animals. In addition, MSCs can target with limited efficiency to the site of injury/inflammation or tumors [9–11].
One challenge in intravenous (i.v.) delivery for therapeutic cells is the massive lung adhesion [12–21]. This obviously impairs the bioavailability of the cells, their targeting to tissues, and most likely the therapeutic outcome. It is possible, however, that i.v. administered MSCs have therapeutic effects by acting from peripheral organs, such as the lung [22] or spleen [23]. Targeting to the injury site and local therapeutic effect are currently still considered critical for MSC therapy.
We have previously observed an influence of cell surface structures on the lung clearance rate of MSCs [24]. In this study we demonstrate for the first time that an alternative enzymatic detachment with pronase increases targeting of MSCs by decreasing unspecific pulmonary retention due to transient changes in the surface protein profile without compromising viability or functionality of the cells.
Materials and Methods
Ethical Considerations
Umbilical cord blood (UCB) collections were performed at the Helsinki University Central Hospital (HUCH) and Helsinki Maternity Hospital (Finland). Bone marrow (BM) was collected from an unaffected bone site from patients operated on for osteoarthritis (>50 years) or scoliosis (<20 years). All donors gave their informed consent prior to sample collection, and the study protocols were approved by the ethical committee of HUCH, the Finnish Red Cross Blood Service (FRCBS), and Oulu University Hospital (Finland).
Cells
Human UCB-MSC derivation and culture was performed as described [25], human and porcine BM-MSCs were isolated and cultured as described [26], and Wistar rat MSCs (Cyagen Biosciences Inc., Sunnyvale, CA, http://www.cyagen.com) were cultured according to the manufacturer's instructions (short description in supplemental online data).
Pronase Detachment and Cell Viability
Pronase is a nonspecific protease mixture isolated from Streptomyces griseus [27]. The subconfluent cells were detached by 0.5% pronase (Roche, Mannheim, Germany, http://www.roche.com) in phosphate-buffered saline-0.25 mM EDTA. Different concentrations of pronase, ranging from 0.05% to 1%, were also tested. Trypsin (TryPLe Express; Life Technologies, Paisley, U.K., http://www.lifetech.com) detachment was always used as a control. As with trypsin detachment, pronase detachment was always stopped with excess culture medium within 4 minutes from the addition of the enzyme to protect the detached cells. Subsequent centrifugation and resuspension were used to remove the enzymes.
Cell viability was determined by trypan blue exclusion or a Nucleocounter NC-100 (Chemometec, Lillerod, Denmark, http://www.chemometec.com). In this study, viability was routinely determined in >30 individual pronase-detached cell samples. Cell morphology was observed and documented by microscopy. For the protein recovery studies, cells were incubated in culture medium for 5–7 hours at +37°C after detachment with subsequent cell viability measurements. Gentle agitation of cells was done to prevent the attachment of cells. In addition, the depolarization of mitochondrial inner membrane potential (ΔΨm) was analyzed from differently detached BM-MSCs as described in the supplemental online data. Readherence to culture vessels and growth kinetics of pronase-detached cells were also investigated and compared with trypsinized cells.
Cell Surface Analysis by Mass Spectrometry
UCB-MSCs were used for mass spectrometry (MS) analysis of cell surface proteins after trypsin and pronase detachment and processed directly or after 5 hours of postdetachment recovery. The cell surface proteins were biotinylated essentially as described [28]. The labeled proteins were harvested with magnetic beads and treated as described [29] prior to liquid chromatography (LC)-MS analysis.
Mass Spectrometry
Digested peptides were loaded to reversed phase precolumn (ProteCol Guard-C18, 150 μm × 10 mm; SGE Analytical Science Pty. Ltd., Ringwood, Victoria, Australia, http://www.sge.com) with 0.1% formic acid and separated in reversed phase analytical column (PepMap100, 75 μm × 150 mm; Thermo Fisher Scientific Inc., Waltham, MA, http://www.thermofisher.com) with linear gradient of acetonitrile. An Ultimate 3000 LC instrument (Thermo Fisher Scientific) was operated in nano-scale with flow rate of 0.3 μl/minute. Eluted peptides were introduced to an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) via an ESI-Chip interface (Advion BioSciences Inc., Ithaca, NY, http://www.advion.com) in positive-ion mode. Peptides were selected for MS/MS fragmentation in the LTQ with collision-induced dissociation according to their intensity.
Data Analysis
Data were processed with Mascot Distiller (version 2.3; Matrix Science Ltd., Boston, MA, http://www.matrixscience.com) and searched with Mascot Server (version 2.2.06; Matrix Science) against human proteins in the UniProtKB database (version 15.12). LC-MS differential expression analysis was performed with Progenesis LC-MS software (version 2.6; Nonlinear Dynamics Ltd., Newcastle upon Tyne, U.K., http://www.nonlinear.com) (detailed data analysis described in the supplemental online data).
Cell Surface Analysis by Flow Cytometry
UCB-MSCs and BM-MSCs were analyzed for the cell surface epitope expression immediately after different detachments and after a 5–7-hours postdetachment recovery. The antibodies against the following proteins were used: CD44, CD49d, CD49e, CD73, CD90, CD105, HLA-DR, CD14, CD19, CD34, CD45, CD13, CD29, CD49c, CD54, CD59, CD106, CD146, CD147 (Abcam, Cambridge, U.K., http://www.abcam.com), CD166, CD184, fibronectin (FN) (Abcam), galectin-1 (Acris Antibodies, Herford, Germany, http://www.acris-antibodies.com), and chondroitin sulfate proteoglycan (CSPG)-4. All antibodies were purchased from BD Biosciences (San Diego, CA, http://www.bdbiosciences.com) unless stated otherwise.
The cells were labeled with 2 μl or 0.5–1 μg (CD147, FN, galectin-1, CSPG4) of the antibodies per 1 × 105 cells. Secondary antibody staining was done for CD147, FN, galectin-1, and CSPG4. The labeled cells were run with a FACSAria (BD Biosciences) flow cytometer, and the results were analyzed with FACSDiva software (BD Biosciences). Adequate isotype control antibodies were also used.
Functionality Assays
T-Cell Proliferation Assay
Peripheral blood mononuclear cells (PBMNCs) were isolated from buffy coats from anonymous blood donors (FRCBS) by density gradient centrifugation (Ficoll-Paque Plus, GE Healthcare, Piscataway, USA) and labeled with 5 μM 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE) solution (Molecular Probes, Eugene, OR, http://probes.invitrogen.com). CFSE-labeled PBMNCs were cocultured with trypsin- or pronase-detached UCB-MSCs (n = 2) in RPMI with 10% fetal bovine serum (FBS), 100 U/ml penicillin + 0.1 mg/ml streptomycin (Life Technologies). MSC:MNC ratios 1:10, 1:20, 1:50, and 1:100 were used. To activate the T-cell proliferation, 100 ng/ml anti-human CD3 antibody clone Hit3a (BioLegend, San Diego, CA, http://www.biolegend.com) was used. Nonstimulated CFSE-labeled cells and stimulated/nonstimulated nonlabeled PBMNCs were used as controls. T-cell proliferation was analyzed after 4 days as dilution of CFSE intensity by FACSAria flow cytometer (BD Biosciences) and FlowJo software (version 7.6.1; Tree Star, Ashland, OR, http://www.treestar.com).
Angiogenesis Assay
Angiogenesis was studied in a validated angiogenesis coculture model [30] using human fibroblasts (American Type Culture Collection, Boras, Sweden, http://www.atcc.org) and human umbilical cord vein endothelial cells (HUVECs). Fibroblasts and HUVECs were cocultured for 7 days before adding differently detached UCB-MSCs. UCB-MSCs were also plated in inserts (Scaffdex, Tampere, Finland, http://www.scaffdex.com). The following media were used: basic test medium (BTM) (Endothelial Basal Medium-2 with 2.0% FBS, 0.1% Gentamicin Amphotericin-B, 1% l-glutamine; Lonza Group Ltd., Basel, Switzerland, http://www.lonza.com), BTM with angiogenic growth factors for positive controls, and UCB-MSC medium. Technical validity was ensured with positive (n = 4) and negative controls (BTM, n = 3; UCB-MSC medium, n = 2). On day 13, tubules were visualized with anti-von Willebrand factor staining and analyzed as described [30].
In Vitro Rolling Assay
A Diaphot TMD inverted-stage microscope (Nikon, Tokyo, Japan, http://www.nikon.com) configured with phase contrast, objective lens (×4), MicroPublisher 5 megapixel camera (QImaging, Surrey, BC, Canada, http://www.qimaging.com) and MCID Core image analysis program (InterFocus Imaging Ltd., Cambridge, U.K., http://www.mcid.co.uk) was used to count the attached cells and their average rolling speed on FN-coated (0.5 μg/μl) capillaries (diameter, 0.93 mm; length, 12.5 cm). An Alaris-Asena syringe pump (CareFusion Corp., Palm Springs, CA, http://www.carefusion.com) was used for perfusion with a calibrated flow speed (5 ml/hour). The cells (20,000 cells per milliliter) were imaged for 2 minutes in each analysis.
Biodistribution of Human and Porcine MSCs In Vivo
Animal Procedures
All animal experiments were approved and authorized by the National Animal Experiment Board in Finland and by the Research Animal Care and Use Committee of the University of Oulu (Oulu, Finland). Anesthesia of the animals is described in the supplemental online data.
Biodistribution of Human MSCs in Mouse Model
Differently detached UCB-MSCs (p4) and BM-MSCs (p6) were labeled with 99mTc-hexamethylpropylene amine oxime (99mTc-HMPAO; Ceretec; GE Healthcare, http://www.gehealthcare.com) (15 minutes at room temperature [RT]) and cell viability was determined. Subsequently, 7–8-week-old female mice received 5 × 105 cells i.v. in 100 μl of saline (UCB-MSC: n = 3; BM-MSC: n = 5). One hour or 15 hours postinjection mice were sacrificed, and lungs, heart, liver, spleen, pancreas, kidneys, gastrointestinal (GI) tract, and femur were collected. Radioactivity of the tissues was determined with a gamma counter (Wallac Wizard 1480; PerkinElmer, Gaithersburg, MD, http://www.perkinelmer.com). The relative radioactivity for each tissue was calculated as percentage from the total amount of radioactivity in all the tissues measured. Additionally, the biodistribution of UCB-MSCs labeled with the fluorescent dye PKH-26 was studied in mice (details described in the supplemental online data).
Biodistribution of Allogeneic Porcine BM-MSCs in a Porcine Model
Twelve pigs (18–27 kg) received 0.5 × 106 BM-MSCs per kilogram. Trypsin- or pronase-detached (n = 6 + 6), 99mTc-HMPAO-labeled BM-MSCs were injected to the inferior vena cava via a Schwan-Ganz pulmonary catheter. A whole-body planar scan and single photon emission computed tomography/computed tomography (SPECT-CT) (Symbia-T2; Siemens Medical Solutions USA, Inc., Malvern, PA, http://usa.healthcare.siemens.com) were performed after the 8-hour follow-up. Hemodynamic data recording and blood sampling were conducted to provide supporting data and assess any adverse effects. SPECT images were analyzed using the Hermes Hybrid PDR software (version 1.4C; Hermes Medical Solutions, Stockholm, Sweden, http://www.hermesmedical.com). Regions of interest (ROIs) were drawn around target organs. The slices were chosen based on repeatability and lack of prominent activity from surrounding organs. Lungs were analyzed by creating a volume of interest (VOI) from the ROIs. Some atelectasis was found from the lungs, confirmed by a radiologist. To assess the impact of atelectasis, the activity in the atelectic areas was subtracted from the whole lung activity. Mean activities of ROIs and VOIs were calculated and correlated to the input activity (details described in the supplemental online data).
Biodistribution of Allogeneic Rat BM-MSCs in a Rat Model of Inflammation
Female Wistar Han rats were used in the study. λ-Carrageenan (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com) was dissolved in saline to give a 4% (wt/vol) solution, and 150 μl was administered into the plantar region of the left hind paw immediately before the cell injection. Subsequently, animals received either trypsin-detached (n = 3) or pronase-detached (n = 4), 99mTc-HMPAO-labeled rat BM-MSCs (1 × 106) into the tail vein. Whole-body SPECT-CT (nanoSPECT/CT; Bioscan Inc., Washington, DC, http://www.bioscan.com) images were acquired 50 minutes, 3 hours, 5 hours, and 24 hours postinjection. Only one animal per group was imaged at the 24-hour time point, and it was excluded from the statistical analysis. The volume of the inflamed paw was measured with a plethysmometer (Ugo Basile, Comerio, Italy, http://www.ugobasile.com) before and 1 minute after the injection and directly after imaging. SPECT images were reconstructed with HiSPECT NG software (Scivis GmbH, Gottingen, Germany, http://www.scivis.de) and fused with CT data sets with InVivoScope software (Bioscan). VOIs were drawn around lungs, kidneys, paw, and liver. The same VOI was used for all subsequent analyses of corresponding organs. The radioactivity in each VOI was calculated and corrected for decay. Results are presented as a percentage from the injected dose (details described in the supplemental online data).
Statistics
Statistical analysis for the biodistribution data of BM-MSCs in mice and rats was done for each organ separately with a linear fixed effect model having factors for time point and treatment, using analysis of variance tests and F tests, and using Tukey's Honestly Significant Difference p value correction where appropriate. In porcine VOI data the difference between the treatments was analyzed with a t test, and the associated background data were analyzed with principal component analysis and linear mixed effects models. In all figures, one asterisk denotes a p value <.05, two asterisks a p value <.01, and three asterisks a p value <.001. Analyses were done with the statistical programming language R, version 2.12. Statistical analysis was not performed for UCB-MSCs because of the low number of animals.
Results
Pronase Detachment Does Not Affect Cell Viability
The pronase detachment protocol produced viable single-cell MSC suspensions without any cell aggregates (supplemental online Fig. 1A, 1B) with >90% viability (n > 30), which is equal to the trypsin control. All tested pronase concentrations (0.05%–1%) detached the cells effectively in 4 minutes. The highest concentration (1%) affected the cell morphology slightly, although cell viability was >90%. A concentration of 0.5% pronase was chosen as a standard concentration as it detached cells rapidly with high viability and effectively cleaved certain cell surface proteins. Pronase-detached cells also adhered normally to culture vessels, proliferated, and reached confluence correspondingly to trypsinized cells (supplemental online Fig. 2A). In addition, one BM-MSC line was selected for the analysis of ΔΨm. The role of ΔΨm in regulation of viability and osteogenic differentiation potency of MSCs has been demonstrated [31]. Trypsin- and pronase-detached cells exhibited identical 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) staining pattern, and most of the cells showed energized or partly depolarized mitochondria compared with control, that is, protonophore carbonyl cyanide m-chlorophenylhydrazone-treated cells exhibiting fully depolarized mitochondria as expected (supplemental online Fig. 1C). Accordingly, all these data indicate that the use of pronase as a substitute for trypsin detachment does not compromise the viability of MSCs.
Pronase Selectively and Transiently Cleaves Cell Surface Protein Epitopes
The MS data and the complete list of identified proteins from trypsin- and pronase-detached MSCs are presented in supplemental online Table 1 and supplemental online Figure 3A. Altogether, in 19 cell surface proteins, pronase treatment decreased the abundance of corresponding peptide features compared with trypsin treatment (e.g., CD166, CD49e, CD44, CSPG4, CD29, CD105, CD49c, CD147, CD146, CD99, and CD90). Also, secreted galectin-1 was digested by pronase. CD29, CD49c, and CD59 had many novel, pronase-generated peptides thus revealing a characteristic cleavage pattern of pronase (supplemental online Table 1). Unfortunately, it was not possible to determine in detail how the cleavage of these peptides affects the actual structure of the proteins. Also, some peptides from cytosolic proteins had diminished peptide feature abundance after the pronase detachment, which is probably due to the cleavage of their interaction partner on the cell surface.
The flow cytometric analysis supported the MS analysis, and there was a clear decrease in the binding of CD166, CD49e, CD44, CSPG4, and CD146 antibodies in the pronase-detached cells compared with trypsin-detached cells (Fig. 1A–1E). The intensity of anti-CD105 binding was also slightly decreased (Fig. 1P). Instead, the cell surface expression of galectin-1, CD29, CD49c, D147, CD59, and CD90 was similar between trypsin- and pronase-detached cells, although the MS analysis had indicated the cleavage of those proteins by pronase (Fig. 1I–1M, 1O). This discrepancy indicates that the antibodies could recognize the protein epitopes that were still left on the cell surface after the pronase detachment. Furthermore, the peptides with glycosylation or other surface modification could not be detected in the MS.
Figure 1.

The expression of selected cell surface proteins in umbilical cord blood (UCB)-mesenchymal stromal/stem cells (MSCs) detached with either trypsin (light gray histograms) or pronase (dark gray histograms). MSCs were stained with antibodies against the indicated antigens and analyzed with flow cytometry. There were several proteins showing reduced expression in pronase-detached cells (A–H) or equal expression compared with trypsin-detached cells (I–O). Percentages of positive cells are also shown for each antibody. Samples were gated according to their respective isotype controls. Abbreviations: APC, allophycocyanin; FITC, fluorescein isothiocyanate; FN, fibronectin; PE, phycoerythrin; pro, pronase; try, trypsin.
The pronase cleavage of additional adhesion or migration-related proteins was studied. We have previously observed significant FN expression on MSCs [24], and this was further verified in immunofluorescence staining of BM-MSCs (supplemental online Fig. 3B). Pronase always consistently cleaved FN from the cell surface (Fig. 1F). Also, CD49d and CD54 expression was lower on the cell surface after the pronase detachment compared with control cells (Fig. 1G, 1H). Their expression levels varied slightly even within the same cell line, most likely reflecting the effect of the culture conditions on the expression. The expression of CD106 and CD184 was low on MSCs (data not shown). The expression of CD73 was similar in both samples (Fig. 1N).
The recovery of cleaved cell surface proteins was studied by flow cytometry. CD44, CD49e, CSPG4, and CD166 showed almost full recovery after a 7-hour recovery period (supplemental online Fig. 3D). Recovery of proteins was also seen in the MS peptide data for both intracellular and membrane proteins (supplemental online Fig. 3C) (described in the supplemental online data).
MSC Functionality Is Not Compromised After Pronase Detachment
To study the effect of pronase on the multipotency or functional properties of MSCs, the differentiation capacity, immunosuppressive properties, and angiogenic potential of pronase-detached cells were compared with those of control cells.
Differentiation
Pronase detachment did not evidently affect the multipotency of MSCs, since both adipogenic and osteogenic differentiation was successful (supplemental online Fig. 2B, 2C) and comparable to results after trypsin detachment (supplemental online Fig. 2D, 2E).
Immunosuppression
Based on the T-cell proliferation assay, the pronase-detached cells inhibited the proliferation of activated T cells in a dose-dependent manner and as effectively as the trypsin-detached cells. This suggests that the pronase detachment does not weaken the immunosuppressive properties of MSCs (Fig. 2A). T-cell proliferation was tested with two UCB-MSC lines against allogeneic PBMNCs from two donors.
Figure 2.

The functionality of MSCs is not compromised with pronase. (A): Pronase-detached umbilical cord blood (UCB)-MSCs were able to inhibit T-cell proliferation as effectively as control cells. T-cell proliferation was determined by measuring the amount of 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE)low cells gated from leukocyte population on the forward and side scatter plot. Responder cells (positive proliferation control) were assigned as 100%. Data are represented as mean (+SD) of two UCB-MSC donors against allogeneic peripheral blood mononuclear cells from two different healthy donors (n = 4). (B): The angiogenic ability of pronase-detached UCB-MSCs in the in vitro assay was comparable to that of control cells. In the coculture setup, the angiogenic ability was as high as or even higher than that of positive controls (assigned as 100%), but in inserts remained on the same level of induction as the medium control. Data are represented as mean (+SD) (n = 3). (C): Different rolling speed profiles of trypsin- and pronase-detached BM-MSCs in flow perfusion assay on a fibronectin-coated capillary. Cells having a rolling speed of greater than 220 μm/second were clearly higher in number in the pronase group (n = 35, mean rolling speed 261 μm/second) compared with the trypsin group (n = 19, mean 221 μm/second). Both human (black dots) and porcine (gray dots) BM-MSCs were analyzed. Abbreviations: MSC, mesenchymal stromal/stem cell; ctrl, control; BM, bone marrow.
Angiogenesis
Both trypsin- and pronase-detached UCB-MSCs were able to strongly stimulate angiogenesis in the in vitro assay, both directly after the detachment and after a 5-hour recovery (Fig. 2B). UCB-MSCs required their own culture medium to stimulate the tubule formation. The medium alone was also favorable for angiogenesis, as it contained growth factors (e.g., platelet-derived growth factor-BB, which is a mild inducer in this assay). In the test medium, the angiogenic effect of MSCs remained rather weak (data not shown). Furthermore, the angiogenic effect was clearly better when MSCs were grown in contact with the angiogenesis assay cells compared with MSCs cultured in inserts.
In Vitro Rolling Assay
Contrary to other functional assays, the pronase detachment had a substantial impact on BM-MSC adherence and rolling speed in an in vitro flow perfusion assay. The number of cells having a rolling speed of greater than 220 μm/second (fast rollers) was clearly higher (p = .056 with t test) in the pronase group (n = 35; mean rolling speed, 261 μm/second) compared with the trypsin group (n = 19; mean, 221 μm/second) (Fig. 2C). No difference was observed between the groups for the slowly moving cells (slow rollers).
Pronase-Detached MSCs Exhibit Faster Clearance From the Lungs
The biodistribution of radioactively labeled BM-MSCs and UCB-MSCs was studied in mice. More than 75% of the total radioactivity was detected in the lungs in both cell detachment groups and for both cell types 1 hour after injection. The rest of the activity was detected in the liver, kidneys, and GI tract, followed by low radioactivity in spleen, pancreas, heart, and femur (Fig. 3A, 3B). At 15 hours postinjection, most of the radioactivity was detected in the liver in both cell detachment groups and for both cell types (49% in the BM-MSC control group vs. 70% in the BM-MSC pronase group, p < .001; Fig. 3A). There was, however, a significant difference in total radioactivity measured in the lungs at this time point with 32% for control BM-MSCs versus 7% for pronase BM-MSCs (p < .001; Fig. 3A). The amount of radioactivity in the lungs was per se lower for control UCB-MSCs (3.6%), but in line with the BM-MSCs there was clearly less radioactivity in the lungs for pronase UCB-MSCs (0.6%) (Fig. 3B). Radioactivity was low in all other organs studied, but generally with slightly more radioactivity observed when using pronase-detached cells.
Figure 3.

Pronase detachment decreases the pulmonary trapping of mesenchymal stromal/stem cells (MSCs) in vivo. (A, B): Biodistribution of trypsin- and pronase-detached, radioactively labeled BM-MSCs (A) (n = 5) and UCB-MSCs (B) (n = 3) in mice at 1 and 15 hours postinjection. The results are presented as mean (+SD) percentage of radioactivity from total amount of radioactivity in all the tissues measured. (C): Biodistribution of trypsin- and pronase-detached, PKH-labeled UCB-MSCs (n = 4) in mice at 1 and 20 hours postinjection. The amount of injected cells was determined per 1 × 106 tissue cells with flow cytometry. Results are presented as means (+SD). *, p < .05; **, p < .01; ***, p < .001. Abbreviations: BM, bone marrow; BMMSC, bone marrow mesenchymal stromal/stem cell; GI, gastrointestinal; UCBMSC, umbilical cord blood mesenchymal stromal/stem cell.
The reduced lung retention for pronase-detached cells was confirmed with fluorescently labeled UCB-MSCs. Twenty hours postinjection, there were one-fifth as many cells in the lungs in the pronase group than in the control group (p < .01, Fig. 3C), in line with the results observed using the radioactively labeled cells. Interestingly, the number of pronase-detached cells was higher in the spleen and peripheral blood compared with control cells 1 hour postinjection (p < .001 and p < .05, respectively). Pronase detachment had no visible effect on cell numbers in spleen, BM, or blood 20 hours postinjection. Unfortunately, it was not possible to analyze the liver with this method because of its high enzymatic activity.
Finally, the effect of pronase detachment on lung clearance was verified in a large-animal model. In line with the mouse data, a significant reduction of lung retention was observed for pronase-detached BM-MSCs in a homologous porcine model (Fig. 4B, p < .05). In line with the rodent data, prominent radioactivity was detected in the lungs, kidneys, liver, and spleen in both cell detachment groups after the 8-hour follow-up (Fig. 4A, 4C). There was, however, more variance in the biodistribution of the cells in the large animals compared with the mouse model. No adverse effects due to cell injections were observed, and the porcine groups had comparable hemodynamic and biochemical data. The lung CT scans revealed perioperative atelectasis in some individuals in both groups. The effect of the potentially damaged areas was evaluated, and it was concluded that the atelectasis did not significantly impact the results.
Figure 4.
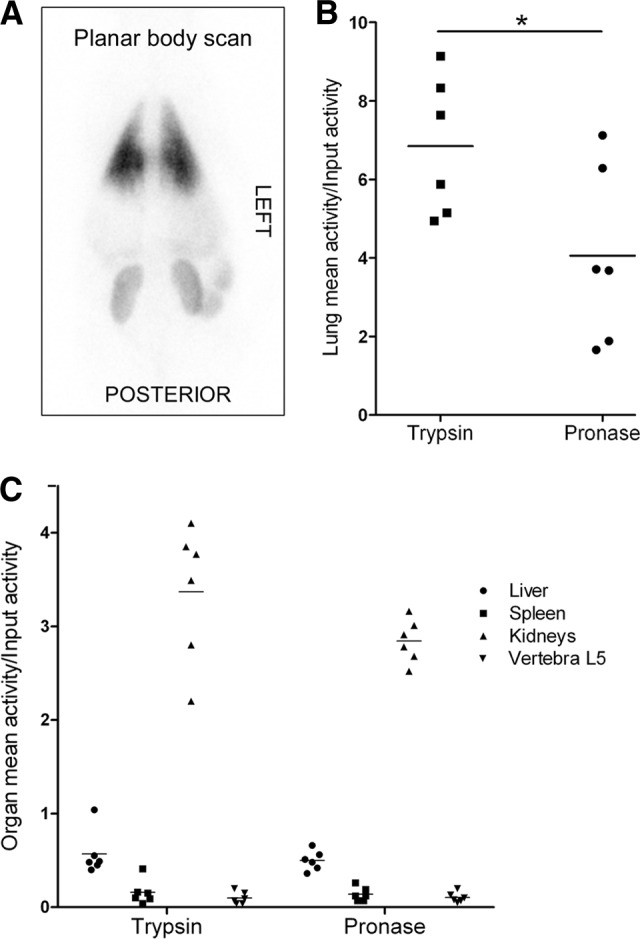
The biodistribution of trypsin- and pronase-detached allogeneic bone marrow-mesenchymal stromal/stem cells in a porcine model 8 hours postinjection. A whole-body planar scan shows prominent radioactivity in the lungs (A). Significantly less radioactivity was detected in the lungs in the pronase group animals compared with the trypsin group (B), whereas in other organs the activities were similar between the groups (C). Results are presented as volume of interest (B) and region of interest (C) activities from single photon emission computed tomography/computed tomography images and correlated to input activity. Horizontal lines represent mean values. *, p < .05 (unpaired t test).
Pronase Detachment Improves MSC Targeting to an Area of Injury
The therapeutic advantage of pronase detachment was further studied in a rat model of carrageenan-induced inflammation. Remarkably, pronase-detached rat MSCs migrated significantly more effectively to the inflamed area compared with control cells (Fig. 5C, 5F, p < .001). Twice as much radioactivity was detected in the inflamed paw in the pronase-group compared with trypsin-group both 50 minutes (0.25% vs. 0.13%, respectively) and 3 hours (0.22% vs. 0.11%) postinjection (Fig. 5F). In line with the previous observations made in mice, significantly (p < .01) less radioactivity was detected in the lungs of the rats receiving pronase-detached MSCs at all time points studied (Fig. 5A–5E; supplemental online Videos 1 and 2). Free 99mTc-HMPAO label accumulated in the GI tract.
Figure 5.

Biodistribution of rat bone marrow (BM)-mesenchymal stromal/stem cells (MSCs) in a peripheral inflammation model. (A–D): Whole body single photon emission computed tomography/computed tomography images 50 minutes and 5 hours postinjection show faster clearance of pronase-detached (n = 4) BM-MSCs from lungs compared with trypsin-detached cells (n = 3). An example of paw uptake at 50 minutes postinjection is presented in the lower corner of (C). Color scale minimum and maximum values were adjusted at the same level for 50-minute and 5-hour images. Analysis was done by measuring the radioactivity of three-dimensional volumes of interest around organs. (E, F): Results are presented as a percentage from injected dose (±SD) and show statistically significant differences in cell distribution between treatment groups both for lungs (E) and paws (F). Only one animal per group was imaged at the 24-hour time point and thus excluded from the analysis. *, p < .05; **, p < .01; ***, p < .001.
Discussion
Pronase was used as a dispersing agent of cultured cells in the 1970s, and it is currently commonly used in protein analytics and antigen retrieval protocols [32]. Pronase and other enzymes have been replaced by trypsin, which is the state-of-art reagent for cell detachment. Our results show that MSCs can be detached with pronase without compromising the MSC fidelity. Interestingly, pronase detachment transiently alters MSC surface adhesion proteins and significantly accelerates the clearance of these therapeutic cells from the lungs, as shown both with human cells in a xenogeneic mouse model and also in homologous rat and porcine models. Moreover, significantly more pronase-detached cells were detected in the inflamed tissue compared with trypsin-detached cells, indicating beneficial effects of pronase detachment on the in vivo targeting of MSCs to areas of injury.
In line with earlier observations [12–14, 17–21, 24], a majority of the MSCs were entrapped in the lungs 1 hour postinjection in all experimental models used in this study, which further emphasizes the major challenge of i.v. delivery of MSCs. Besides reducing cell amounts in target organs, lung retention of MSCs has caused severe lung damage in mice [33, 34]. Still, the i.v. route is considered an optimal method of cell delivery, being rapid, noninvasive, and most suitable for off-the-shelf cell therapy products. Overcoming the challenges of i.v. cell delivery is essential.
MSC lung retention may arise from passive entrapment of cells in small-diameter lung capillaries [19–21] or be an adhesion molecule-mediated phenomenon [9, 21]. We have previously identified cell surface structures, partly explaining the MSC lung retention and clearance [24]. In this study, we show that enzymatic cleavage of certain cell surface structures by pronase significantly induces MSC lung clearance, which is in line with the original hypothesis. UCB-MSCs passed the lungs more rapidly than BM-MSCs. This was also concluded in our previous study, and an influence of both cell size and, notably, cell surface adhesion molecules was hypothesized [24]. A role of cell surface molecules in MSC lung retention is now evident, since pronase accelerated the lung clearance of both cell types, which indicates a specific mechanism linked to protein cleavage on the cell surface. A difference in adhesion properties between pronase- and trypsin-detached cells was further supported by our in vitro findings, since pronase had a substantial impact on the MSC rolling behavior in a flow perfusion assay. The functional effect of pronase detachment on lung clearance was further verified in two homologous animal models. Reduced lung retention was evident in all models studied, but it is noteworthy that initial cell retention was more prominent in a xenogeneic experimental setting.
We hypothesized that the high expression of FN on MSCs could have a role in the adherence of MSCs to lungs, as it plays a major role in cell adhesion and migration and binds to many extracellular matrix (ECM) components and surface receptors [24]. There are several FN binding receptors expressed on lung endothelium that might trap the FN-expressing cells, such as integrins VLA-4 (CD49d/CD29) and VLA-5 (CD49e/CD29). MSCs also express these integrins and can thus bind to FN present in the lung endothelium. In fact, VLA-4/FN interactions have been shown to play a major role in transmigration of MSCs into ECM [35]. Furthermore, the blocking of CD49d on MSCs was shown to increase pulmonary passage [21]. A similar effect has been shown with MSCs dissolved in heparin, a known FN binder [36]. Both FN and CD49d were cleaved by pronase from the cell surface, and that could contribute to the faster clearance of pronase-detached cells from the lungs. Besides FN, CD49d, and CD49e, pronase most effectively cleaved CD44, CSPG4, CD146, and CD166, supported by both MS and flow cytometry data. Interestingly, CD44 and chondroitin sulfate interactions have been shown to mediate leukocyte rolling and adhesion to endothelial cells in vitro and may facilitate the trapping of activated leukocytes and tumor cells in the hepatic sinusoidal walls [37]. Furthermore, CD44 is known to have an important role in MSC migration and adhesion to endothelium [38, 39], thus strengthening it as a potential candidate influencing lung adhesion. Other proteins cleaved by pronase may have a role as well, such as CD54 and CD166, also related to endothelial adhesion of MSCs [39]. Given that multiple molecules are generally required for any type of cell adhesion, lung adhesion is also most likely due to a complex interplay between different molecules on therapeutic cells versus lung endothelial cells. Functional studies are required to verify the exact role of each molecule.
Although systemic effects of MSCs can be beneficial, the targeting of MSCs to the injury site is currently considered critical for their therapeutic effects. Studies have shown that besides the lungs, infused MSCs are commonly found in capillaries within the liver and spleen. MSCs have been shown to target with very low efficiency to sites of inflammation and injury and to, for example, bone marrow and brain [9–11, 13, 40, 41]. These earlier findings were confirmed in this study since healthy animals receiving trypsinized human BM-MSCs still had high numbers of cells in the lungs 15 hours postinjection, followed by the liver, spleen, kidneys, and GI tract. In this study, however, we showed that in a peripheral inflammation model, targeting of MSCs can be significantly increased to the site of injury by pronase detachment of the cells. Remarkably, targeting of pronase-detached MSCs to the inflammation was improved compared with trypsin-detached cells as early as 50 minutes postinjection.
The number of cells did not continuously increase in the injured area after the earliest time point postinjection. In fact, there was a decrease in the number of cells in the paws between 50 minutes and 5 hours postinjection. This was also evident with trypsinized cells and in the control paws, but it was most notable with pronase-detached cells in the inflamed paw. Cleavage of adhesion molecules after enzymatic detachment may have a role in the adherence of MSCs to the injured area. Nevertheless, the amount of pronase-detached cells in the inflamed paw was always the highest at all time points studied as compared with the number of cells detected in the other study groups. The amount of pronase-detached cells remained stabilized from 5 hours onward, suggesting possible engraftment to the tissue and coinciding with the recovery of the cell surface proteins seen in vitro.
The acute nature of carrageenan-induced inflammation can also affect cell recruitment. There is a strong blood flow toward the inflamed paw and rapid formation of edema in the initiation phase of inflammation with lots of cytokines attracting the cells, including MSCs, to the site [42, 43]. It may in fact be advantageous that therapeutic cells are recruited to the inflammation rapidly along with infiltrating neutrophils. After 5 hours, the edema started to decrease, and it is possible that the paracrine nature of MSCs, secreting beneficial factors that reduce the inflammation, is partly also influencing the diminishing edema. Although we have preliminary data to support a beneficial therapeutic effect of pronase-detached cells in an inflammatory condition, further studies should explore this in more detail.
Different strategies to improve the targeting of therapeutic cells have been explored by others. One interesting option to increase the targeting of therapeutic cells would be to add a specific tag on the cell surface. For example, chemical modification of BM-MSC surface with sialyl Lewisx, the moiety for selectin recognition, improved the homing of MSCs to inflamed tissue [44]. Also, a peptide-based tag has been shown to increase the homing of MSCs to ischemic myocardium [45] and adhesion and rolling of MSCs on E-selectin [46]. Moreover, the glycan engineering of CD44 improved the homing of BM-MSCs to the bone marrow [11]. Also, culture conditions can alter the cell surface structures and migratory behavior. Low-density culturing increased the expression of podocalyxin-like protein and CD49f on cell surface, thus improving the targeting of BM-MSCs to the infarcted heart [47]. Furthermore, cytokine pretreatment and low oxygen culturing induced migration, engraftment, and therapeutic potential of MSCs [48–50].
The results of this study show that pronase detachment of MSCs resulted in significantly faster clearance of the cells from the lungs due to transiently altered cell surface. This was detected with both BM-MSCs and UCB-MSCs and using both small- and large-animal models in a xenogeneic and homologous experimental setup. In addition, targeting to inflammation was significantly improved with pronase detachment in a rat model of peripheral inflammation, most likely because of increased bioavailability. Beneficial functional consequences of a transiently modified cell surface by pronase were also supported by the in vitro rolling assay data. Pronase-detached cells retain therapeutically relevant functional properties since both the immunosuppressive and angiogenic potential was equal to that of control cells in vitro. Furthermore, pronase detachment did not affect the multipotentiality of MSCs. No adverse effects due to cell injections were observed in the large-animal model, and the porcine groups had comparable hemodynamic and biochemical data during the follow-up. Combining pronase detachment with other methods, such as optimal culture conditions for migratory properties or adding a tag to induce targeting, could be one way to develop MSC preparations with enhanced targeting capacity in a clinical setting as well.
Conclusion
Pronase detachment could be used to improve the MSC lung clearance and targeting in vivo. Pronase detachment could be easily incorporated in the clinical production processes of adherent cells, since it could replace trypsin. However, there are still many questions surrounding the clinical potential for pronase, especially concerning safety, quality, and the actual improvement of efficacy of therapeutic cells.
Supplementary Material
Acknowledgments
We thank Lotta Sankkila, Teija Kupari, Sirkka Hirschovits-Gerz, Minna Savilampi, Henna Ek, Elisa Lehtilahti, Reeta Viitanen, Mari Rissanen, Oiva Arvola, Henri Haapanen, Fredrik Yannopoulos, Seija Seljänperä, Aira Karjalainen, and Leila Mäkelä for excellent technical assistance and Pauli Ojala for valuable initial data analysis.
Author Contributions
E. Kerkelä: conception and design, data collection, analysis and interpretation of data (main contributor), manuscript writing (main contributor), final approval of manuscript; T.H.: conception and design, collection and analysis of data, manuscript writing; T.M., M.R., O.K., and L.K.: data collection, analysis and interpretation of data, manuscript writing; J. Nikkilä: data analysis and interpretation, manuscript writing; S. Lehtonen, I.R., R.P., M.P.: collection and analysis of data, manuscript writing; R.T.: data collection, analysis and interpretation of data, manuscript writing; T.J.: administrative support, provision of study material; K.B.: collection of data, administrative support; E. Kalso: administrative support, conception and design; L.V.: conception and design, data analysis and interpretation, manuscript writing; S. Laitinen: conception and design, administrative support, data analysis and interpretation, provision of study material; P.L.: conception and design, administrative support, data collection, analysis and interpretation of data, provision of study material, manuscript writing; J. Nystedt: conception and design (main contributor), data collection, analysis and interpretation of data, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
E. Kerkelä, T.H., L.V., P.L., and J. Nystedt have uncompensated intellectual property rights.
References
- 1.Le Blanc K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy. 2003;5:485–489. doi: 10.1080/14653240310003611. [DOI] [PubMed] [Google Scholar]
- 2.Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 3.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 4.Le Blanc K, Frassoni F, Ball L, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 5.Salem HK, Thiemermann C. Mesenchymal stromal cells: Current understanding and clinical status. Stem Cells. 2010;28:585–596. doi: 10.1002/stem.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ley K, Laudanna C, Cybulsky MI, et al. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 7.Rüster B, Gottig S, Ludwig RJ, et al. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood. 2006;108:3938–3944. doi: 10.1182/blood-2006-05-025098. [DOI] [PubMed] [Google Scholar]
- 8.Steingen C, Brenig F, Baumgartner L, et al. Characterization of key mechanisms in transmigration and invasion of mesenchymal stem cells. J Mol Cell Cardiol. 2008;44:1072–1084. doi: 10.1016/j.yjmcc.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Karp JM, Leng Teo GS. Mesenchymal stem cell homing: The devil is in the details. Cell Stem Cell. 2009;4:206–216. doi: 10.1016/j.stem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Devine SM, Cobbs C, Jennings M, et al. Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood. 2003;101:2999–3001. doi: 10.1182/blood-2002-06-1830. [DOI] [PubMed] [Google Scholar]
- 11.Sackstein R, Merzaban JS, Cain DW, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 12.Gao J, Dennis JE, Muzic RF, et al. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs. 2001;169:12–20. doi: 10.1159/000047856. [DOI] [PubMed] [Google Scholar]
- 13.Barbash IM, Chouraqui P, Baron J, et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation. 2003;108:863–868. doi: 10.1161/01.CIR.0000084828.50310.6A. [DOI] [PubMed] [Google Scholar]
- 14.Allers C, Sierralta WD, Neubauer S, et al. Dynamic of distribution of human bone marrow-derived mesenchymal stem cells after transplantation into adult unconditioned mice. Transplantation. 2004;78:503–508. doi: 10.1097/01.tp.0000128334.93343.b3. [DOI] [PubMed] [Google Scholar]
- 15.Kang WJ, Kang H, Kim H, et al. Tissue distribution of 18F-FDG-labeled peripheral hematopoietic stem cells after intracoronary administration in patients with myocardial infarction. J Nucl Med. 2006;47:1295–1301. [PubMed] [Google Scholar]
- 16.Nystedt J, Mäkinen S, Laine J, et al. Human cord blood CD34+ cells and behavioral recovery following focal cerebral ischemia in rats. Acta Neurobiol Exp (Wars) 2006;66:293–300. doi: 10.55782/ane-2006-1618. [DOI] [PubMed] [Google Scholar]
- 17.Tolar J, O'Shaughnessy MJ, Panoskaltsis-Mortari A, et al. Host factors that impact the biodistribution and persistence of multipotent adult progenitor cells. Blood. 2006;107:4182–4188. doi: 10.1182/blood-2005-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyerrose TE, De Ugarte DA, Hofling AA, et al. In vivo distribution of human adipose-derived mesenchymal stem cells in novel xenotransplantation models. Stem Cells. 2007;25:220–227. doi: 10.1634/stemcells.2006-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schrepfer S, Deuse T, Reichenspurner H, et al. Stem cell transplantation: The lung barrier. Transplant Proc. 2007;39:573–576. doi: 10.1016/j.transproceed.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 20.Vilalta M, Degano IR, Bago J, et al. Biodistribution, long-term survival, and safety of human adipose tissue-derived mesenchymal stem cells transplanted in nude mice by high sensitivity non-invasive bioluminescence imaging. Stem Cells Dev. 2008;17:993–1003. doi: 10.1089/scd.2007.0201. [DOI] [PubMed] [Google Scholar]
- 21.Fischer UM, Harting MT, Jimenez F, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells Dev. 2009;18:683–692. doi: 10.1089/scd.2008.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee RH, Pulin AA, Seo MJ, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarting S, Litwak S, Hao W, et al. Hematopoietic stem cells reduce postischemic inflammation and ameliorate ischemic brain injury. Stroke. 2008;39:2867–2875. doi: 10.1161/STROKEAHA.108.513978. [DOI] [PubMed] [Google Scholar]
- 24.Nystedt J, Anderson H, Tikkanen J, et al. Cell surface structures influence lung clearance rate of systemically infused mesenchymal stromal cells. Stem Cells. 2013 doi: 10.1002/stem.1271. (in press) [DOI] [PubMed] [Google Scholar]
- 25.Laitinen A, Nystedt J, Laitinen S. The isolation and culture of human cord blood-derived mesenchymal stem cells under low oxygen conditions. Methods Mol Biol. 2011;698:63–73. doi: 10.1007/978-1-60761-999-4_6. [DOI] [PubMed] [Google Scholar]
- 26.Leskelä HV, Risteli J, Niskanen S, et al. Osteoblast recruitment from stem cells does not decrease by age at late adulthood. Biochem Biophys Res Commun. 2003;311:1008–1013. doi: 10.1016/j.bbrc.2003.10.095. [DOI] [PubMed] [Google Scholar]
- 27.Narahashi Y, Yanagita M. Studies on proteolytic enzymes (pronase) of Streptomyces griseus K-1. I. Nature and properties of the proteolytic enzyme system. J Biochem. 1967;62:633–641. doi: 10.1093/oxfordjournals.jbchem.a128718. [DOI] [PubMed] [Google Scholar]
- 28.Scheurer SB, Rybak JN, Roesli C, et al. Identification and relative quantification of membrane proteins by surface biotinylation and two-dimensional peptide mapping. Proteomics. 2005;5:2718–2728. doi: 10.1002/pmic.200401163. [DOI] [PubMed] [Google Scholar]
- 29.Kinter M, Sherman NE. New York, NY: Wiley-Interscience; 2000. Protein Sequencing and Identification Using Tandem Mass Spectrometry. [Google Scholar]
- 30.Sarkanen JR, Mannerstrom M, Vuorenpaa H, et al. Intra-laboratory pre-validation of a human cell based in vitro angiogenesis assay for testing angiogenesis modulators. Front Pharmacol. 2010;1:147. doi: 10.3389/fphar.2010.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pietilä M, Lehtonen S, Narhi M, et al. Mitochondrial function determines the viability and osteogenic potency of human mesenchymal stem cells. Tissue Eng Part C Methods. 2010;16:435–445. doi: 10.1089/ten.tec.2009.0247. [DOI] [PubMed] [Google Scholar]
- 32.Foley JF, Aftonomos B. The use of pronase in tissue culture: A comparison with trypsin. J Cell Physiol. 1970;75:159–161. doi: 10.1002/jcp.1040750204. [DOI] [PubMed] [Google Scholar]
- 33.Lee JW, Fang X, Gupta N, et al. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci USA. 2009;106:16357–16362. doi: 10.1073/pnas.0907996106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anjos-Afonso F, Siapati EK, Bonnet D. In vivo contribution of murine mesenchymal stem cells into multiple cell-types under minimal damage conditions. J Cell Sci. 2004;117:5655–5664. doi: 10.1242/jcs.01488. [DOI] [PubMed] [Google Scholar]
- 35.Yagi H, Soto-Gutierrez A, Parekkadan B, et al. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010;19:667–679. doi: 10.3727/096368910X508762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deak E, Ruster B, Keller L, et al. Suspension medium influences interaction of mesenchymal stromal cells with endothelium and pulmonary toxicity after transplantation in mice. Cytotherapy. 2010;12:260–264. doi: 10.3109/14653240903401840. [DOI] [PubMed] [Google Scholar]
- 37.Murai T, Sougawa N, Kawashima H, et al. CD44-chondroitin sulfate interactions mediate leukocyte rolling under physiological flow conditions. Immunol Lett. 2004;93:163–170. doi: 10.1016/j.imlet.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Zhu H, Mitsuhashi N, Klein A, et al. The role of the hyaluronan receptor CD44 in mesenchymal stem cell migration in the extracellular matrix. Stem Cells. 2006;24:928–935. doi: 10.1634/stemcells.2005-0186. [DOI] [PubMed] [Google Scholar]
- 39.Brooke G, Tong H, Levesque JP, et al. Molecular trafficking mechanisms of multipotent mesenchymal stem cells derived from human bone marrow and placenta. Stem Cells Dev. 2008;17:929–940. doi: 10.1089/scd.2007.0156. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Chen J, Chen XG, et al. Human marrow stromal cell therapy for stroke in rat: Neurotrophins and functional recovery. Neurology. 2002;59:514–523. doi: 10.1212/wnl.59.4.514. [DOI] [PubMed] [Google Scholar]
- 41.Chapel A, Bertho JM, Bensidhoum M, et al. Mesenchymal stem cells home to injured tissues when co-infused with hematopoietic cells to treat a radiation-induced multi-organ failure syndrome. J Gene Med. 2003;5:1028–1038. doi: 10.1002/jgm.452. [DOI] [PubMed] [Google Scholar]
- 42.Hargreaves K, Dubner R, Brown F, et al. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 43.Morris CJ. Carrageenan-induced paw edema in the rat and mouse. Methods Mol Biol. 2003;225:115–121. doi: 10.1385/1-59259-374-7:115. [DOI] [PubMed] [Google Scholar]
- 44.Sarkar D, Spencer JA, Phillips JA, et al. Engineered cell homing. Blood. 2011;118:e184–e191. doi: 10.1182/blood-2010-10-311464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kean TJ, Duesler L, Young RG, et al. Development of a peptide-targeted, myocardial ischemia-homing, mesenchymal stem cell. J Drug Target. 2012;20:23–32. doi: 10.3109/1061186X.2011.622398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng H, Byrska-Bishop M, Zhang CT, et al. Stem cell membrane engineering for cell rolling using peptide conjugation and tuning of cell–selectin interaction kinetics. Biomaterials. 2012;33:5004–5012. doi: 10.1016/j.biomaterials.2012.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee RH, Seo MJ, Pulin AA, et al. The CD34-like protein PODXL and α6-integrin (CD49f) identify early progenitor MSCs with increased clonogenicity and migration to infarcted heart in mice. Blood. 2009;113:816–826. doi: 10.1182/blood-2007-12-128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hung SC, Pochampally RR, Hsu SC, et al. Short-term exposure of multipotent stromal cells to low oxygen increases their expression of CX3CR1 and CXCR4 and their engraftment in vivo. PLoS One. 2007;2:e416. doi: 10.1371/journal.pone.0000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosova I, Dao M, Capoccia B, et al. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells. 2008;26:2173–2182. doi: 10.1634/stemcells.2007-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hemeda H, Jakob M, Ludwig AK, et al. Interferon-gamma and tumor necrosis factor-alpha differentially affect cytokine expression and migration properties of mesenchymal stem cells. Stem Cells Dev. 2010;19:693–706. doi: 10.1089/scd.2009.0365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.