

Abstract
Coordinated regulation of gene expression relies on transcription factors (TFs) binding to specific DNA sites. Our large-scale information-theoretic analysis of >950 TF-binding motifs demonstrates that prokaryotes and eukaryotes use strikingly different strategies to target TFs to specific genome locations. Although bacterial TFs can recognize a specific DNA site in the genomic background, eukaryotic TFs exhibit widespread, nonfunctional binding and require clustering of sites to achieve specificity. We find support for this mechanism in a range of experimental studies and in our evolutionary analysis of DNA-binding domains. Our systematic characterization of binding motifs provides a quantitative assessment of the differences in transcription regulation in prokaryotes and eukaryotes.
DNA binding and gene regulation
Classical experiments demonstrated that strong binding of a TF to its cognate site in a promoter is sufficient to alter gene expression [1]. Significant effort has been put into experimentally determining [2–6] and computationally inferring [7–10] motifs recognized by TFs and determining the occupancy of promoters by TFs [11]. The motifs and binding locations of a TF have in turn been used to predict which genes it regulates and their expression levels [12]. Such studies rely on linking the binding of TFs to DNA with the regulation of nearby genes.
Although such an association has been strongly established in bacteria, growing experimental evidence in eukaryotes challenges this assumption by showing limited correlation between gene expression and TF binding [12–14]. For example, Gao et al. found no correlation between occupancy patterns and gene expression profiles for the majority (67%) of yeast TFs they studied, suggesting that only a subset of promoters bound by each TF is controlled by it [12]. A more striking example comes from a recent study [13], which demonstrated only 3% overlap between TF occupancy and genes response to TF knock-out. Although this discrepancy can be explained in part by a redundant binding of homologous TFs [15], it might also be evidence of a more fundamental uncoupling between TF binding and gene expression in eukaryotes.
Our analysis of 969 TF-binding motifs provides strong support for the uncoupling hypothesis by demonstrating that eukaryotic TFs do not recognize DNA with sufficient specificity (i.e. do not possess sufficient information) to bind to cognate sites exclusively; instead they occupy tens of thousands of decoy sites throughout a genome. Although managing such promiscuous binding requires several costly mechanisms, its advantages for eukaryotes are yet to be understood.
An information-theoretical approach to binding-motif recognition
To bind its cognate site, a TF has to recognize it among ~106 alternative sites in bacteria or ~109 sites in eukaryotes. Using information theory, we ask whether individual TFs possess enough information for such remarkably precise recognition. The application of information theory to protein–DNA recognition has a rich history [16–18] and provides a theoretical basis for current efforts to characterize motifs recognized by DNA-binding proteins using a range of in vivo and in vitro techniques [6]. The most common use of information theory is to construct ‘sequence logos’ that demonstrate the relative contribution of individual base pair positions to binding specificity (Figure 1). Information theory, however, also allows us to test whether the total information contained in a motif is sufficient to guide a protein to a specific place in a large genome.
Figure 1. Information theory as applied to DNA-binding motifs.

(a) The concepts of minimal information required in theory and in DNA recognition and the consequences of information deficiency, which results in spurious hits. (b) The sequence logos for low- and high- information motifs, and the likelihood of a spurious hit to the motif in a ‘random’ genomic background.
Information theory dictates that finding a unique object among N alternatives requires Imin = log2 N bits of information (Figure 1) [19]. Similarly, a minimum of Imin = log2 N bits of information is needed to specify a unique address in a genome containing N possible sites for a TF to bind (i.e. N bps). For bacteria, with N =106–107 bps this yields Imin = 20–23 bits (Imin = 22 bits for Escherichia coli). For eukaryotic genomes, N =108–1010 bps leading to Imin ≈ 27–33 bits (Imin = 24 bits for Saccharomyces cerevisiae, Imin = 27 bits for Drosophila melanogaster, and Imin = 31 bits for Homo sapiens).
To test whether TF motifs contain enough information to identify unique sites in their corresponding genomes, we calculated the information content of 969 experimentally determined bacterial and eukaryotic motifs. As a measure of information contained in a motif, we applied the commonly used Kullback-Leibler (KL) distance between the motif and the overall genome composition [17, 18]
| (Eqn 1) |
where L is the length of the motif, pi(b) is the frequency of base b at position i in the motif, and q(b) is its background frequency. The information content of a motif quantifies the sensitivity of TF binding affinity to variation in the binding site sequence from the consensus sequence and the probability of a site occurring in a ‘random’ stretch of DNA [16].
Motifs of bacterial and eukaryotic transcription factors are markedly different
Using this metric, we find that the motifs of prokaryotic and eukaryotic TFs are strikingly different (Figure 2, Tables S5–6 in the online supplementary material). The average information content of a prokaryotic motif, I ≈ 23 bits, is slightly above the required Imin = 22 bits, demonstrating that a single cognate site is generally sufficient to address a TF to a specific location in prokaryotes, though there still might be an overlap between the background and weak but functional sites in some cases (Figure S1 in the online supplementary material).
Figure 2. Properties of binding motifs for bacteria, yeast, and multicellular eukaryotes.

(a) The bar chart displays the minimum required information content for bacteria, yeast, and multicellular eukaryotes (red), and the mean information content of TF binding motifs (blue) for 98 bacterial [21], 124 yeast [22] and 123 multicellular [20] eukaryotic motifs. The error bars are ± 1 standard deviation for the information content, and for Imin, the error bars represent the variability in that quantity due to the range of genome sizes N. The blue dots in the chart indicate the average information content from several other transcription factor binding motif databases (Table S6). Below each series in the bar chart, we display an example of sequence logo for a binding motif with close to average information content. The chart demonstrates that bacterial transcription factor binding motifs informative enough to make spurious hits to the genomic background unlikely, in constant to yeast and multicellular eukaryotic motifs.
(b) The distributions of information content of motifs from the three representative databases cited above. The ranges of required information (Imin) are marked in red. Most bacterial motifs have I > Imin, whereas almost all eukaryotic motifs do not.
(c) Average properties of transcription factor binding motifs, the expected number and the spacing between the spurious sites per genome in bacteria, yeast and multicellular eukaryotes.
Although longer eukaryotic genomes require a TF to be more specific, we find that eukaryotic TFs are much less specific than bacterial TFs and do not contain sufficient information to find a cognate site among 109 decoys. The average information content of a multicellular eukaryotic motif is only I ≈ 12.1 bits, falling far below the Imin ≈ 30 bits required to provide a specific address in a eukaryotic genome (Figure 2). Yeast TF motifs have a mean information content of I = 13.8 bits, which is below the required Imin ≈ 24 bits, but represents a smaller information deficiency (Imin – I ≈ 10 bits) than that of the multicellular eukaryotes (Imin – I ≈ 18 bits).
To ensure that the results were not influenced by a poor choice of data, we employ databases [20, 21] that contain motifs for full biological TF units (i.e. dimers when the binding of an individual site is accomplished by a dimer, e.g. LacI, Gal4). We also rely on in vitro experiments [22] that used full-length TFs. In addition, the motifs do not show a significant correlation between the information content and the number of cognate sites used to derive the motif (ρ = −0.27). When motifs with <8 cognate sites in RegTransBase are eliminated, we see a decrease in the mean information content by ~1 bit. Taken together, we conclude the biases due to the number of sites used to construct a TF binding motif do not change our general findings. Finally, these results are consistent for motifs obtained both in vivo and in vitro and for all available data sets (Table S6, in the supplementary material).
Widespread non-functional binding in multicellular eukaryotes
The significant information deficiency in eukaryotes, which emerges because of their large genomes and degeneracy of the motifs, has several biologically important consequences. Primarily, it suggests that numerous sites as strong as the cognate ones are expected to be present in eukaryotic genomes by chance. Using information theory and simulations, we estimate the lower bound of the number of such spurious sites or hits as h ≥ 2Imin–I, with an average spacing s≤ 2I between them (Figure S1c, in the supplementary material). Therefore, an average multicellular eukaryotic TF is expected to have h ≈ 104 – 106 spurious sites per genome, which is reduced to h ≈ 103–105 accessible sites assuming 90% chromatinization of the genome or h ≈ 102–104 assuming 98% chromatinization. For yeast, h ≈ 102–104, assuming 0 to 80% chromatinization.
In multicellular eukaryotes, spurious sites are expected to arise by chance every s ≈ 4000 bp. An important implication of this is that, in eukaryotes, the presence of a site cannot be a distinctive feature of a regulatory region. By contrast, a typical bacterial TF is expected to have few such spurious sites, making the presence of a single high-affinity site a unique event and a distinctive feature of a regulatory region. Consistent with this picture is the atypically low information content of a few bacterial DNA-binding proteins that pack and crosslink DNA: H-NS (histone-like nucleoid structuring protein), Fis (factor for inversion stimulation) and IHF (integration host factor) (I = 7.5, 7.3 and 7.8 bits, respectively). Similarly, and in agreement with Sengupta el al. [8], CRP (catabolism repressor protein) and other global regulators that bind hundreds of sites in the genome have lower information content (CRP: I = 11 bits). The low information content of bacterial global regulator motifs makes it particularly challenging to find their cognate sites [23].
Because the information-theoretic results depend on a rather simple description of the genomic background, we searched real genomic sequences for matches to several well-characterized motifs to verify the validity of the theoretical results. Using a standard bioinformatics approach, we find, in agreement with the theory, >104 spurious sites per genome for degenerate eukaryotic TFs (Table S1). This does not constrain in any way the number of cognate, functional sites a TF has in the genome but demonstrates that, in eukaryotes, cognate sites can be difficult to recognize among 103–105 equally strong spurious sites. This creates a binding landscape with a potential for widespread non-functional binding.
Widespread non-functional binding is consistent with diverse experimental data
Evidence of this landscape has been found in several large-scale experiments. Our estimate of ~103 spurious hits in the chromatinized D. melanogaster genome is consistent with the 103–104 experimentally observed binding events for several TFs [14]. Moreover, our results explain the large number of binding events detected by ChIP-chip [11] and ChIP-seq experiments [24], suggesting that majority of these events reflect the widespread binding to sites that arise by chance and are likely to be non-functional. In agreement with this idea, studies in yeast have shown a decoupling between binding and apparent regulatory function for a nontrivial fraction of TF binding events [12, 13].
Using the estimated frequency of spurious sites in multicellular eukaryotes of once every 4000 bp, and assuming a regulatory (accessible) region of ~1000 bp around the transcription start site of each gene, we estimate that a single TF is expected to bind spuriously to ~25% of all regulatory regions. Consistent with these estimates, ChIP-chip experiments found that NOTCH1 binds to 19%, MYC to 48%, and HES1 to 18% of all human promoters [25]. Our expectation is that most of these binding events have little regulatory effect. The prevalence of widespread, spurious binding events in eukaryotes means that we should be cautious in interpreting all experimentally identified binding events as regulatory interactions.
The abundance of accessible high-affinity spurious sites in eukaryotes has two effects: (i) it sequesters TF molecules; and (ii) it makes it more difficult for the cellular machinery of gene regulation to detect regulatory regions occupied by TFs and discriminate them from occupied spurious sites.
The sequestration of TF molecules by spurious binding sites necessitates a high TF copy number. The number of spurious sites h (or the number of cognate sites to be bound) imposes a lower limit on the TF copy number per cell [26], which is on the order of 1–10 per cell for bacteria, 1000 for yeast, and 103–105 for multicellular eukaryotes. These estimates are consistent with available experimental data: 5–10 copies per cell of LacI repressor in E. coli, an average of approximately 2000 copies per cell of TFs in yeast; and 105 copies per cell of the prototypical multicellular eukaryotic TF p53 (Table S4).
Clustering of cognate sites can provide specificity of eukaryotic TFs
Although high TF copy-numbers are necessary to cope with spurious sites, they are not sufficient to provide specificity (i.e. to allow cellular machinery to distinguish regulatory binding sites from equally strong decoys). However, the presence of multiple sites in proximity of each other can specify a regulatory region. Many regulatory regions in eukaryotes contain multiple sites of the same or different TFs [7, 27–35], a property commonly used in bioinformatics to detect regulatory regions [27, 31]. Using the information content of TF motifs, we can calculate the minimal number of cognate sites (ncluster) in regions of length w ≈ 500–1000 bps needed to determine a unique location in a genome (supplementary methods online, Tables S2, S3). To obtain ncluster, we first calculate how many clusters of n spurious sites are expected to be found in a genome of a given length, E(n). Next we choose ncluster as the minimal number of sites in a cluster such that E(n) <1. In other words, a cluster of sites is unique (i.e. informative) if spurious sites are expected to form less than one such cluster by chance.
In a region of 1000 bp composed of the sites of 3–10 different TFs, we calculate ncluster = 10–20 sites. This lower limit on the number of required binding sites is remarkably consistent with the mean of 18–25 sites per 1000 bp observed in fly developmental enhancers [28]. These results also demonstrate that, beyond the known examples in flies and sea urchins[35], clustering of sites is a common phenomenon applicable to many regulatory regions of multicellular eukaryotes.
We also use an information-theoretical approach to calculate the information content of a cluster of sites and then estimate the minimal number of sites in cluster sufficient to reach the required information Imin. We demonstrate (see the online supplementary material) that for a cluster of sites spanning a region of w bps, the contribution of each site i to the total information content of the cluster (δIi) is approximately
| (Eqn 2) |
where Ii is the information content of motif i. Choosing w = 500 –1000 bps [31, 36] and Ii = 12 bits, we obtain that each site contributes 2–3 bits of information, necessitating 10–15 sites to achieve the ~30 bits of information needed for multicellular eukaryotes.
Eukaryotic and bacterial TF using different repertoire of DNA-binding domains
Our study shows that combinatorial regulation is rooted in the way eukaryotic TFs recognize DNA, but how did this difference from prokaryotes arise? The gradual modifications of the DNA-binding residues, the expansion and/or contraction of the DNA-binding interface, or the re-invention of DNA-binding domains altogether could have contributed to this difference. To investigate the possible evolutionary trajectory, we compared sequences of prokaryotic and eukaryotic DNA-binding domains of TFs available in the PFAM database [37] (Figure 3a). This analysis gives a clear result – prokaryotes and eukaryotes use different sets of DNA-binding domains. Of the 133 known DNA-binding domains, 69 have only eukaryotic members, 49 are totally prokaryotic, and only 15 families have both prokaryotic and eukaryotic members, but are usually dominated by one of two kingdoms (Table S7). This result is consistent with the previous observation of the differing rates of expansion and contraction of DNA binding domain families between prokaryotes and eukaryotes [38]. As a control, we compare this result to domains involved in glycolysis and gluconeogenesis and find that a few of those domains are kingdom specific (Figure 3b). The lack of shared prokaryotic and eukaryotic DNA-binding domain families suggests that the TF machinery employed by eukaryotes might have evolved de novo.
Figure 3. Membership of PFAM protein domain families, by kingdom.
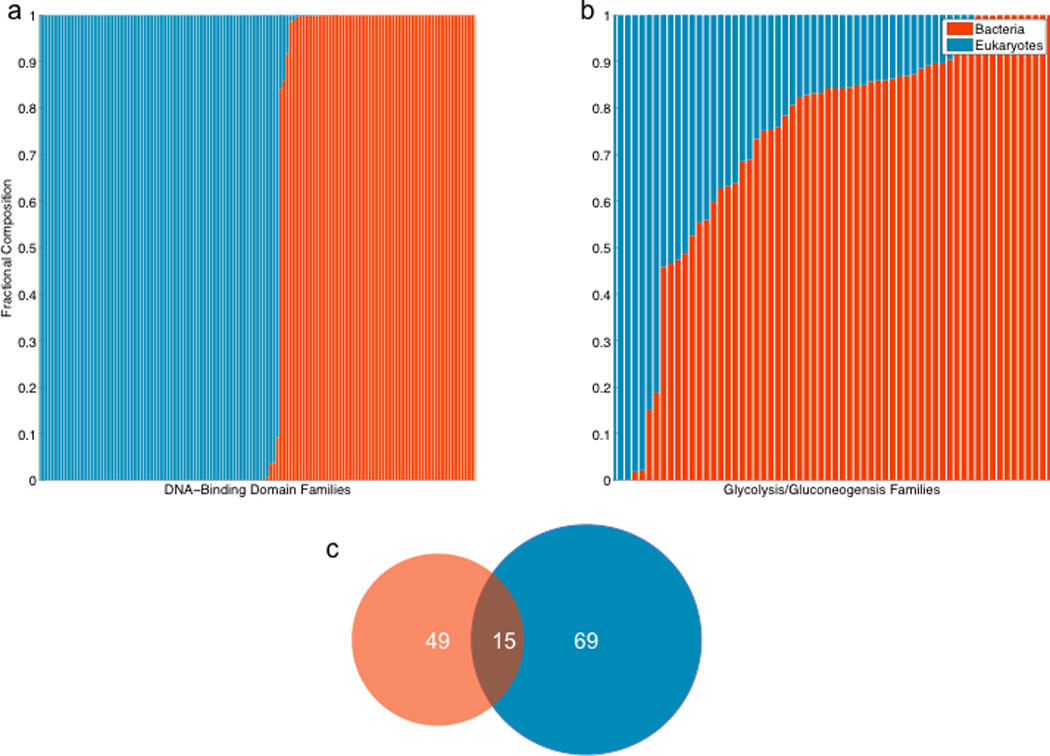
To explore the evolution of DNA-binding domains, we examined the membership of PFAM protein domain families. Each column in (a, b) represents a single PFAM family, and the size of the red or blue bar indicates the proportion of the family’s bacterial and eukaryotic members, respectively. (a), shows the membership of DNA-binding domains, demonstrating that by bacteria and eukaryotes share very few. As a control (b), we plot the composition of PFAM glycolysis and /or gluconeogensis enzyme families, which are shared between kingdoms. In (c), we show a Venn diagram, after removing the weakest 10% of hits to a PFAM family profile.
Energy-based considerations of transcription factor binding
As was demonstrated in the seminal paper by Berg and von Hippel [16] and later papers, for example Ref. [17], this information-theoretical approach is closely related to the energy-based analysis of TF binding motifs. The constraints on the information content of motifs considered here can be interpreted as constraints on the sequence-specific protein-DNA binding energy. Gerland et al. [26] and Lassig [39] have considered these constraints and demonstrated that the energy contribution of each consensus base pair to the sequence-specific binding energy in bacteria should be approximately ε ≈ 2–3 kB T for a motif of L = 15 bps.
The specificity of transcription factor binding can be assessed using an energy-based approach: given a set of cognate sites, how many sites in a genome are expected to have the energy lower than the energy of the cognate sites? A direct answer is provided by our bioinformatics analysis, where such sites are explicitly counted in each genome. We also used the information content of TF motifs to estimate the contribution of each consensus base pair to the sequence-specific binding energy (supplementary methods online), obtaining a range ε ≈ 1.5–3.5 kBT = 1–2 Kcal/mol for both prokaryotes and eukaryotes, which is consistent with recent micro-fluidic measurements [2].
Another important aspect of TF recognition not considered here is the nonspecific binding of proteins to DNA, as our focus was on specific (high affinity) binding. As was demonstrated previously [26, 39, 40], competition between specific binding to cognate sites and non-specific binding to the rest of the DNA determines whether a TF is bound to the cognate site or to non-specific DNA. Using available dissociation constants for specific and non-specific binding [2, 41, 42], we calculate that a bacterial TF binds non-specifically once every 106 bps. Eukaryotic TFs, in contrast, bind non-specifically every 103–104 bps. Therefore, non-specific binding sequesters almost as many TF molecules as the spurious sites, making it difficult for the cell to recognize a regulatory region form the rest of the DNA where TFs are bound specifically and non-specifically.
Concluding remarks
We asked whether individual TF binding motifs possess enough information to find a cognate site in the genome. The promiscuity of eukaryotic TFs leads to widespread, likely non-functional, binding to decoy sites. If supported by direct experimental evidence, this conclusion will challenge our understanding of gene regulation, which was gained largely from experiments in bacterial systems and can be summarized as: one site – one TF – one binding event. In multicellular eukaryotes this paradigm turns into: multiple sites – thousands of copies of each TF – multiple cooperative binding events; making one binding event necessary, but certainly not sufficient to regulate gene expression.
Such a mechanism is consistent with the concept of combinatorial gene regulation in eukaryotes, but goes further by suggesting that not only are several sites required to form a regulatory region, but binding to individual sites is likely to be widespread and possibly non-functional. Cooperative binding [1] and synergetic activation [43] are likely to be some of the mechanisms employed by the cell to differentiate between individual sites and clusters.
Although the apparent paradox of information deficiency in eukaryotes can be resolved by using regulatory regions containing clusters of sites, each TF must nevertheless be present in very high copy-number. Clearly, maintaining the tens of thousands of copies of each TF per cell needed to saturate decoy sites comes at a metabolic cost that is likely outweighed by the advantages of promiscuous binding that are yet to be discovered.
Evolutionary analysis supports our information-theoretical results and shows that the observed differences in DNA recognition are not specific to a few cases but are likely to span across kingdoms and constitute fundamentally different strategies of transcriptional regulation in prokaryotes and eukaryotes. The promiscuity of eukaryotic TFs is likely to constitute one of many eukaryotic evolutionary novelties, which might enable more evolvable gene regulation, and thus be essential for evolution of a variety of structures [44].
Supplementary Material
Acknowledgments
We thank Daniel Fisher, Shamil Sunyaev, Mikahil Gelfand, Shaun Mahoney, Sharad Ramanathan and Alex Shpunt for insightful discussions and Michael Schnall for interpretation of the information cutoff. ZW was supported by a Howard Hughes Medical Institute Predoctoral Fellowship. LM acknowledges support of i2b2, NIH-supported Center for Biomedical Computing at the Brigham and Women’s Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gann A, Ptashne M. Genes & Signals. Cold Spring Harbor Laboratory Press; 2002. [Google Scholar]
- 2.Maerkl SJ, Quake SR. A systems approach to measuring the binding energy landscapes of transcription factors. Science (New York, N.Y. 2007;315:233–237. doi: 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 3.Fields DS, et al. Quantitative specificity of the Mnt repressor. Journal of molecular biology. 1997;271:178–194. doi: 10.1006/jmbi.1997.1171. [DOI] [PubMed] [Google Scholar]
- 4.Noyes MB, et al. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008;133:1277–1289. doi: 10.1016/j.cell.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badis G, et al. Diversity and complexity in DNA recognition by transcription factors. Science (New York, N.Y. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bulyk ML. DNA microarray technologies for measuring protein- DNA interactions. Current opinion in biotechnology. 2006;17:422–430. doi: 10.1016/j.copbio.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siggia ED. Computational methods for transcriptional regulation. Curr Opin Genet Dev. 2005;15:214–221. doi: 10.1016/j.gde.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Sengupta AM, et al. Specificity and robustness in transcription control networks. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2072–2077. doi: 10.1073/pnas.022388499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacIsaac KD, et al. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinney JB, et al. Precise physical models of protein-DNA interaction from high-throughput data. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:501–506. doi: 10.1073/pnas.0609908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harbison CT, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao F, et al. Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data. BMC bioinformatics. 2004;5:31. doi: 10.1186/1471-2105-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Z, et al. Genetic reconstruction of a functional transcriptional regulatory network. Nature genetics. 2007;39:683–687. doi: 10.1038/ng2012. [DOI] [PubMed] [Google Scholar]
- 14.Li XY, et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS biology. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gitter A, et al. Backup in gene regulatory networks explains differences between binding and knockout results. Mol Syst Biol. 2009;5:276. doi: 10.1038/msb.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. Journal of molecular biology. 1987;193:723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- 17.Stormo GD, Fields DS. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem Sci. 1998:109–113. doi: 10.1016/s0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- 18.Schneider TD, et al. Information content of binding sites on nucleotide sequences. Journal of molecular biology. 1986:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 19.Cover TM, Thomas Joy A. Elements of Information Theory. Wiley-Interscience; 1991. [Google Scholar]
- 20.Vlieghe D, et al. A new generation of JASPAR, the open-access repository for transcription factor binding site profiles. Nucleic acids research. 2006;34:D95–D97. doi: 10.1093/nar/gkj115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kazakov AE, et al. RegTransBase--a database of regulatory sequences and interactions in a wide range of prokaryotic genomes. Nucleic Acids Res. 2007;35:D407–D412. doi: 10.1093/nar/gkl865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu C, et al. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res. 2009;19:556–566. doi: 10.1101/gr.090233.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mustonen V, Lassig M. Evolutionary population genetics of promoters: predicting binding sites and functional phylogenies. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15936–15941. doi: 10.1073/pnas.0505537102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson DS, et al. Genome-wide mapping of in vivo protein- DNA interactions. Science (New York, N.Y. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 25.Margolin AA, et al. ChIP-on-chip significance analysis reveals large-scale binding and regulation by human transcription factor oncogenes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:244–249. doi: 10.1073/pnas.0806445106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerland U, et al. Physical constraints and functional characteristics of transcription factor-DNA interaction. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:12015–12020. doi: 10.1073/pnas.192693599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajewsky N, et al. Computational detection of genomic cis-regulatory modules applied to body patterning in the early Drosophila embryo. BMC bioinformatics. 2002;3:30. doi: 10.1186/1471-2105-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berman BP, et al. Computational identification of developmental enhancers: conservation and function of transcription factor binding-site clusters in Drosophila melanogaster and Drosophila pseudoobscura. Genome Biol. 2004;5:R61. doi: 10.1186/gb-2004-5-9-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ochoa-Espinosa A, et al. The role of binding site cluster strength in Bicoid-dependent patterning in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4960–4965. doi: 10.1073/pnas.0500373102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emberly E, et al. Conservation of regulatory elements between two species of Drosophila. BMC bioinformatics. 2003;4:57. doi: 10.1186/1471-2105-4-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berman BP, et al. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc Natl Acad Sci U S A. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markstein M, et al. Genome-wide analysis of clustered Dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc Natl Acad Sci U S A. 2002;99:763–768. doi: 10.1073/pnas.012591199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinha S, et al. Cross-species comparison significantly improves genome-wide prediction of cis-regulatory modules in Drosophila. BMC Bioinformatics. 2004;5:129. doi: 10.1186/1471-2105-5-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hallikas O, et al. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006;124:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 35.Davidson EH. A view from the genome: spatial control of transcription in sea urchin development. Curr Opin Genet Dev. 1999;9:530–541. doi: 10.1016/s0959-437x(99)00013-1. [DOI] [PubMed] [Google Scholar]
- 36.Koudritsky M, Domany E. Positional distribution of human transcription factor binding sites. Nucleic acids research. 2008;36:6795–6805. doi: 10.1093/nar/gkn752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finn RD, et al. The Pfam protein families database. Nucleic acids research. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson D, et al. DBD taxonomically broad transcription factor predictions: new content and functionality. Nucleic acids research. 2007 doi: 10.1093/nar/gkm964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lassig M. From biophysics to evolutionary genetics: statistical aspects of gene regulation. BMC bioinformatics. 2007;8(Suppl 6):S7. doi: 10.1186/1471-2105-8-S6-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kolesov G, et al. How gene order is influenced by the biophysics of transcription regulation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13948–13953. doi: 10.1073/pnas.0700672104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Revzin A. The Biology of Nonspecific DNA Protein Interactions. CRC Press; 1990. [Google Scholar]
- 42.Ozbudak EM, et al. Multistability in the lactose utilization network of Escherichia coli. Nature. 2004;427:737–740. doi: 10.1038/nature02298. [DOI] [PubMed] [Google Scholar]
- 43.Carey M, et al. A mechanism for synergistic activation of a mammalian gene by GAL4 derivatives. Nature. 1990;345:361–364. doi: 10.1038/345361a0. [DOI] [PubMed] [Google Scholar]
- 44.Carroll SB. Evolution at two levels: on genes and form. PLoS biology. 2005;3:e245. doi: 10.1371/journal.pbio.0030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.