

Abstract
Brain contains a highly diversified complement of molecular species of a mitochondria-specific phospholipid, cardiolipin (CL), which - due to its polyunsaturation - can readily undergo oxygenation. Here, we used global lipidomics analysis in experimental traumatic brain injury (TBI) and showed that TBI was accompanied by oxidative consumption of polyunsaturated CL and accumulation of more than 150 new oxygenated molecular species in CL. RNAi-based manipulations of CL-synthase and CL levels conferred resistance of primary rat cortical neurons to mechanical stretch - an in vitro model of traumatic neuronal injury. By applying the novel brain permeable mitochondria-targeted electron-scavenger, we prevented CL oxygenation in the brain, achieved a substantial reduction in neuronal death both in vitro and in vivo, and markedly reduced behavioral deficits and cortical lesion volume. We conclude that CL oxygenation generates neuronal death signals and that its prevention by mitochondria-targeted small molecule inhibitors represents a new target for neuro-drug discovery.
Every year in the United States alone, an estimated 1.7 million people sustain acute brain injury from trauma and of those, 52,000 die and 85,000 suffer from long term disabilities1,2. This includes injuries from many etiologies such as road traffic accidents, falls, assaults, and sports concussion. Also, the RAND Corporation estimates that over 400,000 US soldiers have suffered TBI in Operation Iraqi Freedom and Operation Enduring Freedom3. A specific therapy for TBI does not yet exist and standard treatment remains supportive in nature. Like many forms of acute brain injury, TBI involves primary injury that is felt to be refractory to treatment and secondary injury, characterized by a cascade of biochemical and cellular events contributing to neuronal damage over time4. Secondary injury involves multiple pathways of neuronal death which represent viable therapeutic targets5,6.
The complexity of brain function requires sophisticated coordination of highly diversified operational signals. Numerous small molecule-signals formed from a relatively limited number of metabolic precursors are accountable for the precision and effectiveness of the brain’s operational language. Among these are polyunsaturated lipids capable of undergoing enzymatic oxidation and other metabolic conversions yielding oxygenated signaling entities - from eicosanoids, prostanoids and resolvins to cannabinoids and neuroprotectins. There is substantial archaeological evidence that exploitation of abundant food resource enriched with polyunsaturated lipids was essential for sustaining the comparatively large size, the apparent unique complexity and high level of interconnectivity in the modern human brain and provided the advantage in multi-generational brain development, thus making possible the advent of H. sapiens7. The demand for diverse polyunsaturated lipid precursors is so large that mitochondria-specific CL is represented in the brain—but not in other tissues—by hundreds of polyunsaturated individual molecular species8. Our previous work demonstrated that in fatally injured cells, oxidation of polyunsaturated CL species by cytochrome c (cyt c) generates death signals essential for mitochondrial permeability transition and release of death factors into the cytosol9.
RESULTS
TBI induces selective oxidation of CL
While lipid peroxidation has been long associated with acute brain injury, its specific role in mediating damaging pathways and signaling cascades is not well understood10,11. We reasoned that aberrant CL peroxidation may be an important pathogenic pathway in acute brain injury. Using a newly developed 2-dimensional liquid chromatography mass spectrometry (2D-LC-MS) protocol, we performed global lipidomics analysis of phospholipids (Fig. 1a), which revealed almost 190 individual molecular species of CL in normal rat brain, of which only 10 were oxygenated. Notably, experimental TBI - controlled cortical impact (CCI) - induced oxidation of the majority of polyunsaturated molecular species of CL; the number of non-oxidized CL species decreased to ~100, while that of oxygenated species increased to 166 (Fig. 1b). Quantitatively, the content of oxidized CL species increased 20-fold at the expense of decreased amounts of non-oxidized CL (Fig. 1c). Major CL species that underwent oxidation and oxidation products generated after TBI are presented in Supplementary Table 1. This oxidation effect was specific to CL, as other, more abundant polyunsaturated phospholipids, phosphatidylcholine (PC) and phosphatidylethanolamine (PE), remained non-oxidized (Supplementary Fig. 1). Clusters of oxidized CL were also observed in the 2D-LC-MS lipidomics profile in a human brain sample after TBI (Supplementary Fig. 2). The scale of CCI-induced oxidative changes in CL far exceeded the detectable but much less pronounced decreases of the levels of glutathione (GSH) and protein sulfhydryls (PSH) in the rat brain (Supplementary Fig. 3a,b). The decrease in GSH levels after CCI could be associated with the responses to accumulation of CL hydroperoxides by mitochondrial GSH peroxidase IV known to catalytically reduce CL-hydroperoxides at the expense of GSH12,13.
Figure 1.

Assessment of molecular species of CL and its oxidation products by 2D-LCMS after TBI.
(a) Typical spectra of CL obtained from brain cortex. Upper panel shows the presence of multiple non-oxidized (8-10 major clusters of mass ions shown in blue) and few oxidized (shown in red, CLox) clusters of CL in naïve rat. Left upper insert: 1st dimension chromatographic separation of phospholipids in lipid extracts of the ipsilateral cortex. CL eluted with the 10–12 min retention time window. Right inserts: 2nd dimension chromatographic separation of non-oxidized and oxidized CL. The latter eluted during the 5-6 min retention time window. Middle panel demonstrates non-oxidized (blue) and the appearance of numerous oxidized (red) CL species after TBI. Lower panel illustrates the effect of XJB-5-131 administration after TBI on the profile of non-oxidized (blue) and oxidized (red) CL species. (b) Evaluation of the number of non-oxidized and oxidized molecular species of CL in the brain.(c) Quantification of CL oxidation by 2D-LCMS. Increased content of CLox at 3 h after CCI and its attenuation by XJB-5-131. *P < 0.05 vs. naïve and XJB-5-131; error bars, standard deviation, n = 4 rats per group.
To gain further insight into the role of CL and its oxidation in CCI induced damage, we utilized an in vitro TBI model of mechanical stretch injury in rat primary cortical neurons. Mechanical stretch triggered robust and selective peroxidation of CL (Supplementary Fig. 3c) but not PC or PE, which comprise 49% and 30%, respectively, of total phospholipids in primary neurons. Stretch decreased neuronal viability assessed by: i) LDH, MTT and Trypan blue exclusion assays (Fig. 2a and Supplementary Fig. 4, 5a), ii) microtubule-associated protein 2 (MAP-2, neuronal cytoskeleton marker), and cleaved caspase 3 immunostaining (Supplementary Fig. 5b,c), iii) and caspase 3/7 activity measurements (Fig. 2b). Of note, exposure of primary neurons to oxidized CL (but not to non-oxidized CL) caused dose-dependent cell death (P < 0.01) (Fig. 3).
Figure 2.
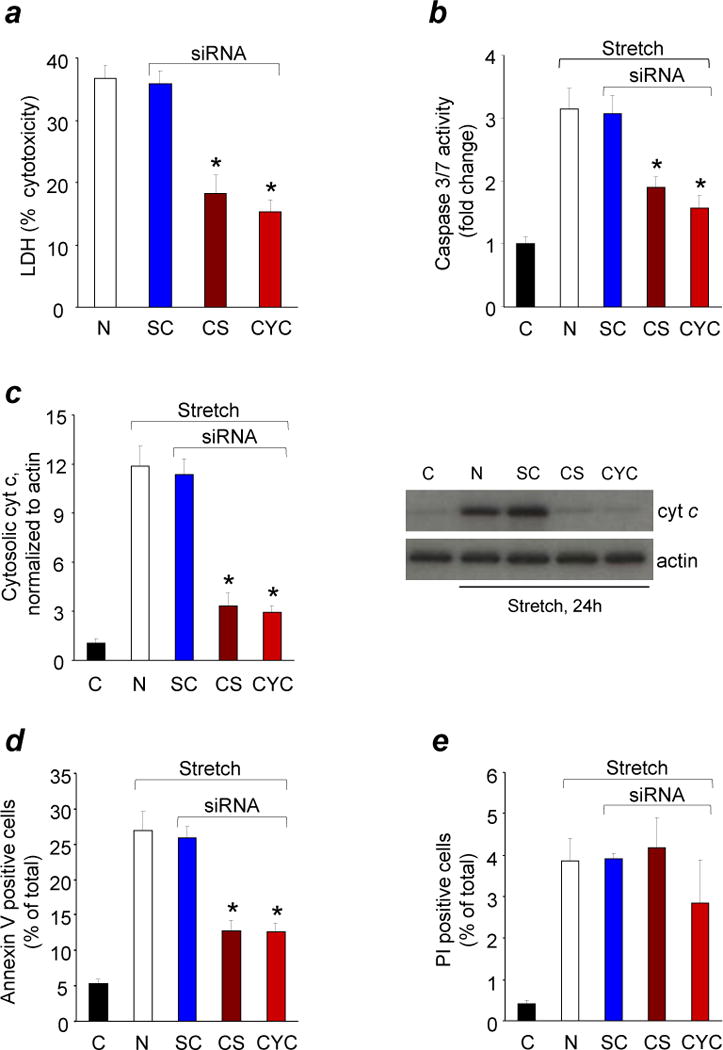
Response of CL or cyt c deficient neurons to in vitro TBI.
Quantification of cytotoxicity (lactate dehydrogenase (LDH) release) relative to Triton exposure (a), caspase 3/7 activity (b), cyt c release from mitochondria into cytosol (c), annexin V (d) and propidium iodide (PI) positivity (e) in rat cortical neurons transfected with Cardiolipin synthase (CS) or Cytochrome c (CYC) or scrambled control (SC) siRNA after mechanical stretch. Rat cortical neurons were transfected 72 h before mechanical stretch and measurements were obtained at 24 h after stretch injury. C: control normal neurons; N: non-transfected neurons.* P < 0.01 vs. N and SC; error bars, standard deviation; n = 4 experiments. Stretch induced PI positivity did not change in CL and cyt c deficient neurons (P > 0.05).
Figure 3.

Neuronal cell death in response to non-oxidized and oxidized cardiolipin.
(a) Quantification of Annexin V and/or propidium iodide (PI) positivity, (b) cytotoxicity (lactate dehydrogenase (LDH) release) relative to Triton exposure, and (c) caspase 3/7 activity in rat cortical neurons exposed to non-oxidized tetralinoleyl cardiolipin (TLCL) and oxidized TLCL (TLCLox) in the form of liposomes. At three tested concentrations (5, 10, 25uM), TLCLox caused dose-dependent cell death in contrast to non-oxidized TLCL. * P < 0.01 vs. 0 μM and TLCL; error bars, standard deviation; n = 3 experiments.
Knock down of CL synthase or cytochrome c attenuates mechanical stretch induced neuronal death
We further utilized an RNAi approach to manipulate the content of CL in neurons by knocking-down CL-synthase (Supplementary Fig. 6a), the key-enzyme of de novo CL biosynthesis, and assessed their responses to stretch. The CL content decreased to 44% of its level in parental cells (Supplementary Fig. 6b), whereas the ATP content did not change (Supplementary Fig. 6c). Notably, characteristic markers of stretch-induced cell death – elevated caspase 3/7 activity, release of cyt c from mitochondria into the cytosol, increased annexin V-positivity, and release of LDH– were all attenuated and cell survival was increased in CL-deficient cells (Fig. 2a–d and Supplementary Fig. 5). Our previous work demonstrated that cell death-associated CL oxidation was catalyzed by peroxidase activity of the cyt c/CL complex9. With this in mind, we generated cyt c-deficient primary neuronal cells (Supplementary Fig. 6d) and examined their vulnerability to stretch-induced damage compared to control cells. Sensitivity of cyt c-deficient neurons to stretch was lower than that of parental cells as evidenced by caspase 3/7 activity, cyt c release, annexin V positivity, and cell viability (Fig. 2a–d and Supplementary Fig. 5). Taken together, these in vitro results are compatible with our hypothesis that CL oxidation, possibly catalyzed by cyt c, was involved in TBI induced damage of neurons.
Targeted electron scavenger XJB-5-131 is effectively delivered into neuronal mitochondria and crosses blood brain barrier
Because mutation or suppression of CL synthesis and cyt c is therapeutically impractical, we hypothesized that inhibition of CL peroxidation could attenuate neuronal death and brain injury. This might be achieved by targeting electron scavengers which will prevent formation of H2O2 - the fuel for cyt c/CL peroxidase - in the mitochondria. Therefore, we designed novel mitochondria-targeted nitroxide pay-loads conjugated to selective transporters into mitochondria14. We reasoned that these novel mito-nitroxides may represent effective therapeutic agents for brain injury, if they i) accumulate in mitochondria and prevent oxidative damage; ii) penetrate into the central nervous system (CNS) and accumulate in the brain tissue, and iii) display low toxicity and high neuroprotective potential. To this end, we used XJB-5-131 (Supplementary Fig. 3d) – a conjugate of 4-amino TEMPO and a chemically modified segment of a bacterial membrane targeting antibiotic, Gramicidin S (GS) – that effectively delivered the nitroxide into mitochondria. The characteristic triplet electron paramagnetic resonance (EPR) signal of GS-nitroxide was detected almost exclusively in the mitochondrial fraction of primary cortical neurons treated with XJB-5-131 (Fig. 4a), without affecting cell viability (Supplementary Fig. 4).
Figure 4.

Analysis of XJB-5-131 distribution in neurons and brain.
(a) XJB-5-131 (10 μM) partitions into mitochondria in primary cortical neurons. Recovered nitroxide radicals in whole cells, mitochondria, and cytosol fractions were suspended in phosphate buffered saline in the presence or absence of 1.5 mM ferricyanide (K3Fe(CN)6). Insert: representative EPR spectra of XJB-5-131 in different fractions in the presence of ferricyanide. *P < 0.05 vs. without ferricyanide; error bars, standard deviation; n = 4 experiments. (b) EPR-based analysis of XJB-5-131 in CSF in naïve rats. A typical ascorbate radical signal (top) is detected by EPR in the absence of ferricyanide. After addition of ferricyanide, a typical nitroxide signal of XJB-5-131 is detected (bottom). (c) Imaging of XJB-5-131 in the brain of naïve rats by L-Band EPR spectroscopy. For optimal positioning of the head, micro-CT was utilized (upper panel). Lower panels demonstrate typical EPR images of XJB-5-131 distribution in the brain obtained at 5 min and 25 min after its i.v. injection (50 mg/kg). Arrows indicate two nitroxide radical standards (2.5 and 5 μL of 10 mM 3-carboxy-proxyl solution) placed in proximal portions of capillary tubes. (d) Distribution of XJB-5-131 in rat brain assessed by mass spectrometry imaging (MSI) and corresponding Hematoxylin-and-Eosin (H&E) staining of the frozen section. XJB-5-131 was detected as the lithium adduct of its hydroxylamine form at 966 m/z in positive mode TOF/TOF MSI with DHB matrix. The white scale bar is 2 mm. The pixels are a heat map with red being the highest intensity.
To evaluate CNS penetration of XJB-5-131, we measured the drug concentration in cerebrospinal fluid (CSF) by EPR spectroscopy. Typical nitroxide signals were detected in CSF when XJB-5-131 (10 mg/kg) was administered intravenously (i.v.) to naïve post-natal day 17 (PND17) rats (Fig. 4b). At 30 min after injection, total concentration of nitroxide and its reduced form (revealed in the presence of ferricyanide) in CSF was 1.4 ± 0.1 (mean ± s.d.) nM. By employing micro computerized tomography (CT) and L-Band in vivo EPR imaging, we documented a time- and dose-dependent distribution of XJB-5-131 in naïve PND17 rat brains after i.v. (Fig. 4c) injection. We further utilized mass spectrometric imaging and confirmed the presence of XJB-5-131 in the brain tissue after a single i.v. dose (Fig. 4d). Finally, direct quantification of XJB-5-131 in the brain tissue by LC-MS also demonstrated its accumulation to a level of 16.5 ± 4.3 pmole/gram of tissue (mean ± s.d.) at 3 h after a single 10 mg/kg i.v. injection.
XJB-5-131 inhibits CL oxidation and improves behavioral outcome after TBI
We further examined whether XJB-5-131 would inhibit TBI-induced CL oxidation. We found that CL oxidation was almost completely blocked by XJB-5-131 administered 10 min after TBI (Fig. 1). The number of non-oxygenated and oxygenated species was comparable to those in control brain. Quantitatively, the amounts of non-oxidized and oxidized CLs remained also similar to those in the non-traumatized brain (Fig. 1c). In addition, XJB-5-131 protected brain thiols - GSH and PSH – oxidized by TBI15,16(Supplementary Fig. 3a).
Based on the ability of XJB-5-131 to protect against CL oxidation, we evaluated its neuroprotective potential in TBI and performed neurobehavioral testing on PND 17 rats subjected to CCI injury. We assessed motor function with the well-established beam-balance and inclined platform tasks and evaluated cognitive performance using novel object recognition (NOR) and Morris water maze tests, which are sensitive to motor and cognitive function/dysfunction after TBI17–19. Balancing ability or inclined platform performance did not differ among groups prior to surgery (Fig. 5a, b) suggesting that pre-training was consistent among all groups. However, after surgery, a significant improvement in motor performance (with both tasks) was observed on days 3 to 5 in the sham and CCI + XJB-5-131 groups compared to the CCI + vehicle group (Fig. 5a, b). Although all groups began at a similar level in the water maze, both the CCI + XJB-5-131 and sham groups performed progressively better vs. the CCI + vehicle group. This cognitive benefit was observed during both the acute and sub-chronic phases of training, which correspond to post-injury days 11–15 and 30–34, respectively (Fig. 5c and Supplementary Fig. 7a) (P < 0.001). While the rats treated with XJB-5-131 found the platform significantly faster than vehicle treated rats (P < 0.001, N=7-10 per group), the time spent searching in the target quadrant during a single probe trial was not statistically different between the two groups (P = 0.09, N=7-10 per group). To further assess memory retention, we utilized the NOR memory test on post-operative day 29. XJB-5-131 treated rats spent significantly more time exploring the novel object vs. the vehicle treated rats (P < 0.01, Fig. 5d). Swim speed did not differ among groups (P = 0.12), indicating that water maze or NOR test performance were not influenced by differences in swimming or motor ability or motivational deficits.
Figure 5.
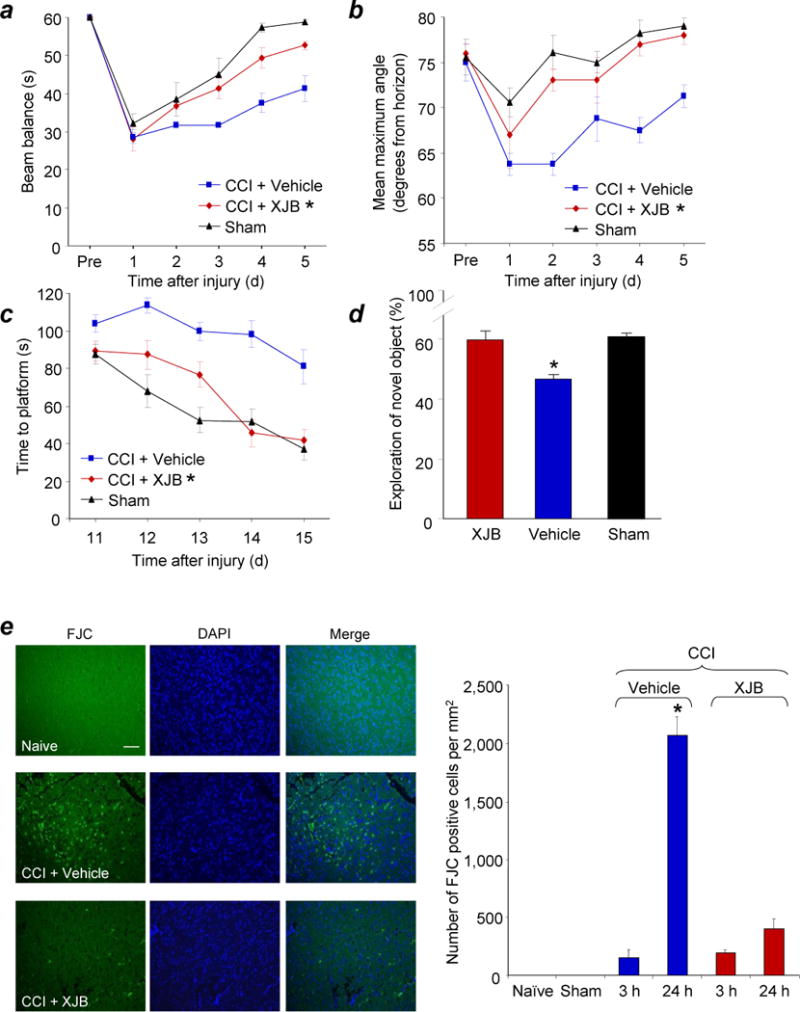
Assessments of neurobehavioral and histological outcome in PND 17 rats treated with XJB-5-131 after TBI.
(a) Ability of rats to remain (seconds) on the beam balance apparatus before and after CCI or sham injury. A repeated measures ANOVA revealed a significant group (F2,15 = 14.452, P = 0.0003), day (F5,75 = 47.631, P < 0.0001), and group by day interaction effect (F10,75 = 2.186, P = 0.028). Bonferroni post hoc analyses revealed that the CCI + XJB group performed significantly better than the CCI + vehicle group (*P = 0.001; error bars, standard error; n = 7-10 rats per group). (b) Maximum angle (degrees) for rats to remain on an inclined platform. A repeated measures ANOVA revealed significant group (F2,15 = 13.164, P = 0.0005), day (F5,75 = 18.085, P < 0.0001), and group by day interaction (F10,75 = 2.902, P = 0.004). Bonferroni post hoc analyses revealed that the TBI + XJB group performed significantly better than the TBI + vehicle group (*P < 0.001; error bars, standard error; n = 7-10 rats per group). (c) Latency (seconds) for rats to locate a hidden (submerged) platform on post-TBI days 11-15. A repeated measures ANOVA revealed significant group (F2,15 = 19.753, P < 0.0001), day (F4,120 = 22.126, P < 0.0001), and group by day interaction (F8,120 = 2.437, P = 0.018). Bonferroni post hoc analyses revealed that the TBI + XJB group performed significantly better than the TBI + vehicle group (P < 0.001; error bars, standard error; n = 7-10 rats per group) and did not differ from the sham controls (P = 0.08). (d) NOR task performance 29 days after sham or CCI injury. TBI + XJB rats exhibited a better 24-hour delay NOR task performance score compared to TBI + vehicle rats (P < 0.001, ANOVA, F2,27 = 14.736; error bars, standard error; n = 9-10 rats per group). (e) Assessment of neurodegeneration by Fluoro-Jade C (FJC) staining. Neurodegeneration observed in the pericontusional area at 24 h after CCI was attenuated by XJB-5-131. The white scale bar is 40 μm. *P < 0.05 vs. naïve and sham controls, CCI 3h + Vehicle, and CCI + XJB-5-131; error bars, standard deviation; n = 4 rats per group.
XJB-51-131 attenuates TBI-induced apoptotic neuronal death and decreases cortical lesion volume
To evaluate the potency of XJB-5-131 in preventing neuronal death, we employed Fluoro-Jade C staining (Fig. 5e), commonly used to detect degenerating neurons regardless of the specific cell death pathway20. The number of Fluoro-Jade C positive cells increased at 24 h vs. 3 h after TBI and naïve/sham. Fluoro-Jade C positivity was observed predominantly in the pericontusional area and was significantly attenuated by treatment with XJB-5-131. Furthermore, cortical lesion volume was significantly decreased at 5 weeks in the TBI group treated with XJB -5-131 vs. the TBI group that received vehicle (P < 0.05, Supplementary Fig. 7b).
To better define possible cell death pathways responsible for neuronal degeneration and the effect of XJB-5-131 in vivo, we examined caspase 3/7 activity. Ipsilateral cortical caspase-3/7 activity was increased at 24 h (but not at 3 h) after CCI (Supplementary Fig. 8). This effect was attenuated by XJB-5-131, pointing to its anti-apoptotic mechanism of action. Similarly, treatment with XJB-5-131 (5, 10, and 25 μM) attenuated stretch-induced neuronal death (Supplementary Fig. 4 and 5) via suppression of mitochondrial superoxide production (Supplementary Fig. 9a) and stretch-induced cyt c release into the cytosol (Supplementary Fig. 9b). 4-Amino-TEMPO - the active nitroxide part of XJB-5-131 - without mitochondria-targeting hemigramicidin moiety, failed to protect against stretch-induced neuronal death (Fig. 6), suggesting that mitochondrial damage was involved in neuronal cell death.
Figure 6.

Response of neurons treated with 4-Amino-TEMPO (4-AT) to in vitro TBI.
(a) Percent cytotoxicity (lactate dehydrogenase (LDH) release relative to Triton exposure (corrected for background LDH). (b) 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as a percentage of normal control conditions. Vehicle or 4-AT was added to the medium 10 min before stretch. Mechanical stretch induced approximately 35% neuronal death assessed by MTT and LDH at 24 hours. *P < 0.01 vs. stretch neurons, error bars, standard deviation; n = 3 experiments.
XJB-5-131 acts as an electron scavenger
The two likely mechanisms of XJB-5-131’s action in the brain are either as a SOD-mimetic - catalyzing the dismutation of superoxide radicals into H2O2 - or as an electron scavenger - preventing one-electron reduction of molecular oxygen to superoxide. In the former case, XJB-5-131 would retain its radical state; while in the latter case, XJB-5-131 would be reduced to hydroxylamine. In different redox-environments, dynamic inter-conversions between the nitroxide and hydroxylamine forms take place resulting in establishment of equilibrium between the two. This equilibrium is readily detectable by EPR spectra of the nitroxides (Fig. 7). Nitroxide radicals elicit a characteristic EPR signal with hyperfine splitting constants of 16.4 G (aN). In contrast, hydroxylamines are EPR-silent and produce no signals in the spectrum. A characteristic triplet EPR signal (Fig. 6aA) from nitroxide radicals disappeared and was substituted by a typical doublet signal of ascorbate radicals (with hyperfine splitting constants of 1.84 G) upon reduction of nitroxides (B, C). This signal of ascorbate radicals disappeared upon addition of ascorbate oxidase (D). As potent reductants, hydroxylamines are readily oxidized by metalloproteins in their higher valency states (e.g. Fe(III)-cyt c) and even more so by their reactive intermediates (eg, oxo-ferryls Fe(IV)=O produced in the presence of H2O2). Indeed, addition of oxidized ferri-cytochrome c (E) or cyt c/H2O2 (F) – to hydroxylamine (produced by the ascorbate-driven reduction of the nitroxide with subsequent elimination of excess ascorbate by ascorbate oxidase) resulted in one-electron oxidation of hydroxylamines and re-appearance of the characteristic nitroxide EPR signals.
Figure 7.
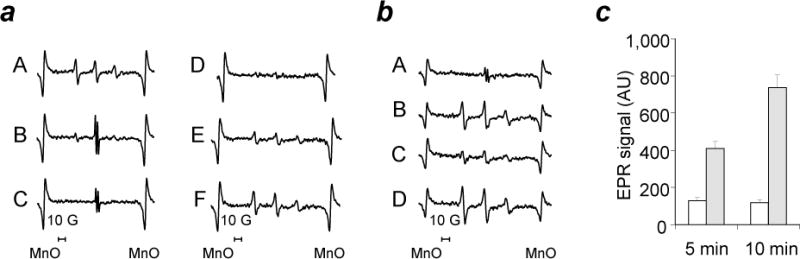
Electron paramagnetic resonance (EPR) spectroscopy assessment of inter-conversions of nitroxides and hydroxylamines.
(a) Effect of Ascorbate, Ascorbate oxidase and Cyt c/H2O2 on the EPR signal of XJB 5-131. A: XJB-5-131 alone in 35 μL of PBS plus 35 μL of DMSO. B: A plus 250 μM Ascorbate, 1 min. C: A plus 250 μM Ascorbate, 5 min. D: A plus 250 μM Ascorbate, 5 min plus ascorbate oxidase (1U, 15 min). E: D plus10 μM Cyt c. F: D plus10 μM Cyt c/200 μM H2O2. (b) Effect of ascorbate, ascorbate oxidase, and NADH on the EPR signal of XJB-5-131 in brain homogenates. A: 35 μL of Brain Homogenate (350 μg protein) in PBS plus 7 μM XJB-5-131 plus 35μL of DMSO. B: A plus Ascorbate Oxidase (2U), 30 min. C: B plus NADH (200 μM), 30 min. D: C plus K3Fe(CN)6 (100 μM). (c) Effect of pH on the EPR signal of XJB-5-131-OH. 50 μL of 7 μM XJB-5-131-OH in DMSO were mixed with 50 μL of 50 μM HEPES pH 7.45 (open bars) or with 50 μL of 50 μM HEPES pH 8.25 (closed bars). Error bars, standard deviation.
We also observed these inter-conversions between hydroxylamines and nitroxides in brain homogenates (Fig. 7b). Addition of the nitroxide to brain homogenate produced a typical ascorbate radical signal due to the reaction with endogenous ascorbate (A) 16. After pretreatment of the homogenate with ascorbate oxidase, the addition of the nitroxide immediately yielded the characteristic triplet signal of the nitroxide radical (B). Notably, triggering NADH-dependent transport of electrons resulted in significant suppression of the EPR signal of nitroxides due to their reduction by electron transport chain (ETC) (C). Finally, the redox inter-conversion between the nitroxide (> N–O*) and hydroxylamine (> N–OH) functionalities is a H+-dependent process, whereby, in the pH range of 6 -7, deprotonation of the hydroxylamine (> N+H–OH) markedly accelerates its auto-oxidation to nitroxide21,22. This suggests that in basic environment of mitochondria (pH~8.0), targeted XJB-5-131 hydroxylamine will spontaneously undergo autooxidation to the respective nitroxide (Fig. 7c). Furthermore utilizing LC/MS-based quantification, we found that nearly all XJB-5-131 (91.7 ± 14.5%) was present in the hydroxylamine form and the nitroxide represented only a small fraction of total XJB-5-131. Given that the hemigramicidin S-derived alkene peptide isostere sequence in XJB-5-131 is responsible for its predominant localization in mitochondria, its action is likely through scavenging of electrons leaking from discoordinated electron carriers, rather than through involvement of SOD-like activity (see also Fig.7).
DISCUSSION
The central role of oxidative stress in acute brain injury has been implicated for decades10,23,24 and prompted some of the earliest randomized controlled trials in the field of TBI25. However, limitations with specificity, potency, blood-brain barrier and/or cell penetration with conventional antioxidant approaches may have contributed to these past treatment failures26. This work identified selective peroxidation of CL as an important pathogenic mechanism for TBI. The peroxidation process yields a high diversity of oxygenated CL products that may be required to activate the neuronal death program.
Redistribution of CL from the inner mitochondrial membrane to the outer mitochondrial membrane and subsequent accumulation of CL oxidation products, catalyzed by cyt c, are required stages in the execution of the intrinsic apoptotic program9,27–31. Oxidized CL (but not CL) causes release of pro-apoptotic factors, including cyt c, from mitochondria into the cytosol and caspase activation32. The exact mechanisms of mitochondrial membrane permeabilization triggered by oxidized CL are currently under study and likely involves interactions with several outer membrane proteins (eg, Bax/Bak andVDAC) 33–35. It is likely that CL peroxidation is not an aftermath of cell death but rather is causative to it, thus offering an opportunity for a targeted therapy. Indeed, we found that mitochondrial delivery of small molecule inhibitors – electron scavengers – lead to prevention of both accumulation of a large number of CL oxidation products as well as brain damage. The use of the mitochondria-targeted nitroxides offers considerable therapeutic advantages compared to strategies used in the past, in terms of getting to the source of oxidative stress at an early point with the highest specificity and the least toxicity, and possibly limiting deleterious effects on oxidative signaling in other brain lipids.
Stable nitroxide radicals combine electron and free radical scavenging actions with superoxide dismutase (SOD)-mimicking and recycling capacities. One of these nitroxides, Tempol, has been shown to improve motor function after experimental TBI36; however, high millimolar concentrations were required to attain this improvement. Dependent on the redox environment, nitroxides may be present in three major forms: (1) oxidized - oxoammonium cation, RN+=O, (2) semi-reduced - the nitroxide radical, RN-O•, and (3) reduced - the hydroxylamine, RN-OH 37. Accordingly, there are three major biochemical mechanisms of action for nitroxides: i) Superoxide anion-radical-driven inter-conversions between 1↔2 underlie the SOD-mimicking activity of nitroxides 38. This catalytic activity regenerates the nitroxide form of the compound; no reduced form, hydroxylamine, is produced in this reaction. ii) Electron scavenging from other reductants, particularly reduced respiratory complexes of mitochondria. This mechanism prevents leakage of electrons from electron transport chains to molecular oxygen hence the formation of superoxide radicals and yields hydroxylamines via a one-electron reduction process. In extra-mitochondrial compartments, ascorbate is the most significant low molecular weight reductant of nitroxides whereas the reactivity of GSH and other thiols is much lower39. iii) Finally, interactions of hydroxylamines with oxidizing radicals, oxidized intermediates of peroxidases or oxidized forms of transition metals or metalloproteins yield the nitroxide. For example, reduction of hydroxylamines by oxidizing radicals – peroxyl, alkoxyl, or ferri-cytochrome c, etc – will convert hydroxylamines back to their nitroxide forms22,40,41. We performed EPR spectroscopy to more clearly illustrate these inter-conversions of nitroxides and hydroxylamines. Importantly, these inter-conversions between hydroxylamines and nitroxides can be observed in brain homogenates.
It is known that the immature brain has compromised antioxidant defenses compared to adults42, implying that the beneficial effects of XJB-5-131 could potentially be magnified in the developing brain. Given the enormous burden of TBI in pediatrics, our use of a developmental TBI model may be of additional clinical relevance. Germane to our findings, considerable mitochondrial dysfunction with altered bioenergetics43, increased oxidative stress markers44, membrane permeabilization45 and release of proapoptotic factors46 have been demonstrated both in experimental models and in humans after severe TBI. There is accumulating evidence in support of the role of TBI as a trigger for a number of neurodegenerative diseases such as dementia47, Amyotrophic lateral sclerosis, and Parkinson’s disease48. Given the role of oxidative stress across the field of brain injury, our work suggests the possibility that CL peroxidation may be characteristic of other types of acute brain injury, such as that seen in stroke and cardiac arrest. Therefore, this mechanism and the linked new therapeutic approach are relevant to other forms CNS injury.
METHODS
Methods and any associated references are available in the online version of the paper.
Supplementary Material
Acknowledgments
This study was supported in part by grants from NIH (NS061817 (HB), NS060005 (AEK), HL70755 (VEK), ES020693 (YYT & VEK), U19AIO68021 (HB & VEK)); NIOSH (OH008282 (VEK)) and the US Army (W81XWH-09-0187 (PMK)). A.S.A. is a recipient of a research fellowship from La Junta de Extremadura y el Fondo Social Europeo (2010063090). The authors would like to thank Jesse Lewis for the technical assistance of unbiased stereology for cortical lesion volume, Dr. Jennifer Davoren for the preparation of XJB-5-131, and Dr. Yves-Michel Frapart (Laboratoire de Chimie Biochimie Pharmacologique et Toxicologique, Université Paris Descartes) for providing an L-band EPR spectrometer for in vivo imaging.
Footnotes
AUTHOR CONTRIBUTIONS
J.J. designed and performed experiments, analyzed data and wrote the manuscript; A.E.K and J.P.C. contributed to the neurocognitive outcome assessment and writing; A.A. ,Y.Y.T and A.S.A. contributed to the assessment of CL oxidation by 2D-LCMS; L.J.S. contributed to the MS imaging; V.A.T. contributed to the EPR measurements; B.F. contributed to the in vivo EPR imaging; M.D.M. contributed to the unbiased stereology for cortical lesion volume. A.M.P. and D.O.O. contributed to evaluation of human TBI tissue and writing. H.A. performed CCI model; R.S.B.C. and P.M.K. contributed to the data analysis and manuscript writing; P.W. contributed to the preparation of XJB-5-131 and manuscript writing; V.E.K. and H.B. initiated and directed the entire study, designed experiments and wrote the manuscript.
References
- 1.Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths 2002-2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- 2.Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: A public health perspective. J Head Trauma Rehabil. 1999;14:602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 3.RAND. RAND Invisible Wounds of War Study. 2011 Jun 26; www.rand.org/multi/military/veterans.html.
- 4.Ghajar J. Traumatic brain injury. Lancet. 2000;356:923–929. doi: 10.1016/S0140-6736(00)02689-1. [DOI] [PubMed] [Google Scholar]
- 5.Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7:115–126. doi: 10.1016/j.nurt.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broadhurst CL, et al. Brain-specific lipids from marine, lacustrine, or terrestrial food resources: potential impact on early African Homo sapiens. Comp Biochem Physiol B Biochem Mol Biol. 2002;131:653–673. doi: 10.1016/s1096-4959(02)00002-7. [DOI] [PubMed] [Google Scholar]
- 8.Kiebish MA, et al. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J Neurochem. 2008;106:299–312. doi: 10.1111/j.1471-4159.2008.05383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kagan VE, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 10.Chan PH, Fishman RA. Transient formation of superoxide radicals in polyunsaturated fatty acid-induced brain swelling. J Neurochem. 1980;35:1004–1007. doi: 10.1111/j.1471-4159.1980.tb07100.x. [DOI] [PubMed] [Google Scholar]
- 11.Verweij BH, et al. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg. 2000;93:815–820. doi: 10.3171/jns.2000.93.5.0815. [DOI] [PubMed] [Google Scholar]
- 12.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ran Q, et al. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem. 2004;279:55137–55146. doi: 10.1074/jbc.M410387200. [DOI] [PubMed] [Google Scholar]
- 14.Wipf P, et al. Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J Am Chem Soc. 2005;127:12460–12461. doi: 10.1021/ja053679l. [DOI] [PubMed] [Google Scholar]
- 15.Bayir H, et al. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- 16.Bayir H, et al. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51:571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Hamm RJ, et al. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma. 1992;9:11–20. doi: 10.1089/neu.1992.9.11. [DOI] [PubMed] [Google Scholar]
- 18.Kline AE, Massucci JL, Ma X, Zafonte RD, Dixon CE. Bromocriptine reduces lipid peroxidation and enhances spatial learning and hippocampal neuron survival in a rodent model of focal brain trauma. J Neurotrauma. 2004;21:1712–1722. doi: 10.1089/neu.2004.21.1712. [DOI] [PubMed] [Google Scholar]
- 19.Scafidi S, Racz J, Hazelton J, McKenna MC, Fiskum G. Neuroprotection by acetyl-L-carnitine after traumatic injury to the immature rat brain. Dev Neurosci. 2010;32:480–487. doi: 10.1159/000323178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 21.Stoyanovsky DA, Cederbaum AI. ESR and HPLC-EC analysis of ethanol oxidation to 1-hydroxyethyl radical: rapid reduction and quantification of POBN and PBN nitroxides. Free Radic Biol Med. 1998;25:536–545. doi: 10.1016/s0891-5849(98)00081-1. [DOI] [PubMed] [Google Scholar]
- 22.Zhang R, Goldstein S, Samuni A. Kinetics of superoxide-induced exchange among nitroxide antioxidants and their oxidized and reduced forms. Free Radic Biol Med. 1999;26:1245–1252. doi: 10.1016/s0891-5849(98)00328-1. [DOI] [PubMed] [Google Scholar]
- 23.Siesjo BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1:155–185. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- 24.Wei EP, Kontos HA, Dietrich WD, Povlishock JT, Ellis EF. Inhibition by free radical scavengers and by cyclooxygenase inhibitors of pial arteriolar abnormalities from concussive brain injury in cats. Circ Res. 1981;48:95–103. doi: 10.1161/01.res.48.1.95. [DOI] [PubMed] [Google Scholar]
- 25.Muizelaar JP, et al. Improving the outcome of severe head injury with the oxygen radical scavenger polyethylene glycol-conjugated superoxide dismutase: a phase II trial. J Neurosurg. 1993;78:375–382. doi: 10.3171/jns.1993.78.3.0375. [DOI] [PubMed] [Google Scholar]
- 26.Lo EH, Singhal AB, Torchilin VP, Abbott NJ. Drug delivery to damaged brain. Brain Res Brain Res Rev. 2001;38:140–148. doi: 10.1016/s0165-0173(01)00083-2. [DOI] [PubMed] [Google Scholar]
- 27.Sorice M, et al. Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ. 2004;11:1133–1145. doi: 10.1038/sj.cdd.4401457. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa Y. Initiation of apoptotic signal by the peroxidation of cardiolipin of mitochondria. Ann N Y Acad Sci. 2004;1011:177–184. doi: 10.1007/978-3-662-41088-2_18. [DOI] [PubMed] [Google Scholar]
- 29.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Z, et al. Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology. 2010;151:3038–3048. doi: 10.1210/en.2010-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrosillo G, Moro N, Paradies V, Ruggiero FM, Paradies G. Increased susceptibility to Ca(2+)-induced permeability transition and to cytochrome c release in rat heart mitochondria with aging: effect of melatonin. J Pineal Res. 2010;48:340–346. doi: 10.1111/j.1600-079X.2010.00758.x. [DOI] [PubMed] [Google Scholar]
- 32.Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett. 2006;580:6311–6316. doi: 10.1016/j.febslet.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 33.Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW. Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem. 2011;286:26334–26343. doi: 10.1074/jbc.M110.188516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Betaneli V, Petrov EP, Schwille P. The role of lipids in VDAC oligomerization. Biophys J. 2012;102:523–531. doi: 10.1016/j.bpj.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rostovtseva TK, Kazemi N, Weinrich M, Bezrukov SM. Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J Biol Chem. 2006;281:37496–37506. doi: 10.1074/jbc.M602548200. [DOI] [PubMed] [Google Scholar]
- 36.Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008;28:1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- 37.Wilcox CS, Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev. 2008;60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitchell JB, et al. Biologically active metal-independent superoxide dismutase mimics. Biochemistry. 1990;29:2802–2807. doi: 10.1021/bi00463a024. [DOI] [PubMed] [Google Scholar]
- 39.Bobko AA, Kirilyuk IA, Grigor’ev IA, Zweier JL, Khramtsov VV. Reversible reduction of nitroxides to hydroxylamines: roles for ascorbate and glutathione. Free Radic Biol Med. 2007;42:404–412. doi: 10.1016/j.freeradbiomed.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soule BP, et al. The chemistry and biology of nitroxide compounds. Free Radic Biol Med. 2007;42:1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trnka J, Blaikie FH, Smith RA, Murphy MP. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic Biol Med. 2008;44:1406–1419. doi: 10.1016/j.freeradbiomed.2007.12.036. [DOI] [PubMed] [Google Scholar]
- 42.Fan P, Yamauchi T, Noble LJ, Ferriero DM. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma. 2003;20:437–445. doi: 10.1089/089771503765355513. [DOI] [PubMed] [Google Scholar]
- 43.Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16:783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- 44.Singh IN, Sullivan PG, Hall ED. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J Neurosci Res. 2007;85:2216–2223. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- 45.Alessandri B, et al. Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J Neurotrauma. 2002;19:829–841. doi: 10.1089/08977150260190429. [DOI] [PubMed] [Google Scholar]
- 46.Lewen A, et al. Oxidative stress-dependent release of mitochondrial cytochrome c after traumatic brain injury. J Cereb Blood Flow Metab. 2001;21:914–920. doi: 10.1097/00004647-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 47.DeKosky ST, Ikonomovic MD, Gandy S. Traumatic brain injury--football, warfare, and long-term effects. N Engl J Med. 2010;363:1293–1296. doi: 10.1056/NEJMp1007051. [DOI] [PubMed] [Google Scholar]
- 48.McKee AC, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.