

Abstract
Transgenic mice overexpressing human progastrin (hGAS) show colonic crypt hyper-proliferation and elevated susceptibility to colon carcinogenesis. We aimed to investigate effects of p53 mutation on colon carcinogenesis in hGAS mice. We show that introducing a p53 gene mutation further increases progastrin dependent BrdU labeling and results in markedly elevated number of aberrant crypt foci (ACF) and colonic tumors. We demonstrate that hGAS/Lgr5-GFP mice have higher number of Lgr5+ colonic stem cells per crypt when compared to Lgr5-GFP mice indicating that progastrin changes crypt biology through increased stem cell numbers and additional p53 mutation leads to more aggressive phenotype in this murine colon cancer model.
Keywords: Progastrin, p53, Colon cancer, Azoxymethane, Lgr5
INTRODUCTION
Colorectal cancer (CRC) is a leading cause of cancer related morbidity and mortality worldwide (1). In the United States, CRC remains the third most common cancer in both sexes and contributes to approximately 9% of all cancer related deaths (1) and (2). Work from number of laboratories has elucidated a series of somatic mutations involved in colon cancer development including inactivation of tumor suppressor genes such as p53, APC, and activation of the KRas oncogene (3–5).
p53 gene mutations are found in more than 50% of sporadic cases of CRC (6). p53 plays an important role as a cell cycle regulator and tumor suppressor by recognizing DNA damage, then triggering repair, apoptosis or cellular senescence (7). p53 maintains genomic stability and prevents the accumulation of multiple genetic changes, which are prerequisites for the development of neoplasia. Inactivation of p53 results in attenuated apoptosis and accelerated tumor growth suggesting that loss of p53-mediated apoptosis is an important step in tumor progression (6) and (7). Notably, these mutations may confer dominant-negative or gain-of-function properties to p53 leading to functional loss of the wild type allele (8).
Studies by Hu et al. have shown that p53 heterozygous knockout (p53+/−) mice develop spontaneous and azoxymethane (AOM) induced colon tumors at higher rate than WT animals secondary to attenuated apoptosis in response to AOM. In human cancers, most p53 gene mutations are point (structural) mutations. A transgenic mouse bearing a structural p53 mutation (p53R172H) was originally generated to determine the physiological effects of p53 point mutation in a model of Li-Fraumeni syndrome (8). This structurally mutated form of p53 is commonly found in spontaneous human tumors. Studies have shown that these p53 mutant mice (p53R172H) demonstrate significant differences in their tumor spectra and develop carcinomas more frequently when compared to p53+/− knockout mice (8–10). Previous studies have shown that hyper-proliferation of the colonic epithelium is the most common alteration in high risk conditions for development of CRC and is believed to be the first step in a sequence of events leading to uncontrolled growth and malignant transformation (11) and (12).
Gastrin exists in a number of molecular forms. The most abundant and well-studied forms of gastrin, G17 and G34, are amidated at the C-terminus after post-translational processing of the 101-amino acid precursor molecule, named preprogastrin (13). Prior to its conversion to the amidated forms, preprogastrin undergoes cleavage of a signal peptide to yield progastrin. The livers of adult hGAS mice express abundant human gastrin mRNA and human progastrin (hGAS), but are unable to process this peptide to the mature amidated form. As a result, elevated serum progastrin levels and normal amidated gastrin levels are observed (14). In vitro, incompletely processed gastrins (progastrin and glycine-extended gastrin) exert significant mitogenic effects and several studies using CRC cell lines demonstrated stimulating effect of progastrin on cellular proliferation (2), (14), and (15). Importantly, progastrin and glycine-extended gastrin (G-Gly) are the predominant forms of gastrin found in many human cancers, including colon (16) and (17), lung (18) and ovarian (19). Several studies using colorectal cancer cell lines demonstrated stimulating effect of progastrin on cellular proliferation (20–24). We have reported that elevated levels of circulating progastrin observed in hGAS mice result in a significant increase in the incidence and multiplicity of colorectal tumors in response to AOM when compared to wild-type mice. Previous studies were conducted using mice in an FVB/N background (15) and (17). Here, we present mice in a C57BL6 genetic background to study the effect of progastrin on colonic proliferation and tumorigenesis. The effects of progastrin are not dependent on other forms of gastrin because hGAS mice show a similar phenotype when crossed to a gastrin knockout background (25). Furthermore, studies have shown patients with CRC have significantly elevated levels of nonamidated gastrins when compared to control subjects. This suggests that these hormones are secreted by colonic tumors in humans (26–28). Interestingly, gastrin gene expression is upregulated early in the adenoma-carcinoma sequence supported by the detection of gastrin mRNA and progastrin in a majority of adenomatous polyps (29).
p53R172H mice in a C57BL6 background have never been investigated for their susceptibility to AOM induced colon tumor formation. With prior studies showing that progastrin contributes to colon carcinogenesis, we set out to study its effects on p53 mutant mice with the hypothesis that the hGAS/p53R172H(+/−) mice would display accelerated tumor growth with histologically more aggressive and invasive tumors. In the AOM colon cancer model, hGAS/p53R172H(+/−) double transgenic mice indeed displayed a more aggressive biological behavior when compared to controls. Additionally, we wanted to clarify the effect of progastrin on colonic stem cells. Therefore, we crossed hGAS mice to Lgr5-GFP (30). Notably, hGAS/Lgr5-GFP mice had a significant higher number of Lgr5 cells per colonic crypt when compared to Lgr5-GFP mice. Our studies suggest that progastrin increases cancer susceptibility by increasing the number of active stem cells in the colonic crypt and that mutations in p53 can accelerate and further promote colonic tumor formation in hGAS mice.
MATERIALS AND METHODS
Animals
hGAS transgenic mice (F9 on FVB/N) on C57BL/6 background were crossed with p53R172H+/− mice. Breeding resulted in littermates that included hGAS/+, hGAS/p53R172H, p53R172H+/−, and WT (C57BL6) animals. p53R172H+/− mice were a gift from Dr. Tyler Jacks (Massachusetts Institute of Technology, Cambridge, MA, USA). The generation and characteristics of the p53R172H+/−mice have previously been described (8). Furthermore, age matched hGAS mice were crossed with Lgr5-GFP mice to generate hGAS/Lgr5-GFP double transgenic mice. The Lgr5-GFP mice were a gift from Dr. Hans Clevers (Hubrecht Institute and University Medical Center, Utrecht, the Netherlands). Mice were bred and maintained under specific pathogen free conditions at the animal facility of Columbia University Medical Center (CUMC). All experiments were approved by the Subcommittee on the Research and Animal Care in the Irving Cancer Research Center at CUMC.
Induction and analysis of ACF using the chemical carcinogen azoxymethane
The AOM (Sigma-Aldrich; molecular weight, 36,000–50,000) used in the experiments was purchased 2 weeks before the start of the experiment and diluted in PBS. All animals received the same concentration of AOM from the same batch originally purchased. Six-week-old sex-matched mice in each of the four study groups (n = 10) were given weekly intraperitoneal injections of 12 mg/kg AOM for two weeks. Results of previous studies from our lab and others have shown that AOM injections of 10–12 mg/kg body weight induced the formation of an optimal number of aberrant crypt foci (ACF) in the colonic mucosa without being overly toxic to the mice (11) and (31). Four mice in each group received 200 μL of PBS as control. After the injections, mice were placed in a laminar flow hood in the animal facility at CUMC and monitored for signs of sickness or adverse effects. Two weeks after the last injection, mice were sacrificed and full-length colons were removed within 5 min. The colons were distended under mild pressure by filling them with saline, slit open by mid-line incision and fixed flat with their mucosal side facing up on Whatman filter paper in 70% ethanol, as described previously (31) and (32). Specimens were stored at 4°C until further analysis.
After 24 hr, the colon specimens were removed from 70% ethanol and stained with 0.25% methylene blue. ACF are detected as a cluster of abnormally large darkly stained and slightly elevated mucosal crypts with slit-like or dilated openings. The total number of ACF was recorded using Nikon TE2000 light microscopy. ACF that involved one, two, and three crypts per foci were called singlets, doublets, and triplets, respectively. More than three crypts present in a focus were designated as multiplets.
Colon tumor induction using the chemical carcinogen azoxymethane
Six-week-old sex-matched mice in each of the four groups (n = 8) received weekly intraperitoneal injections of 12 mg/kg AOM in phosphate buffered saline (PBS) for 6 weeks. Six mice in each group received 200 μL of PBS as control. After the injections, animals were handled as described above. After 24 weeks, mice were sacrificed, and colons were dissected out within 5 min, flushed with PBS and slit open longitudinally through the mid-line. Tumor size (millimeters), number, and locations within the colons were recorded. Photographs of gross tumors were taken. Full-length colons from the anus to cecum were removed, dissected longitudinally, and rolled on a plastic bar using the “Swiss roll” technique. For histological examination, colons were incubated in 10% neutral buffer formalin (VWR) at 4°C overnight; paraffin sections of colonic mucosa five-micrometer in thickness were made. From each paraffin block, five-micrometer thick sections were cut serially for each colon and stained with Hematoxylin and Eosin.
Labeling index (LI)
Four mice from each of the four groups that were injected with AOM for colon tumor induction were used for 5-Bromo-2-deoxyuridine (BrdU) labeling. Animals were 24 weeks old when the experiment was performed. The animals were injected intraperitoneally with 10 mg/kg BrdU (Sigma-Aldrich) dissolved in 200 μL PBS 1 hr before being sacrificed. Mice were sacrificed exactly 60 min after the BrdU injection to ensure uniformity in staining. Colons were dissected out as described above.
Histopathological and immunohistochemical analyses
Formalin-fixed, paraffin-embedded tissue slides were rehydrated using xylene to alcohol washings. For immunohistochemical detection of BrdU, DCAMKL-1, and p53 positive cells, deparaffinized tissue was incubated in hydrogen peroxide (3%) for 5 min and blocked with 2% BSA (Sigma) for 1 hr, followed by incubation in optimal dilutions of anti-BrdU rat monoclonal antibody (Abcam Inc.), anti-p53 (mouse; Abcam Inc., Cambridge, Massachusetts, USA), and anti-DKAMKL-1 rabbit polyclonal antibody (Abgent Inc.) for 1 hr. Samples were washed with distilled water and incubated with HRP-conjugated anti-rat IgG (DAKO Inc.) and anti-rabbit IgG (DAKO Inc., Carpinteria, California, USA). Immune complexes were visualized with peroxidase substrate DAB kit (DAKO Inc.). Finally, slides were counterstained with hematoxylin, dehydrated and a cover slip was placed on top. The percentage of nuclei stained with BrdU and cytoplasm stained with DCAMKL-1 in the proximal, middle, and distal part of colon crypts was calculated using a light microscope. For quantification of BrdU positive cells, the number of positive cells was expressed in relation to the total number of epithelial cells from each crypt column as described previously (29). The epithelial cell proliferation rate was expressed as the number of BrdU positive cells divided by the total number of cells in each crypt. Thirty crypts in different sites of each colon were randomly chosen. Histological changes and proliferation were evaluated and compared between all study groups.
Quantification of Lgr5+ stem cell population
Six-week-old sex-matched mice in each of the hGAS/Lgr5-GFP and Lgr5-GFP groups (n = 3) were sacrificed. The full-length colons from the anus to cecum were removed, dissected longitudinally, and rolled on a plastic bar using the “Swiss roll” technique as described previously. For histological examination, colons were fixed in 4% paraformaldehyde for freezing in Tissue-Tek, OCT compound (Sakura Finetek USA, Inc., Torrance, California, USA). The colon tissues were cut in 5-μm slices, stained with DAPI, and analyzed for GFP positive cells under a Nikon TE2000 microscope.
Measurement of progastrin in plasma
At the time of sacrificing the animals from the colon tumor group, blood was collected by cardiac puncture on anesthetized mice prior to cervical dislocation, followed by centrifugation of the samples (3000 rpm for 10 min) and separation of the serum. A radioimmunoassay (RIA) for progastrin was performed as described previously (33). Serum progastrin was extracted from the serum using ethanol extraction in a 1:2 ratio of serum to ethanol, and was measured using antiserum 1137. The antiserum 1137 was raised using hGAS 92–101 conjugated to keyhole limpet hemocyanin with bis-diazotized benzidine and injected into rabbits.
Statistical analysis
Values were compared across all the four study groups and statistical differences were computed using 1-way ANOVA followed by a Tukey or Dunnet’s test. p-values found to be less than .05 were considered statistically significant. The group measures are expressed as mean ± SD for all parameters determined, unless otherwise indicated.
RESULTS
P53 mutation increases ACF formation in hGAS mice after AOM treatment
ACF are considered to be preneoplastic lesions of the colonic epithelium (31). In order to evaluate the role of p53 mutation in the formation of ACF, AOM was given to all four groups (WT, p53R172H, hGAS, hGAS/p53R172H) of mice. The majority of ACF were seen in the distal part of the colon and all animals that were injected with AOM developed ACF within the time frame of the study. No animals in the control groups (200 μL PBS injection) developed ACF within the same period. ACF were classified according to multiplicity into three different morphologic types of ACF: single crypt (singlet), double crypt (doublet), triple crypts (triplets), or multi-crypt (multiplets) ACF (Figure 1). Singlets were most common in all groups. While doublets were more common than triplet ACF, multiplets were rare (Figure 1A). ACF numbers were highest in the hGAS/p53R172H group with an average of 10.78 ± 3.02. This observation reached statistical significance when compared to the p53R172H group (p < .02) and WT group (p < .003) (Figure 1B). We observed a higher total number and more advanced ACF in the hGAS/p53R172H in comparison to the hGAS mice but this trend did not reach statistical significance (n = 0.31).
Figure 1.

Progastrin exerts significant proliferative effects on the colonic epithelia leading to ACF formation. (A) Representative pictures of the three different types of ACF (A1-single, A2-double, and A3-multiple crypts) observed in the colon of AOM-treated mice. Colons were removed 3 weeks after being treated with AOM, fixed with ethanol overnight and analyzed for aberrant crypts after methylene blue staining (×200). (B) Multiplicity of ACF across the four groups. (*p < .03, **p < .003). (C) Average number of ACF seen in all the four study groups (N = 4 pergroup). All values represent the mean ± SD. (*p < .02)
P53 mutation increases colonic cell and tumor cell proliferation in hGAS mice
The BrdU-labeling assay determines the percentage of cells in the “S” phase of the cell cycle and can be used to determine the proliferative index of epithelial cells within a crypt (16). Animals from the AOM group were 24 weeks old and had received six doses of AOM when they were sacrificed. BrdU positive cells contained darkly stained nuclei (Figure 2D). Photomicrographs of BrdU stained nuclei of each of the four groups are shown in Figure 4(A). The BrdU-labeling index (LI) was calculated as the proportion of BrdU positive cells to the total number of cells within the colonic crypt (Figure 2). This enabled us to compare the proliferation indices between tumors and normal areas of the colon in the same animal.
Figure 2.

Inactivation of the p53 gene accelerates progastrin-dependent colonic proliferation (BrdU-Labeling Index). (A) Table showing the different rates of BrdU-labeling indices among the four study groups compared with their respective controls, and also shows the difference in proliferation rates between the tumor area and uninvolved area of the colon in AOM treated C57Bl6 mice. (B) Graphical representation of the percentage labeling indices across the four study groups. (∞ p = .46; *p < .03; **p < .01). (C) Graphical comparison of the LI of tumor area and uninvolved areas of the colon in the study groups (∞ p = .46, *p < .03). (D) Photographs showing immunohistochemical staining of BrdU positive cells in the colon from tumor area (1) and normal crypt (2) of an AOM injected hGAS/p53RH double mutant mouse.
Figure 4.
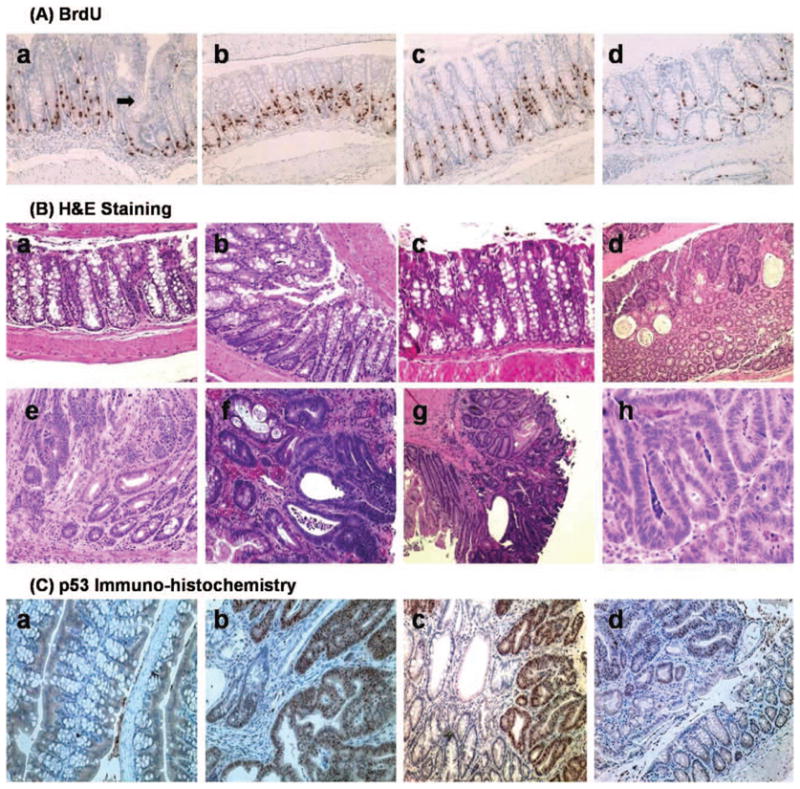
(A) Immunohistochemical staining of BrdU labeling (200×) shown in representative groups (a) hGAS colon. Arrow demonstrates an area of crypt fission, which may eventually transform into adenoma, (b) hGAS/p53RH crypt also demonstrating a high degree of BrdU labeling, (c) p53RH colon, and (d) Wild type C57BL6 colon showing a relatively lower LI and crypt depth. (B) Hematoxylin and eosin staining shows the differences in histopathology across the four study groups. (a) Normal area of colon showing uninvolved crypts in a WT untreated mouse (100×). (b) HE staining of an hGAS mouse distal colon (age approx. 10 weeks) showing aberrant crypt foci (ACF) and increased crypt depth (200×). (c) Hypertrophic crypts in the double mutant hGAS/p53RH animal that was not treated with AOM (control) at thirty weeks of age. (d) Note the large vacuolization and increased cellular debris, a characteristic finding secondary to rapid cell turnover in colonic adenocarcinoma. (e) Is an adenoma (200×) developing in a WT mice that was AOM treated. Noteworthy is the loss of crypt architecture and increase in immature crypts with increase in stromal matrix. (f) Shows an advanced moderately differentiated tumor in an hGAS mouse (400×). (g) Represents a moderately differentiated adenocarcinoma (100×) that is invading the serosa in an AOM treated double mutant mouse. (h) Tubular adenoma in a p53RH mutant mouse at 200× magnification respectively. (C) p53 immunohistochemistry done on representative sections in all the four study groups: (a) Wild type colon showing no cellular localization of p53 protein. Intranuclear accumulation of p53 protein is clearly visualized in Figures(b) and (c) respectively, magnified to 200× here. The tumors in the hGAS/p53RH colon in (b) and p53RH colon in (c) have the accumulation of intranuclear p53 protein. In Figure(c) areas of normal colon are also visible (lightly stained) but the areas showing the abnormal crypts (tumor) contain dark brown stained nuclei, which suggest p53 protein. (d) Colonic adenoma in an hGAS mouse with no immunohistochemical evidence of p53 accumulation.
The hGAS/p53R172H group showed the highest LI with 21.05% of cells labeled within a crypt. The difference in labeling indices was found to be statistically significant when compared to the WT group. Interestingly, tumors in the hGAS/p53R172H group had a 50% increase in labeling when compared to uninvolved areas of the colon. This finding was consistent in the hGAS and p53R172H group as well. Furthermore, when untreated WT mice were compared with the AOM treated WT group there was a demonstrable increase in LI. In the progastrin overexpressing mice, BrdU labeled cells were found distributed in the lower and middle third of the crypt. In contrast, the untreated AOM mice (p53R172H and WT) showed a lower amount of positive cells, which were confined mainly to the lower third of the crypt. The significant increase in ACF formation in hGAS/p53R172H and hGAS compared to that in p53R172H and WT mice likely reflects the higher LI seen in the tumor and normal areas of the hGAS and hGAS/p53R172H double transgenic mice.
P53 mutation increases colon tumor formation in hGAS mice
After AOM treatment, animals were carefully monitored for signs of rectal bleeding and rectal prolapse. A majority of the hGAS/p53R172H and a significant number of hGAS mice exhibited signs of rectal bleeding and rectal prolapse before the end of the aging period (30 weeks). These animals were promptly identified and euthanized for analysis. One hGAS/p53R172H, two hGAS, and three p53R172H mice died before reaching the end point of the study (28–30 weeks). For analysis, animals were sacrificed and the colons were dissected out from the anal to cecal end, and their photographs were digitally captured (Figure 3A). 100% hGAS/p53R172H mice treated with AOM developed tumors. Tumors were also seen in 89% of the hGAS and p53R172H mice and in 78% of the animals in the WT group.
Figure 3.
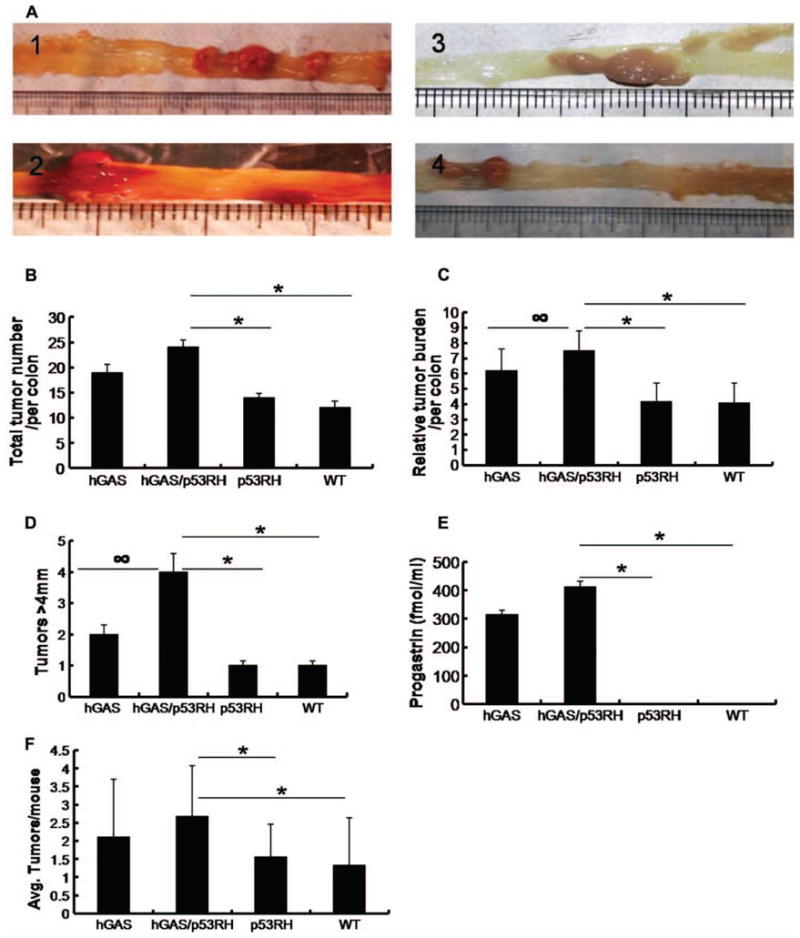
Colon tumor study. (A) Macroscopic changes and tumor formation in colons of mice from each group. Colons were removed at 30 weeks age after 6 weeks of AOM treatment. Representative results from four independent animals are shown: (1) hGAS/+, (2) hGAS/p53RH, (3) p53RH, and (4) WT mice. (B) Total tumor numbers in the colons of experimental mice. (*p < .03). (C) Relative tumor burden per animal colon (∞p = 0.11, *p < .03). (D) Graph showing the distribution of tumors larger than 5 mm (∞ p = 0.13, *p < .03). (E) Concentration of progastrin levels in the serum of AOM treated mice (*p < .03). Serum was collected at around thirty weeks of age and progastrin levels were calculated by radioimmunoassay. (F) Average tumor numbers per colon in all four groups (*p < .03).
Total tumor numbers in all the four groups are shown in Figure 3(B). The differences in total tumor numbers between hGAS/p53R172H and p53R172H groups (p < .03) and also between hGAS/p53R172H and WT groups (p < .03) were statistically significant. Furthermore, hGAS mice displayed a higher number of tumors when compared to WT animals (p < .04). To calculate the tumor burden per mouse the size (in mm) of all tumors found in a particular group was added up and divided by the total number of animals in that group. The results are summarized in Figure 3(C). The hGAS/p53R172H group had the highest tumor burden among all the groups. This finding achieved statistical significance (p < .03) when compared to the p53R172H and WT groups. The hGAS group had tumor burden significantly higher than the WT group (p < .03). Differences in tumor burden between hGAS mice and the p53R172H mice were observed but did not reach statistical significance with p = .11. Based on the relative size (diameter), the tumors were divided into small (<2 mm), medium (2–4 mm), and large (>4 mm) tumors. Medium sized tumors were most common in all the groups comprising 65–80% of all tumors. However, the hGAS/p53R172H group had the largest percentage (17%) of tumors that were greater than 4 mm in diameter, with the largest tumor measuring approximately 7 mm in diameter (Figure 3D). This particular tumor was highly vascularized and caused frank rectal bleeding. In order to evaluate the effect of progastrin overexpression in this study the serum levels of progastrin were measured. As shown in Figure 3(E), progastrin levels were found to correlate positively with the findings as the serum levels in hGAS animals (hGAS and hGAS/p53RH groups) were between 300–400 fold higher when compared to p53RH and WT groups (p < .03). Higher progastrin levels seen in the hGAS/p53RH group and the hGAS group are consistent with the results observed particularly to the tumor burden in each of the groups studied. Not only were the numbers of animals with tumors higher in the AOM treated hGAS/p53R172H group (2.67 ± 1.14), but these mice also had more tumors per colon than AOM treated mice in the hGAS (2.11 ± 1.75), p53R172H (1.56 ± 0.88), and WT group (1.33 ± 1.11) (Figure 3F). Paraffin sections of colon tumors and normal areas of colon were stained by Hematoxylin and Eosin [Figure 4(B)]. Normal colonic mucosa and tumors in various stages of development in the adenoma-carcinoma sequence are digitally captured and shown in Figure 4[B (a–h)].
Tumors in p53R172H and hGAS/p53R172H mice show an increase in nuclear p53 accumulation
p53 activity is kept low in normal cells so that the cell cycle is not disrupted and cells do not undergo untimely death. This is achieved through operation of a negative feedback loop consisting of the p53 and MDM2 genes. In the case of DNA damage, the p53-mediated pathways are activated leading to cell cycle arrest and repair of the DNA (7). In order to examine possible nuclear accumulation of p53 in p53R172H mice, immunohistochemistry for p53 was performed on tumors from the aging studies. Sections of normal and tumor areas were stained with polyclonal antibody directed against p53. Interestingly, as shown in Figure 4(C), there was substantial nuclear accumulation of p53 protein in the tumors of the hGAS/p53R172H mice. This was also observed in the p53R172H group but to a lesser degree. There were few areas of intracellular accumulation of p53 protein in the colonic adenocarcinomas of WT and hGAS mice. No p53 immunoreactivity was observed in normal colonic crypt cells of mice from any of the four groups. An integral component of the Wnt signaling pathway is the activation of β-catenin. Activation of β-catenin is a critical early step in colonic proliferation and progression to colon cancer (34). Strong β-catenin staining was evident in the cytoplasm and nuclei of the adenocarcinoma cells and adenomas and dysplastic cells also showed positive immunoreactivity for β-catenin though the intensity of staining was weaker than in the neoplastic cells (data not shown).
Progastrin overexpression and p53 mutation induces an increase in colonic Lgr5 and DCAMKL-1 cells
Lgr5 has recently been described as a stem cell marker in the gastrointestinal tract (30). Therefore, we wanted to investigate the effect of high progastrin levels on the number of Lgr5 cells in the murine colon. For this purpose, we crossed Lgr5-GFP mice to hGAS mice. In hGAS/Lgr5-GFP double transgenic mice, an increase in the number of GFP-positive cells at the crypt base was observed when compared to Lgr5-GFP controls. An average of 81 GFP positive crypts/section was seen in hGAS/Lgr5-GFP mice versus 58 GFP positive crypts per section in WT/Lgr5-GFP mice. Each group consisted of three mice and three sections per colon were examined. This difference in the number of Lgr5+ cells between Lgr5-GFP and hGAS/Lgr5-GFP mice reached statistical significance with a p < .01 (Figure 6).
Figure 6.

Lgr5-GFP localization is increased in hGAS compared with WT mice. (A) The localization of Lgr5-GFP positive cells in hGAS/Lgr5-GFP and Lgr5-GFP mice colon. (B) The average of Lgr5-GFP positive cells in crypts in each of group were measured under a immunofluorescence microscope. We had total of three mice in each group and three sections were examined per colon. All values represent the mean ± SD. **p < .01.
Doublecortin and CaM kinase-like-1 (DCAMKL-1) was recently reported as a transcript specifically upregulated in gut entero-endocrine cells or “tuft cells” (35) and (36), and has been suggested to mark putative stem cell or progenitor cells in the intestine (37). Furthermore, expansion of DCAMKL-1 positive cells is seen in gastric inflammation and carcinogenesis (38). Therefore, a possible increase in DCAMKL-1 positive cells in our model of injury-induced colon cancer was investigated. Mice were AOM treated (two doses of 10 mg/kg given a week apart) at 24 weeks of age and sacrificed 2 weeks later for analysis (2 per group). Fifteen crypts were counted randomly in the proximal, middle, and distal parts of the colon of each mouse. Figure 5 shows average number of DCAMKL-1 stained cells per crypt.
Figure 5.
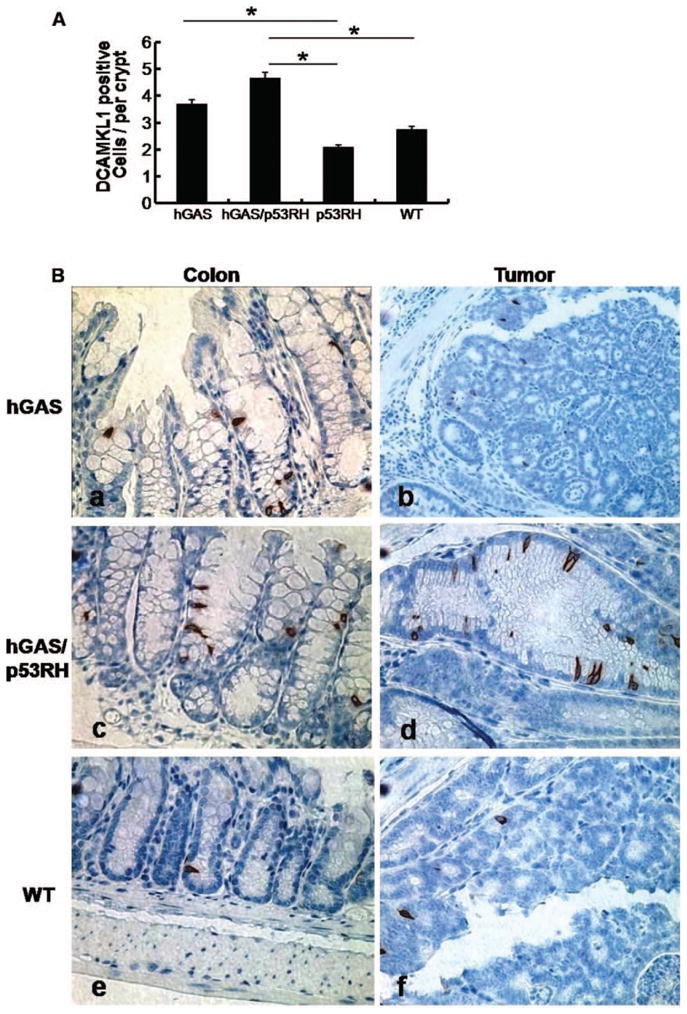
Progastrin overexpression promotes the expression of colonic cells positive for DCAMKL-1. (A) The average percentage of DCAMKL-1 positive cells in the colon crypts of the study groups observed per high power field (400×) in each of the four groups of mice (n = 4 per group). All values represent the mean ± SD (*p < .03). (B) Immunohistochemistry for DCAMKL-1 in colonic mucosa and colon tumors in mice from each group treated with AOM (hGAS; hGAS/p53RH; p53RH and WT mice). Representative results from four different groups are shown in different magnifications: (a) hGAS mouse colon showing crypt fission and DCAMKL-1 positive cells (400×). (b) Expression of the progenitor cells is seen in an hGAS mouse colon tumor (200×). (c) DCAMKL-1 positive cells are present in the hyper-proliferative epithelium of hGAS/p53RH double mutant mouse (400×). (d) Colon tumor in hGAS/p53RH colon showing abundance of DCAMKL-1 positivity (200×). (e) WT uninvolved mucosa showing a single cellular DCAMKL-1 expression, and (f) shows a colon tumor in AOM treated WT animal with low DCAMKL-1 positivity.
DCAMKL-1 positive cells were found to be located close to the isthmus in colonic crypts of wild type mice. In AOM treated hGAS mice, additional DCAMKL-1 positive cells tended to appear toward the bottom of colonic crypts, in contrast to the crypts of mice not overexpressing progastrin (p53RH and WT) [Figure 5(B)]. Interestingly, the absolute number of DCAMKL-1 positive cells was higher in the colonic crypts of hGAS and hGAS/p53RH mice compared to WT mice (p < .02) and p53RH mice (p < .03). In tumors, DCAMKL-1 positive cells were found to be widely distributed throughout the tumors. The highest number of DCAMKL-1 positive cells was observed in colonic tumors of hGAS/p53RH mice followed by tumors in hGAS mice.
DISCUSSION
In the current study, we provide evidence for a synergy between the known proliferative and carcinogenic effects of progastrin and a dysfunctional p53 gene in a murine model of colon cancer. Crosstalk between tumor cells and their microenvironment is critical for tumor formation and progression. In a recent study by Goldstein et al. knock-in mice harboring the p53R172H mutant, which is an equivalent to the human hot-spot mutant p53R175H, no p53 accumulation was found in the normal tissues of p53H/H homozygote mice (39). In this study, we show that there is immunohistochemical evidence of intranuclear p53 accumulation particularly in AOM treated p53 mutant mice overexpressing progastrin (hGAS/p53RH). This nuclear accumulation may be due to a progastrin-mediated destabilization of p53. p53 mutations are very common (>50%) in human colon cancers (40). The hGAS/p53RH and hGAS mice had significantly increased numbers of ACF, a surrogate marker for progression to colon cancer when compared with p53RH and WT mice. The ACF system can be used as a simple, efficient, and economical quantitative assessment tool of colon carcinogenesis (41). Originally described by McLellan and Bird, ACF are putative preneoplastic lesions in the colon (31) and (42). ACF are induced by all colon carcinogens in a species and dose-dependent manner and modulators of colon carcino-genesis modify their growth (43) and (44). The differences in ACF formation between the hGAS and hGAS/p53RH did not reach statistical significance. This finding leads to the argument that in the early stages of disease (i.e., preneoplastic stage) the effect of excessive progastrin leads to the formation of more ACF in hGAS and hGAS/p53R172H mice than the other groups (p53R172H and WT mice) that do not over-express progastrin. Furthermore, the progastrin expressing mice displayed increased multiplicity of their ACF, which correlates with an increased risk for the development of colon cancer (43). Earlier studies done by measuring rates of apoptosis and proliferation have demonstrated that p53 status has no effect on crypt cellularity (10) and (45). Combining these results with the data presented here indicates that progastrin exerts significant proliferative effects on the colonic epithelium and functions as a potent co-carcinogen in the presence of a genotoxic agent.
Colonic crypt homeostasis is maintained by tissue resident stem cells. Lgr5 is an established marker of gastrointestinal stem cells (30). Expansion of colonic stem cells is thought to promote colonic growth in part through crypt fission, and studies have reported that intestinal crypts divide in response to a doubling of stem cell number (37). Crypt fission is a physiologic mechanism of crypt reproduction that increases in pathophysiologic situations when crypt regeneration is required. Importantly, we observed an expansion of Lgr5 colonic stem cells in hGAS mice, which might explain the hyperproliferation and increased susceptibility to colon cancer seen in these animals. These findings support the idea that progastrin promotes the initiation of cancer through an expansion of stem cells, while additional mutation of p53 changes the biological behavior of established tumors. The data presented in this study support this concept. While BrdU labeling and ACF formation in the hGAS and hGAS/p53RH groups did not markedly differ, we observed higher total numbers in colonic tumors hGAS/p53R172H double transgenic mice. These tumors were found to be larger and more invasive when compared to neoplasms in WT animals, p53R172H and hGAS mice. Interestingly, progastrin overexpression also caused an expansion of DCAMKL-1 positive cells. Within tumors, we observed a widely scattered pattern of expression of DCAMKL1-positive cells in hGAS and hGAS/p53RH mouse colon tumors, compared with only very few DCAMKL1-positive cells in WT mouse tumors [Figure 5(B)]. Therefore, DCAMKL1 expression seems to correlate with progastrin dependent tumor growth. These findings are in line with other studies were DCAMKL1 was found to be overexpressed during inflammation and metaplasia (38–36). While the connection between the expansion of DCAMKL-1 positive cells in injury and the development of cancer has been established in a variety of models the significance of this observation and the role of DCAMKL-1 cells need to be further investigated in the future.
Obviously, the p53 status does not only play a significant role at the time of AOM administration. The fate of the remaining wild-type p53 allele during all subsequent steps of malignant transformation is also of major importance. All of the previous studies on the molecular mechanisms of p53 gene mutations in colon cancer have used p53 heterozygous and homozygous knockout [p53 (+/−), p53 (−/−)] strains. Some studies found that loss of the wild-type allele of p53 is correlated with attenuated apoptosis and accelerated tumor growth (49), while other studies have failed to reveal a major role for p53 in modulating apoptosis in tumor growths (50). Here, we present data on the effects of the structural mutant p53R172H (knock-in) in the murine model of AOM induced colon carcinogenesis. It has been described earlier that the p53 knockout mice developed a distinctive tumor spectra compared to the p53R172H mice (8) and (10). We aimed to investigate the combination of a known co-carcinogen (progastrin) and a p53R172H gene mutation in one transgenic animal model (hGAS/p53R172H group) and separately (p53R172H, hGAS, and WT groups). By comparing outcome (colon tumor formation) along with the intermediate markers of colonic tumorigenesis namely, ACF formation and BrdU-labeling assay in all the four groups, we were able to examine the effects of progastrin signaling and p53 mutations. We present evidence that a high proportion of animals in the p53RH group mice retain an intact, functional, wild type p53 allele shown by their rates of ACF and colon tumor formation that is comparable to the WT group. However, in the presence of progastrin overexpression, this wild type allele is either lost or destabilized since hGAS/p53R172H double transgenic mice show the most aggressive tumor behavior among all groups.
In conclusion, we provide direct evidence that progastrin leads to crypt hyperproliferation and increased susceptibility to malignant transformation by increasing the number of colonic stem cells. Additional mutation in the p53 gene allows for a more aggressive behavior of the resulting tumors. The mechanisms underlying the progastrin-mediated expansion of the Lgr5+ compartment and the role of DCAMKL-1 cells in injury and malignant transformation awaits further investigation.
Acknowledgments
We would like to express our gratitude to Prof. Arthur Shulkes (Department of Surgery, University of Melbourne, Austin campus, Heidelberg, Victoria, Australia) for performing the mice serum progastrin radioimmunoassay. This work was supported by the NIH through a grant (No. 5 R01 DK052778) to Timothy C. Wang.
Footnotes
DECLARATION OF INTEREST
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.American Cancer Society. Cancer Facts & Figures 2010. Atlanta, GA: ACS; 2010. [Google Scholar]
- 2.Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Hamilton SR, Vogelstein B. Clonal analysis of human colorectal tumors. Science. 1987;238:193–197. doi: 10.1126/science.2889267. [DOI] [PubMed] [Google Scholar]
- 4.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 5.Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G, Kinzler KW, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990;247:49–56. doi: 10.1126/science.2294591. [DOI] [PubMed] [Google Scholar]
- 6.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 7.Bose I, Ghosh B. The p53-MDM2 network: from oscillations to apoptosis. J Biosci. 2007;32:991–997. doi: 10.1007/s12038-007-0103-3. [DOI] [PubMed] [Google Scholar]
- 8.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 10.Hu Y, Le Leu RK, Young GP. Absence of acute apoptotic response to genotoxic carcinogens in p53-defficient mice is associated with increased susceptibility to azoxymethane-induced colon tumours. Int J Cancer. 2005;115:561–567. doi: 10.1002/ijc.20876. [DOI] [PubMed] [Google Scholar]
- 11.Singh P, Velasco M, Given R, Varro A, Wang TC. Progastrin expression predisposes mice to colon carcinomas and adenomas in response to a chemical carcinogen. Gastroenterology. 2000;119:162–171. doi: 10.1053/gast.2000.8527. [DOI] [PubMed] [Google Scholar]
- 12.Lipkin M, Newmark H. Application of intermediate biomarkers and the prevention of cancer of the large intestine. Prog Clin Biol Res. 1988;279:135–150. [PubMed] [Google Scholar]
- 13.Dockray GJ. Gastrin-releasing peptide: an identity crisis at 30? Lancet. 2001;358:778–779. doi: 10.1016/s0140-6736(01)06008-1. [DOI] [PubMed] [Google Scholar]
- 14.Wang TC, Koh TJ, Varro A, Cahill RJ, Dangler CA, Fox JG, Dock-ray GJ. Processing and proliferative effects of human progastrin in transgenic mice. J Clin Invest. 1996;98:1918–1929. doi: 10.1172/JCI118993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh P, Velasco M, Given R, Wargovich M, Varro A, Wang TC. Mice overexpressing progastrin are predisposed for developing aberrant colonic crypt foci in response to AOM. Am J Physiol Gastrointest Liver Physiol. 2000;278:G390–G399. doi: 10.1152/ajpgi.2000.278.3.G390. [DOI] [PubMed] [Google Scholar]
- 16.Chen D, Zhao CM, Dockray GJ, Varro A, Van Hoek A, Sinclair NF, Wang TC, Koh TJ. Glycine-extended gastrin synergizes with gastrin 17 to stimulate acid secretion in gastrin-defficient mice. Gastroenterology. 2000;119:756–765. doi: 10.1053/gast.2000.16480. [DOI] [PubMed] [Google Scholar]
- 17.Koh TJ, Field JK, Varro A, Liloglou T, Fielding P, Cui G, Houghton J, Dockray GJ, Wang TC. Glycine-extended gastrin promotes the growth of lung cancer. Cancer Res. 2004;64:196–201. doi: 10.1158/0008-5472.can-03-2112. [DOI] [PubMed] [Google Scholar]
- 18.Kochman ML, DelValle J, Dickinson CJ, Boland CR. Post-translational processing of gastrin in neoplastic human colonic tissues. Biochem Biophys Res Commun. 1992;189:1165–1169. doi: 10.1016/0006-291x(92)92326-s. [DOI] [PubMed] [Google Scholar]
- 19.Koh TJ, Dockray GJ, Varro A, Cahill RJ, Dangler CA, Fox JG, Wang TC. Overexpression of glycine-extended gastrin in transgenic mice results in increased colonic proliferation. J Clin Invest. 1999;103:1119–1126. doi: 10.1172/JCI4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baldwin GS, Hollande F, Yang Z, Karelina Y, Paterson A, Strang R, Fourmy D, Neumann G, Shulkes A. Biologically active recombinant human progastrin(6–80) contains a tightly bound calcium ion. J Biol Chem. 2001;276:7791–7796. doi: 10.1074/jbc.M009985200. [DOI] [PubMed] [Google Scholar]
- 21.Brown D, Yallampalli U, Owlia A, Singh P. pp60c-Src Kinase mediates growth effects of the full-length precursor progastrin1–80 peptide on rat intestinal epithelial cells, in vitro. Endocrinology. 2003;144:201–211. doi: 10.1210/en.2002-220501. [DOI] [PubMed] [Google Scholar]
- 22.Hollande F, Lee DJ, Choquet A, Roche S, Baldwin GS. Adherens junctions and tight junctions are regulated via different pathways by progastrin in epithelial cells. J Cell Sci. 2003;116:1187–1197. doi: 10.1242/jcs.00321. [DOI] [PubMed] [Google Scholar]
- 23.Singh P, Lu X, Cobb S, Miller BT, Tarasova N, Varro A, Owlia A. Progastrin1–80 stimulates growth of intestinal epithelial cells in vitro via high-affinity binding sites. Am J Physiol Gastrointest Liver Physiol. 2003;284:G328–G339. doi: 10.1152/ajpgi.00351.2002. [DOI] [PubMed] [Google Scholar]
- 24.Pannequin J, Delaunay N, Buchert M, Surrel F, Bourgaux JF, Ryan J, Boireau S, Coelho J, Pelegrin A, Singh P, Shulkes A, Yim M, Baldwin GS, Pignodel C, Lambeau G, Jay P, Joubert D, Hollande F. Beta-catenin/Tcf-4 inhibition after progastrin targeting reduces growth and drives differentiation of intestinal tumors. Gastroenterology. 2007;133:1554–1568. doi: 10.1053/j.gastro.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 25.Ottewell PD, Varro A, Dockray GJ, Kirton CM, Watson AJ, Wang TC, Dimaline R, Pritchard DM. COOH-terminal 26-amino acid residues of progastrin are sufficient for stimulation of mitosis in murine colonic epithelium in vivo. Am J Physiol Gastrointest Liver Physiol. 2005;288:G541–G549. doi: 10.1152/ajpgi.00268.2004. [DOI] [PubMed] [Google Scholar]
- 26.Seva C, Dickinson CJ, Yamada T. Growth-promoting effects of glycine-extended progastrin. Science. 1994;265:410–412. doi: 10.1126/science.8023165. [DOI] [PubMed] [Google Scholar]
- 27.Siddheshwar RK, Gray JC, Kelly SB. Plasma levels of progastrin but not amidated gastrin or glycine extended gastrin are elevated in patients with colorectal carcinoma. Gut. 2001;48:47–52. doi: 10.1136/gut.48.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konturek PC, Bielanski W, Konturek SJ, Hartwich A, Pierzchalski P, Gonciarz M, Marlicz K, Starzynska T, Zuchowicz M, Darasz Z, Gotze JP, Rehfeld JF, Hahn EG. Progastrin and cyclooxygenase-2 in colorectal cancer. Dig Dis Sci. 2002;47:1984–1991. doi: 10.1023/a:1019652224424. [DOI] [PubMed] [Google Scholar]
- 29.Chang WW, Leblond CP. Renewal of the epithelium in the descending colon of the mouse. I. Presence of three cell populations: vacuolated-columnar, mucous and argentaffn. Am J Anat. 1971;131:73–99. doi: 10.1002/aja.1001310105. [DOI] [PubMed] [Google Scholar]
- 30.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 31.McLellan EA, Bird RP. Specificity study to evaluate induction of aberrant crypts in murine colons. Cancer Res. 1988;48:6183–6186. [PubMed] [Google Scholar]
- 32.BoseDasgupta S, Ganguly A, Roy A, Mukherjee T, Majumder HK. A novel ATP-binding cassette transporter, ABCG6 is involved in chemoresistance of Leishmania. Mol Biochem Parasitol. 2008;158:176–188. doi: 10.1016/j.molbiopara.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Piard F, Martin L, Chapusot C, Ponnelle T, Faivre J. Genetic pathways in colorectal cancer: interest for the pathologist. Ann Pathol. 2002;22:277–288. [PubMed] [Google Scholar]
- 34.Giannakis M, Stappenbeck TS, Mills JC, Leip DG, Lovett M, Clifton SW, Ippolito JE, Glasscock JI, Arumugam M, Brent MR, Gordon JI. Molecular properties of adult mouse gastric and intestinal epithelial progenitors in their niches. J Biol Chem. 2006;281:11292–11300. doi: 10.1074/jbc.M512118200. [DOI] [PubMed] [Google Scholar]
- 35.Gerbe F, van Es JH, Makrini L, Brulin B, Mellitzer G, Robine S, Romagnolo B, Shroyer NF, Bourgaux JF, Pignodel C, Clevers H, Jay P. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J Cell Biol. 2011;192:767–780. doi: 10.1083/jcb.201010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerbe F, Brulin B, Makrini L, Legraverend C, Jay P. DCAMKL-1 expression identifies Tuft cells rather than stem cells in the adult mouse intestinal epithelium. Gastroenterology. 2009;137:2179–2180. doi: 10.1053/j.gastro.2009.06.072. author reply 2180–2181. [DOI] [PubMed] [Google Scholar]
- 37.May R, Riehl TE, Hunt C, Sureban SM, Anant S, Houchen CW. Identification of a novel putative gastrointestinal stem cell and adenoma stem cell marker, doublecortin and CaM kinase-like-1, following radiation injury and in adenomatous polyposis coli/multiple intestinal neoplasia mice. Stem Cells. 2008;26:630–637. doi: 10.1634/stemcells.2007-0621. [DOI] [PubMed] [Google Scholar]
- 38.Saqui-Salces M, Keeley TM, Grosse AS, Qiao XT, El-Zaatari M, Gumucio DL, Samuelson LC, Merchant JL. Gastric tuft cells express DCLK1 and are expanded in hyperplasia. Histochem Cell Biol. 2011;136:191–204. doi: 10.1007/s00418-011-0831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein I, Marcel V, Olivier M, Oren M, Rotter V, Hainaut P. Understanding wild-type and mutant p53 activities in human cancer: new landmarks on the way to targeted therapies. Cancer Gene Ther. 2011;18:2–11. doi: 10.1038/cgt.2010.63. [DOI] [PubMed] [Google Scholar]
- 40.Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, Van Dyke T. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–711. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 41.Wargovich MJ, Chen CD, Harris C, Yang E, Velasco M. Inhibition of aberrant crypt growth by non-steroidal anti-inflammatory agents and differentiation agents in the rat colon. Int J Cancer. 1995;60:515–519. doi: 10.1002/ijc.2910600415. [DOI] [PubMed] [Google Scholar]
- 42.McLellan EA, Bird RP. Aberrant crypts: potential preneoplastic lesions in the murine colon. Cancer Res. 1988;48:6187–6192. [PubMed] [Google Scholar]
- 43.Corpet DE, Tache S. Most effective colon cancer chemopreventive agents in rats: a systematic review of aberrant crypt foci and tumor data, ranked by potency. Nutr Cancer. 2002;43:1–21. doi: 10.1207/S15327914NC431_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karlin DA, McBath M, Jones RD, Elwyn KE, Romsdahl MM. Hypergastrinemia and colorectal carcinogenesis in the rat. Cancer Lett. 1985;29:73–78. doi: 10.1016/0304-3835(85)90125-9. [DOI] [PubMed] [Google Scholar]
- 45.Takayama T, Katsuki S, Takahashi Y, Ohi M, Nojiri S, Sakamaki S, Kato J, Kogawa K, Miyake H, Niitsu Y. Aberrant crypt foci of the colon as precursors of adenoma and cancer. N Engl J Med. 1998;339:1277–1284. doi: 10.1056/NEJM199810293391803. [DOI] [PubMed] [Google Scholar]
- 46.Hu Y, Martin J, Le Leu R, Young GP. The colonic response to genotoxic carcinogens in the rat: regulation by dietary fibre. Carcinogenesis. 2002;23:1131–1137. doi: 10.1093/carcin/23.7.1131. [DOI] [PubMed] [Google Scholar]
- 47.Gryfe R, Swallow C, Bapat B, Redston M, Gallinger S, Couture J. Molecular biology of colorectal cancer. Curr Probl Cancer. 1997;21:233–300. doi: 10.1016/s0147-0272(97)80003-7. [DOI] [PubMed] [Google Scholar]
- 48.Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 49.Pan H, Griep AE. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes Dev. 1995;9:2157–2169. doi: 10.1101/gad.9.17.2157. [DOI] [PubMed] [Google Scholar]
- 50.Nemeth J, Taylor B, Pauwels S, Varro A, Dockray GJ. Identification of progastrin derived peptides in colorectal carcinoma extracts. Gut. 1993;34:90–95. doi: 10.1136/gut.34.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]