

Abstract
The generation of chimeras, which is now a standard technology for producing gene modified mutant mice, was originally developed as a tool for developmental biology. However, the application of conventional single marker chimeric mice for developmental study was initially limited. This situation has been dramatically changed by development of multicolor chimeric mice using various kinds of fluorescent proteins. Now using our technology, up to ten different clones could be distinguished by their colors, which enable us to perform more accurate statistical analyses and lineage tracing experiments than by conventional methods. This method could be applied to visualize not only cell turnover of normal stem cells but also cancer development of live tissues in vivo. In the present review, we will discuss how these methods have been developed and what questions they are now answering by mainly focusing on intestinal stem cells and intestinal tumors.
Keywords: Chimeras, Mosaic, Intestinal stem cells, Lineage tracing, Intestinal tumors
Developmental studies by generating chimeras
Chimeric analysis is one of the classical methods for cell lineage tracing by generating mice having more than one lineages of cells. If chimeras are introduced in early embryos, they could be used to analyze cell lineage relationships of early embryonic tissues. In mammals, chimeras are basically experimental. However, in rare cases, human chimeras could be naturally generated. One typical example is an XY/XO mosaic. The mechanisms of generation of naturally occurring chimeras is unclear [1]. One possible mechanism is that two embryos fuse together at an early embryonic stage and develop as a single embryo (Fig. 1). It is known that even if two cell lineages have distinct major histocompatibility complexes (MHCs), immune tolerance is induced during development and rejection does not occur after birth. Because it is ethically impossible to generate experimental chimeras in humans, the naturally occurring chimeras have been used for analyzing the origins of human tumors.
Fig. 1.

Case of a human African-Caucasian chimera [1]. A case of a human chimera boy who has mosaic skin colors reported in [1]
The first artificial method to create mammalian chimeras was developed by Tarkowski in 1961 [2, 3]. He removed zona pellucida from two early embryos and mechanically fused them together. Interestingly, when fused embryo is transferred to the uterus of a surrogate mother, it starts developing as a single large embryo, but is born at a normal size [4]. If two lineages of cells that compose chimeric mice could be distinguished, the mice could be used for lineage tracing experiments. Later, Gardner developed another method to generate mammalian chimeras. He isolated cells from inner cell mass of blastocysts, and transferred to the blastocyst cavity of another embryo. The injected blastocysts developed as chimeras [5, 6]. Evans and Martin developed the method to culture cells from inner cell mass in vitro, and established embryonic stem (ES) cells [7, 8]. This method, blastocyst injection, is now a standard method to generate knockout mice.
Another line of generating chimeras is via methods that make use of the X chromosome inactivation or the genome imprinting of [9–11]. In this method, a marker gene, such as LacZ is put into the genome locus susceptible to gene inactivation. During the early embryonic stage, one of the two alleles is inactivated at random in each cell. In heterozygous mice, one of the two alleles has LacZ gene. As a result of gene inactivation, LacZ positive/negative two-color chimeras are formed, which could be used for lineage tracing [9–11].
What kind of past experiments have been done using chimeras? Early studies on developmental biology using mammalian chimeras was described in detail in the book by MacLaren [12]. The simplest application of original chimera methods was most likely to analyze if the origin of the tissue of interest is monoclonal or polyclonal. Classical two-color chimeras were composed of marker positive/negative cells, i.e. they were two-color chimeras (Fig. 2). If the origin of the tissue is monoclonal, the resulting chimeric tissue always shows single color, i.e. all white or all blue. However, if origin of the tissue is generally multiple, the resulting tissue should usually be two-color [13]. By using the method, it was shown that epithelial cells in crypts of adult small intestine or colon are monoclonal in origin [14, 15]. This is one successful example of the analyses. However, early studies had shown that the origin of most of the tissues in mice is polyclonal [12]. In the past, some attempts were made to calculate the number of progenitors that generate the tissue of interest from the size of the patches in two-color chimeras. However, it was difficult to draw definitive conclusions from these studies [12, 16]. It means that application of two-color chimeras to developmental studies was limited, and to widen their application, technical improvement to increase the number of colors would be required.
Fig. 2.
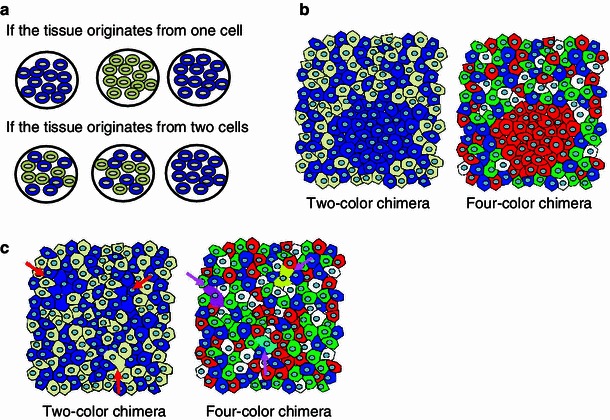
Advantages of using multicolor chimeras [13]: a Clonal analysis using conventional marker positive/negative chimeras (see text). b By using four-color chimeras, monoclonal areas could be more easily detected than conventional chimeras. c By using four-color chimeras, cells that underwent cell fusion could be detected, whereas it is not possible by using conventional marker positive/negative chimeras
Multicolor lineage tracing methods and their application to study of intestinal epithelial cells
Initially, markers used to form chimeras were not easy to visualize [17], however, technical improvement has changed the situation. LacZ has been long used for these analyses because it is relatively easy to visualize. However, to detect LacZ positive cells, tissues must be fixed, and therefore it was not possible to observe live LacZ positive cells. The discovery of green fluorescent protein (GFP) from A. victoria made it possible to detect marker positive cells alive, and time-lapse imaging of cells and tissues has been made possible. If only GFP is used, the resulting chimeras are two-color chimeras, and conclusions that can be drawn are basically the same as those from LacZ positive/negative chimeras. However, by increasing the number of colors that form chimeras, the outcome of chimeric tissues should be much more complicated, which would increase the accuracy of the statistical and clonal analyses. Isolation of DsRed, a red fluorescent protein from the coral Discosoma sp., and development of mutants of GFP and DsRed have created various fluorescent proteins that cover almost the entire spectrum of visible light [18, 19]. Based on these available markers, we developed the method to generate four-color chimeras (tetrachimeras) in 2006 [20]. The strategy involved knocking EGFP, ECFP and mRFP1 [18] cDNAs into the Rosa26 genome locus with a CAG promoter [21], and thus Rosa26-EGFP, Rosa26-ECFP, and Rosa26-mRFP1 knock-in mouse ES cell clones were established. The colored ES cells were injected into host colorless blastocysts as a mixture. The injected blastocysts were put back into the uteri of surrogate mothers. The resulting embryos and mice have four lineages of cells that could be distinguished by their colors (green, red, blue and no color). Confirming the previous findings, epithelial cells within each crypt always have single colors in tetrachimeric mice [13, 20] (Fig. 3). But by using multiple fluorescent markers, the boundary of clonal areas is much easier to identify, which makes it possible to detect finer patches in the chimeric tissues. Moreover, in the multicolor chimeras, fused cells could be easily detected by finding cells that co-express more than one color.
Fig. 3.
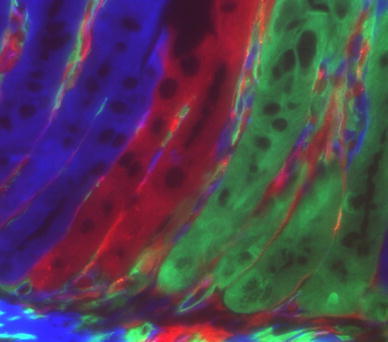
Chimera analysis reveals that intestinal crypts are monoclonal. This picture is a citation from our previous report [20]
To search for tissues that are generated by monoclonal progenitors, we analyzed various tissues from adult tetrachimeric mice [20]. In this experiment, monoclonal tissues were almost exclusively observed in endodermal epithelial and acinar tissues. Mesodermal and ectodermal tissues are generally polyclonal in origin (our unpublished findings).
As described, estimating the number of progenitors that generate the tissue/organ of interest by two-color chimeras was not successful. Interestingly, we found that in tetrachimeric mice, organs or tissues that are derived from a limited number of progenitors do not always have four colors. For example, if a group of cells always originates from four cells, it does not always happen that the resulting cells have all the four colors, but it is more likely to have two or three colors (Fig. 4) [13]. Mouse spermatogenic progenitors in testis represented one such example, as they usually generate two or three colors in tetrachimeric mice. The observation that germ cells in left and right testes always have the same colors indicates that the colors were selected before PGCs separate to left and right testes. By utilizing this phenomenon, we estimated the number of progenitors that generate the whole germ cells of mouse testes by statistical analysis. The result was that mouse male germ cells were generally derived from approximately four progenitors [22]. Blimp1 is one of the earliest markers of PGCs so far, and it was reported that the number of the first Blimp1 positive PGC progenitors was around six that appeared in epiblast [23], matching with our estimate. This is an interesting application of multicolor chimeras.
Fig. 4.
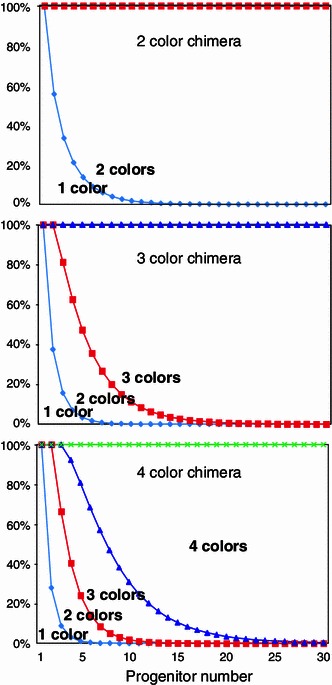
Probabilities of “fewer” color tissues in 2- and 4-color chimeras [13]. By increasing the number of colors that form chimeras, probabilities of generation of tissues/organs that have “fewer” color than the whole mouse increase. X-axis number of progenitors, Y-axis probabilities of fewer color organs/tissues [13]
By these methods, the chimeras are generated at early embryonic stages before gastrulation, therefore application of these methodologies to clonal analyses has a limitation. To analyze clonality in adult tissues, methods to induce chimera generation at the adult stage was needed. To this end, we modified reported methods for inducible chimeras by using the cre-loxp system [24, 25], so that the system could be applied to all the tissues in the mouse body. We generated Rosa26-rainbow mice, in which four fluorescent protein cDNAs are separated by three sets of loxp mutant sequences (Fig. 5). By cre recombinase, one of the three sets of lox sites is recombined and the cell having the construct changes its color from green to red, blue or orange. The fluorescent cDNA is expressed under the CAG promoter, and the whole construct is knocked into the Rosa26 locus [26, 27, 28]. As described, by increasing the number of colors, the accuracy of clonal analysis could be improved. The Rosa26-rainbow construct has four fluorescent cDNAs. By using homozygous mice, a combination of four colors can create up to ten colors (Fig. 6).
Fig. 5.

Schema of the construct of the Rosa26-rainbow system [28]. By using three sets of loxp mutant sequences, the cre-lox mediated DNA recombination changes the color of the cell that has the construct from green (EGFP) to one of three colors, blue (mCerulean), orange (mOrange) and red (mCherry)
Fig. 6.

Ten-color chimeric analysis by using homozygous Rosa26-rainbow mice. a Homozygous rainbow mice can create 10 different combination of colors. b Salivary glands of a Shhcre/+/Rosa26rainbow/rainbow mouse
By expressing an inducible form of cre, CreER and its mutant CreERT2, which is activated by tamoxifen but not endogenous estrogen [29–32] under tissue specific promoter, multicolor chimeras can be introduced in a time and space specific manner even in the tissues of interest. Of course, the system can be used for lineage tracing method [33]. It is not only a very powerful tool to follow the fate of stem cell-derived cells, but it adds the important information regarding clonality to the conventional lineage tracing method [26–28].
Two different models of intestinal stem cells
There has been a debate for decades regarding intestinal stem cells, which exist at the bottom of crypts [34, 35] (Fig. 7). Two major models were proposed in 1970’s; first, Cheng et al. [36] found by electron microscopic analyses that there are immature cells between Paneth cells, and they designated them as crypt base columnar cells (CBCs). On the other hand, Potten et al. [37] found that some cells are labeled by [3H]thymidine but retain the label for a long period after surrounding cells lose the label as they proliferate. The label retaining cells (LRCs) tended to locate at around position +4 from the bottom of the crypt. These cells were proposed as intestinal stem cells. In 2007, Barker et al. [38] identified a Wnt upregulated gene Lgr5 as a specific marker for CBCs, and showed by lineage tracing that Lgr5 positive CBCs include long-term intestinal stem cells. In 2008, however, Capecchi et al. [39] showed that Bmi1 positive cells that tend to exist at around position +4 also include long-term intestinal stem cells. Later, a more sensitive method was developed and confirmed the model [40], however, it was found that some of the cells co-express Lgr5 and Bmi1, and expression of Bmi1 is not limited to position +4 cells, but is expressed in TA cells up to position +10. Lgr5 is expressed not only in CBCs but also in position +4, and TA cells. However, Bmi1 expression is most strong at around position +4, and Lgr5 is at CBCs.
Fig. 7.
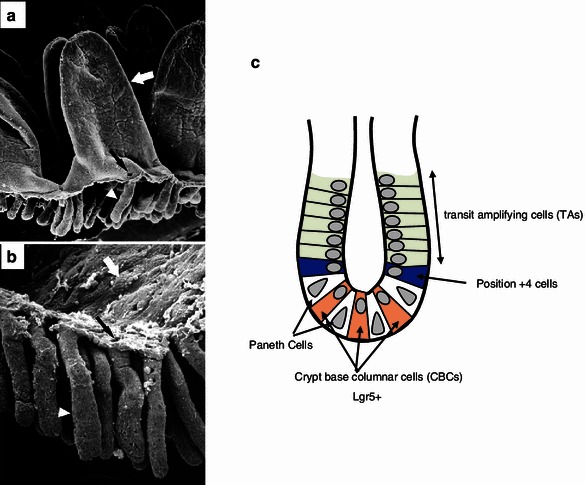
Intestinal crypts and two models of intestinal stem cells. a, b Scanning electron microscopic analyses of intestinal crypts. These images are cited from the past report [34]. c Schema of two models of intestinal stem cells in intestinal crypts
Basically, Lgr5 strong positive CBCs are cycling and Bmi1 strong positive cells are slow growing or resting within crypts. However, when Lgr5 positive CBCs are ablated, Bmi1-expressing stem cells compensate for the loss of Lgr5-expressing cells and Bmi1-expressing cells give rise to Lgr5-expressing cells, suggesting that they can compensate with each other [41]. If all the intestinal stem cells are cycling, they are likely to be very susceptible to DNA damage and could thus easily acquire mutations that can lead to cancers. However, human epithelial malignant tumors are extremely rare in small intestine, suggesting that there is a kind of safety system to protect against tumors in small intestine. In other tissues such as hematopoietic tissue in bone marrow and hair follicle, it has been established that long-term and short-term stem cells have distinct roles in maintaining the tissue. Short-term stem cells are cycling and mainly supply differentiated tissue cells but are short-lived. On the other hand, long-term stem cells are usually resting or slow growing, and occasionally enter the cell cycle to supply short-term stem cells [35]. In this model, long-term stem cells are protected from DNA damage. Short-term stem cells are susceptible for DNA damage but they are short-lived and therefore they would not get cancers unless they are immortalized. From the past reports, Lgr5 positive CBCs that are cycling have the characteristics of short-term stem cells [38]. On the other hand, Bmi1 positive stem cells tend to locate at position +4 and are slow growing, matching with the characteristics of long-term stem cells. However, as described, these two populations are in part overlapping, and at least Bmi1 positive stem cells can generate Lgr5-positive CBCs, making interpretation of the data complicated. Therefore, it is not clear whether Lgr5 positive CBCs are short-term stem cells with a capacity to generate long-term stem cells, or whether they all have a capacity to be long-term stem cells from the beginning. To further investigate the issue, multicolor lineage tracing experiments have been done.
Clonal analysis of intestinal stem cells
As described above, it has been shown by chimera analysis that epithelial cells within each crypt originate from a single progenitor during development. This raises a question as to when the single cell progenitor appears during fetal development. Interestingly, it was reported that epithelial cells in neonatal crypts are polyclonal by chimeric analyses, and gradually acquire monoclonality within the first 10–14 days [42, 43]. This means that adult type stem cells appear in the crypts during the neonatal period. Because by germ line chimeras, it is not possible to control the timing of generation of chimeras. However, to accurately examine the timing of generation of stem cells during development, inducible chimeras are required. The methods for inducible chimeras via the cre-loxp system had already been developed [24, 25]. We and others improved the methods so that they could be used for all the tissues in the mammalian body [26–28, 38], and applied the method to analyses of maintenance of intestinal progenitor cells in the crypts. In the experiments, crypt epithelial cells within one crypt are initially labeled with multiple colors; however, they gradually acquired monoclonality after 1 month (our unpublished data) [44]. To explain the results, two models were proposed. One is that each crypt has one long-term stem cell and multiple short-term stem cells, and that a hierarchy exists between stem cells. Another model is that all the Lgr5 positive CBCs have the capacity as long-term stem cells, but constantly compete for limited space in the niche, and only a single clone survives in the long term (neutral drift model) [44]. However, the study hypothesized that all the Lgr5 positive CBCs (approximately 15 cells per crypt) possess equal ability as long-term stem cells, and competition occurs between these equal stem cells, one of which survives. Now, it has been widely accepted that there exist two types of stem cells in crypts as described above [35, 45], and that Bmi1 positive stem cells can generate Lgr5 positive stem cells [41]. Taking this into consideration, the model would not be so simple. These issues have not reached consensus and debate is currently ongoing.
Maintenance of crypt number by crypt fission
It is known that intestinal crypts increase in number by crypt fission [46] (Fig. 8). Crypt fission happens in adult intestine in a physiological condition at a low frequency, suggesting that some of the crypts are constantly destroyed and are supplemented by newly generated crypts. Frequency of crypt fission increases in situations such as during the neonatal period, upon injury or upon Wnt activation. In a physiological condition, one crypt basically generates two crypts by fission [46–48]. The signal that induces crypt fission is unknown, however, activation of Wnt apparently increases the frequency of crypt fission. It is not clear if a function of crypt fission is to maintain a proper number of intestinal stem cells within crypts.
Fig. 8.

Crypt fission. a, b Images of crypt fission (cited from the past report [46])
In vitro culture of intestinal epithelial cells
To accurately examine different features of two classes of intestinal stem cells, it is important to observe the fate of each stem cell in vivo. However, so far it is not possible to directly observe fluorescence expressing intestinal stem cells from outside the body. One alternative method is to observe them in vitro by culturing intestinal epithelial cells, which until very recently was not possible. Sato et al. developed a method to culture intestinal epithelial cells in Matrigel. EGF, Noggin (a BMP inhibitor), and R-spondin1 (Wnt activator) were essential factors for the culture. Both cells from a single crypt or a single Lgr5 positive stem cell can generate organoid in the culture (Fig. 9) [49]. The existence of mesenchymal cells that form the niche enhances the growth of the organoid culture, but it is not indispensable [49]. The technique has made it possible to directly observe intestinal stem cells in vitro; however, so far, R-spondin1 (a Wnt activator) is required for the culture, and the addition of R-spondin1 modulates the normal physiological cell turnover. Therefore, it is important to establish methods to culture intestinal epithelial cells without Wnt agonists to accurately observe the dynamics of intestinal stem cells in vitro in the future.
Fig. 9.
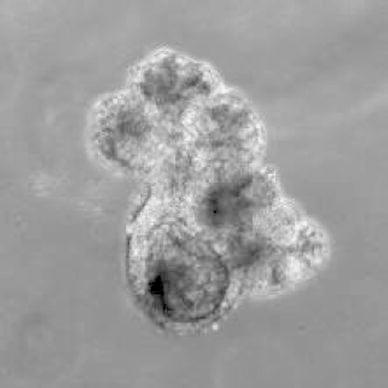
In vitro culture of intestinal epithelial cells. An image of organoid culture is shown
Wnt signaling pathway and cancer development
It has been established that the Wnt signaling pathway is very important for the growth of intestinal stem cells, and abnormalities in the Wnt pathway leads to the formation of intestinal tumors. In the canonical Wnt signaling pathway, Wnt ligands bind to the Frizzled receptor and its co-receptor, Lrp5/6. Without ligands, the destruction complex composed of APC, Axin, and GSK3-β ubiquitinate and destroy β-catenin, and thus the amount of intracellular β-catenin decreases [50]. When Wnt binds to the Frizzled, Lrp5/6 receptor complex, it inhibits degradation of β-catenin by the destruction complex and induces accumulation of intracellular β-catenin and its nuclear translocation. Then β-catenin binds to transcription factors Tcf·LEF, and upregulates various kinds of Wnt target genes [50] (Fig. 10). Some of the Wnt and Frizzled members are expressed differentially in tissues. By in situ hybridization, it has been shown that, in intestine, mainly Wnt-3, 6, and 9b are expressed in epithelial cells, and Wnt-5a is expressed in interstitial cells. Frizzled5, 7 are expressed in crypt epithelial cells, and Frizzled6 is expressed in villus and crypt epithelial cells [51].
Fig. 10.
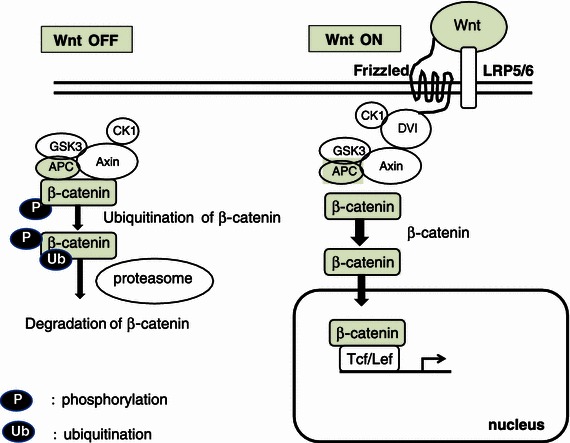
Schematic presentation of the Wnt signaling pathway
APC is an inhibitor of the Wnt signaling pathway. Mutations in the APC gene cause both genetic and sporadic adenomas. Familial adenomatous polyposis (FAP) is an inherited disease in which numerous polyps are formed mainly in epithelium of the colon, due to mutations of the APC gene [52–54]. It has been shown that APC inhibits transformation of colon epithelial cells [55, 56].
Lgr5 and its homologues are known to bind to Wnt receptors and mediate Wnt signaling pathway [57]. It has also been shown that Lgr5 activates the Wnt signaling pathway by competing with Dkk1, the inhibitor of the canonical Wnt signaling pathway, and inhibits internalization of the LRP co-receptor [58, 59]. R-spondin1 is a ligand for Lgr4 and Lgr5 and thus activates the Wnt signaling pathway [60, 61]. Interestingly, the whole genome sequencing of human colon tumors has identified recurrent abnormal fusion genes involving R-spondin2 and R-spondin3 that enhance the Wnt signal, raising the possibility that it could be a new target for cancer therapy [62].
The mechanism of how activation of the Wnt signaling pathway immortalizes cancer cells, or cancer stem cells has not been clear, but it was shown that activation of the Wnt/β catenin pathway activates telomerase activity [63], as the accumulation of β catenin stabilizes telomerase [64–66, 67].
Clonal analyses of the origin of intestinal tumors
Clonal origin of tumors has been one of the most important themes in cancer biology. Chimera analyses should be very useful for these studies. Basically it has been imagined that, regardless of whether they are benign or malignant, tumors develop from a single cell that undergoes oncogene mutations. To examine this, the clonal origin of tumors were studied by using naturally occurring human chimeras [1] or by analyzing glucose-6-phosphate dehydrogenase (G6PD). In these studies, the clonal origin of sporadic tumors was studied, revealing that the origin of tumors such as chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), carcinomas in the cervix, and leiomyoma are monoclonal [68–71]. Moreover, by using experimental models, research has found that the origin of mouse colon tumors generated by azoxymethane [72] or DMH [73] is monoclonal.
However, regarding benign tumors, there have been conflicting reports. In the case of hereditary neurofibromatosis, both benign and malignant neural tumors are generated. By chimera analysis with G6PD, the clonal origin of benign (neurofibroma, neuroma) and malignant (primary neurofibrosarcoma, hereditary neurofibrosarcoma) tumors was examined. Interestingly, benign tumors were polyclonal and malignant tumors were monoclonal in origin [74].
In the case of human colon tumors, it is well known that a benign adenoma is formed first, followed by growth of adenocarcinoma from a part of the adenoma. Multiple genes are known to involved in carcinogenesis of the colon, including APC, ras, DCC, p53. The multistep hit model involving these genes has been proposed and is well established. The APC gene mutation by itself generates benign adenomas. However, activation of proliferation by Wnt activation leads to additional mutations in essential genes for colon carcinogenesis, and generates cancers.
In 1983, Hsu et al. [75] analyzed adenomas in Gardner syndrome patients by using genome imprinting of the G6PD marker, and reported that they are generally polyclonal in origin. On the other hand, in 1987, Vogelstein [76] utilized RFLPs (restriction fragment length polymorphism) for analyzing the clonal origin of human colon tumors (adenoma and adenocarcinoma), and reported that both adenomas and adenocarcinomas are monoclonal in origin. In 1996, Novelli et al. [77] analyzed adenomas in an FAP patient with XY/XO chimerism, and reported that they are generally polyclonal independent of their size. The mechanism of forming polyclonal tumors is not clearly understood. It was proposed in these reports that in FAP models, multiple adjacent polyps, each of which is monoclonal, form a single tumor by “collision”, resulting in tumors in which multiple clones participate [72, 77]. However, it has not been made clear if it is also the case with sporadic adenomas without a germ line mutation in APC. One difficulty of the analysis is that we don’t yet have a good mouse model for sporadic adenomas.
Future directions
In this review, we have presented an overview of how chimeric analyses were originally developed, as well as how new methods using multicolor chimeras were applied to studies of tissue specific stem cell maintenance and the development of cancers. Newer technologies are now helping to expand research on these fundamental questions. They should be especially powerful when combined with in vitro organ cultures, and time lapse imaging of live animals, which at this time are not easy to perform, but will be possible with future technical developments.
Acknowledgments
The authors thank members of the Department of Stem Cell Pathology, Kansai Medical University for their helpful discussion. We acknowledge financial support from the following sources: Funding Program for Next Generation World-Leading Researchers, The Mochida Memorial Foundation, The Naito Memorial Foundation, The Cell Science Research Foundation, The Uehara Memorial Foundation, The Mitsubishi Foundation and The Yasuda Memorial Foundation to H.U.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Zuelzer WW, Beattie KM, Reisman LE. Generalized unbalanced mosaicism attributable to dispermy and probable fertilization of a polar body. Am J Hum Genet. 1964;16:38–51. [PMC free article] [PubMed] [Google Scholar]
- 2.Tarkowski AK. Experiments on the development of isolated blastomers of mouse eggs. Nature. 1959;184:1286–1287. doi: 10.1038/1841286a0. [DOI] [PubMed] [Google Scholar]
- 3.Tarkowski AK. Experimental studies on regulation in the development of isolated blastomeres of mouse eggs. Acta Theriologica. 1959; 3:191–267.
- 4.Tarkowski AK. Mouse chimaeras developed from fused eggs. Nature. 1961;190:857–860. doi: 10.1038/190857a0. [DOI] [PubMed] [Google Scholar]
- 5.Gardner RL. Mouse chimeras obtained by the injection of cells into the blastocyst. Nature. 1968;220(5167):596–597. doi: 10.1038/220596a0. [DOI] [PubMed] [Google Scholar]
- 6.Gardner RL, Rossant J. Investigation of the fate of 4–5 day post-coitum mouse inner cell mass cells by blastocyst injection. J Embryol Exp Morphol. 1979;52:141–152. [PubMed] [Google Scholar]
- 7.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 8.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA. 1981;78(12):7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tam PP, Tan SS. The somitogenetic potential of cells in the primitive streak and the tail bud of the organogenesis-stage mouse embryo. Development. 1992;115(3):703–715. doi: 10.1242/dev.115.3.703. [DOI] [PubMed] [Google Scholar]
- 10.Tan SS, Williams EA, Tam PPL. X-chromosome inactivation occurs at different times in different tissues of the post-implantation mouse embryo. Nat Genet. 1993;3(2):170–174. doi: 10.1038/ng0293-170. [DOI] [PubMed] [Google Scholar]
- 11.Tan SS, Breen S. Radial mosaicism and tangential cell dispersion both contribute to mouse neocortical development. Nature. 1993;362(6421):638–640. doi: 10.1038/362638a0. [DOI] [PubMed] [Google Scholar]
- 12.McLaren A. Mammalian chimeras. Cambridge University Press, Cambridge; 1976.
- 13.Ueno H, Weissman IL. The origin and fate of yolk sac hematopoiesis: application of chimera analyses to developmental studies. Int J Dev Biol. 2010;54(6–7):1019–1031. doi: 10.1387/ijdb.093039hu. [DOI] [PubMed] [Google Scholar]
- 14.Ponder BA, et al. Derivation of mouse intestinal crypts from single progenitor cells. Nature. 1985;313(6004):689–691. doi: 10.1038/313689a0. [DOI] [PubMed] [Google Scholar]
- 15.Griffiths DF, et al. Demonstration of somatic mutation and colonic crypt clonality by X-linked enzyme histochemistry. Nature. 1988;333(6172):461–463. doi: 10.1038/333461a0. [DOI] [PubMed] [Google Scholar]
- 16.McLaren A. Numerology of development. Nature. 1972;239(5370):274–276. doi: 10.1038/239274a0. [DOI] [PubMed] [Google Scholar]
- 17.Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the mouse embryo: a laboratory manual. 3rd ed. New York: Cold Spring Harbor Laboratory Press; 2003.
- 18.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22(12):1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 19.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2(12):905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 20.Ueno H, Weissman IL. Clonal analysis of mouse development reveals a polyclonal origin for yolk sac blood islands. Dev Cell. 2006;11(4):519–533. doi: 10.1016/j.devcel.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Miyazaki J, et al. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene. 1989;79(2):269–277. doi: 10.1016/0378-1119(89)90209-6. [DOI] [PubMed] [Google Scholar]
- 22.Ueno H, Turnbull BB, Weissman IL. Two-step oligoclonal development of male germ cells. Proc Natl Acad Sci USA. 2009;106(1):175–180. doi: 10.1073/pnas.0810325105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohinata Y, et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature. 2005;436(7048):207–213. doi: 10.1038/nature03813. [DOI] [PubMed] [Google Scholar]
- 24.Zong H, et al. Mosaic analysis with double markers in mice. Cell. 2005;121(3):479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Livet J, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450(7166):56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- 26.Red-Horse K, et al. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464(7288):549–553. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rinkevich Y, et al. Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature. 2011;476(7361):409–413. doi: 10.1038/nature10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H, et al. Experimental evidence showing that no mitotically active female germline progenitors exist in postnatal mouse ovaries. Proc Natl Acad Sci USA. 2012;109(31):12580–12585. doi: 10.1073/pnas.1206600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metzger D, et al. Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc Natl Acad Sci USA. 1995;92(15):6991–6995. doi: 10.1073/pnas.92.15.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feil R, et al. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci USA. 1996;93(20):10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feil R, et al. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237(3):752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- 32.Branda CS, Dymecki SM. Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6(1):7–28. doi: 10.1016/S1534-5807(03)00399-X. [DOI] [PubMed] [Google Scholar]
- 33.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 34.Bjerknes M, Cheng H. Intestinal epithelial stem cells and progenitors. Methods Enzymol. 2006;419:337–383. doi: 10.1016/S0076-6879(06)19014-X. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327(5965):542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. I. Columnar cell. Am J Anat. 1974;141(4):461–479. doi: 10.1002/aja.1001410403. [DOI] [PubMed] [Google Scholar]
- 37.Potten CS, Kovacs L, Hamilton E. Continuous labelling studies on mouse skin and intestine. Cell Tissue Kinet. 1974;7(3):271–283. doi: 10.1111/j.1365-2184.1974.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 38.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 39.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40(7):915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Itzkovitz S, et al. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat Cell Biol. 2012;14(1):106–114. doi: 10.1038/ncb2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian H, et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478(7368):255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt GH, et al. Clonal analysis of intestinal crypt populations in mouse aggregation chimaeras. J Embryol Exp Morphol. 1985;85:121–130. [PubMed] [Google Scholar]
- 43.Schmidt GH, Winton DJ, Ponder BA. Development of the pattern of cell renewal in the crypt-villus unit of chimaeric mouse small intestine. Development. 1988;103(4):785–790. doi: 10.1242/dev.103.4.785. [DOI] [PubMed] [Google Scholar]
- 44.Snippert HJ, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143(1):134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 45.Yan KS, et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA. 2012;109(2):466–471. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Booth C, Potten CS. Gut instincts: thoughts on intestinal epithelial stem cells. J Clin Invest. 2000;105(11):1493–1499. doi: 10.1172/JCI10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cairnie AB, Millen BH. Fission of crypts in the small intestine of the irradiated mouse. Cell Tissue Kinet. 1975;8(2):189–196. doi: 10.1111/j.1365-2184.1975.tb01219.x. [DOI] [PubMed] [Google Scholar]
- 48.Totafurno J, Bjerknes M, Cheng H. The crypt cycle. Crypt and villus production in the adult intestinal epithelium. Biophys J. 1987;52(2):279–294. doi: 10.1016/S0006-3495(87)83215-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 50.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8(5):387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 51.Gregorieff A, et al. Expression pattern of Wnt signaling components in the adult intestine. Gastroenterology. 2005;129(2):626–638. doi: 10.1016/j.gastro.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 52.Leppert M, et al. The gene for familial polyposis coli maps to the long arm of chromosome 5. Science. 1987;238(4832):1411–1413. doi: 10.1126/science.3479843. [DOI] [PubMed] [Google Scholar]
- 53.Kinzler KW, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253(5020):661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 54.Nishisho I, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 55.Korinek V, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275(5307):1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 56.Morin PJ, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 57.de Lau W, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. 2011;476(7360):293–297. doi: 10.1038/nature10337. [DOI] [PubMed] [Google Scholar]
- 58.Binnerts ME, et al. R-Spondin1 regulates Wnt signaling by inhibiting internalization of LRP6. Proc Natl Acad Sci USA. 2007;104(37):14700–14705. doi: 10.1073/pnas.0702305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim KA, et al. R-Spondin family members regulate the Wnt pathway by a common mechanism. Mol Biol Cell. 2008;19(6):2588–2596. doi: 10.1091/mbc.E08-02-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim KA, et al. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science. 2005;309(5738):1256–1259. doi: 10.1126/science.1112521. [DOI] [PubMed] [Google Scholar]
- 61.Carmon KS, et al. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc Natl Acad Sci USA. 2011;108(28):11452–11457. doi: 10.1073/pnas.1106083108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seshagiri S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012; 448:660–4. [DOI] [PMC free article] [PubMed]
- 63.Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet. 2007;8(4):299–309. doi: 10.1038/nrg2047. [DOI] [PubMed] [Google Scholar]
- 64.Mosimann C, Hausmann G, Basler K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol. 2009;10(4):276–286. doi: 10.1038/nrm2654. [DOI] [PubMed] [Google Scholar]
- 65.Park JI, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature. 2009;460(7251):66–72. doi: 10.1038/nature08137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoffmeyer K, et al. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336(6088):1549–1554. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- 67.Greider CW. Molecular biology. Wnt regulates TERT—putting the horse before the cart. Science. 2012; 336(6088): 1519–20. [DOI] [PubMed]
- 68.Fialkow PJ, Gartler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci USA. 1967;58(4):1468–1471. doi: 10.1073/pnas.58.4.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wiggans RG, et al. Probable clonal origin of acute myeloblastic leukemia following radiation and chemotherapy of colon cancer. Blood. 1978;52(4):659–663. [PubMed] [Google Scholar]
- 70.Smith JW, Townsend DE, Sparkes RS. Genetic variants of glucose-6-phosphate dehydrogenase in the study of carcinoma of the cervix. Cancer. 1971;28(2):529–532. doi: 10.1002/1097-0142(197108)28:2<529::AID-CNCR2820280235>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 71.Linder D, Gartler SM. Glucose-6-phosphate dehydrogenase mosaicism: utilization as a cell marker in the study of leiomyomas. Science. 1965;150(3692):67–69. doi: 10.1126/science.150.3692.67. [DOI] [PubMed] [Google Scholar]
- 72.Ponder BA, Wilkinson MM. Direct examination of the clonality of carcinogen-induced colonic epithelial dysplasia in chimeric mice. J Natl Cancer Inst. 1986;77(4):967–976. [PubMed] [Google Scholar]
- 73.Griffiths DF, et al. The clonal origin of experimental large bowel tumours. Br J Cancer. 1989;59(3):385–387. doi: 10.1038/bjc.1989.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Friedman JM, et al. Probable clonal origin of neurofibrosarcoma in a patient with hereditary neurofibromatosis. J Natl Cancer Inst. 1982;69(6):1289–1292. [PubMed] [Google Scholar]
- 75.Hsu SH, et al. Multiclonal origin of polyps in Gardner syndrome. Science. 1983;221(4614):951–953. doi: 10.1126/science.6879192. [DOI] [PubMed] [Google Scholar]
- 76.Fearon ER, Hamilton SR, Vogelstein B. Clonal analysis of human colorectal tumors. Science. 1987;238(4824):193–197. doi: 10.1126/science.2889267. [DOI] [PubMed] [Google Scholar]
- 77.Novelli MR, et al. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science. 1996;272(5265):1187–1190. doi: 10.1126/science.272.5265.1187. [DOI] [PubMed] [Google Scholar]