

Abstract
Recently, IgG4-related disease (IgG4-RD) has been recognized as a novel clinical entity with multiorgan involvement and unknown origin, associated with abundant infiltration of IgG4-positive cells. The Japanese research committee, supported by the Ministry of Health, Labor and Welfare of Japan, unified many synonyms for these conditions to the term “IgG4-RD” in 2009. The international symposium on IgG4-RD endorsed the comprehensive nomenclature as IgG4-RD, and proposed the individual nomenclatures for each organ system manifestations in 2011. Although the criteria for diagnosing IgG4-RD have not yet been established, proposals include the International Pathological Consensus (IPC) and the Comprehensive Diagnostic Criteria (CDC) for IgG4-RD for general use, and several organ-specific criteria for organ-specialized physicians, e.g., the International Consensus Diagnostic Criteria (ICDC) and the revised clinical diagnostic criteria in 2011 by the Japan Pancreas Society (JPS-2011) for type1 AIP; the Clinical Diagnostic Criteria 2012 for IgG4-sclerosing cholangitis (IgG4-SC-2012); the diagnostic criteria for IgG4-positive Mikulicz’s disease by the Japanese Society for Sjogren’s syndrome; and diagnostic criteria for IgG4-related kidney disease by the Japanese Society of Nephrology. In cases of probable or possible IgG4-RD diagnosed by the CDC, organ-specific diagnostic criteria should be concurrently used according to a diagnosis algorithm for IgG4-RD, with referral to a specialist.
Keywords: IgG4, IgG4-related disease (IgG4-RD), Autoimmune pancreatitis, Mikulicz’s disease, Diagnostic criteria
The history of IgG4-related disease: before and after discovery of IgG4
Recently, IgG4-related disease (IgG4-RD) has been recognized as a novel clinical entity with multiorgan involvement and an unknown origin, associated with abundant infiltration of IgG4-positive cells [1–8]. IgG4-RD has been found to affect the pancreas [9, 10], bile duct [10, 11], lacrimal glands [10, 12], salivary glands [10, 12], central nervous system [10, 13, 14], thyroid [10, 15, 16], lungs [10, 17, 18], liver [10, 19, 20], gastrointestinal tract [10, 21–24], kidney [10, 25, 26], prostate [10, 27, 28], retroperitoneum [10, 29], arteries [10, 30], lymph nodes [10, 31], skin [10, 32], and breast [10, 33]. However, before the disease was identified, each organ lesion was described independently.
In 1892, Mikulicz et al. [34] first observed a patient with symmetrical swelling of the lachrymal, parotid and submandibular glands, with massive infiltration of mononuclear cells. The condition was called Mikulicz’s disease (MD); however, it has since been classified as an atypical type of Sjögren’s syndrome, which also presents with bilateral, painless, and symmetrical swelling of the lachrymal, parotid, and submandibular glands. Küttner [35] reported a tumor-like enlargement of the submandibular gland that was sometimes a result of stones in the Wharton duct, which indicated that the underlying cause had not been identified. In 1961, Sarles et al. [36] first observed a case of particular pancreatitis with hypergammaglobulinemia, a prototype of autoimmune pancreatitis (AIP). The concept of AIP was first proposed by Yoshida et al. [37] in 1995. Following the histopathological description of lymphoplasmacytic sclerosing pancreatitis (LPSP) in 1991, from the resected pancreas of tumor-forming pancreatitis, which are clinically difficult to distinguish from pancreatic cancer, has been regarded as a characteristic histopathological finding of IgG4-related AIP (type 1 AIP) [38]. In 1967, Comings et al. [39] reported the first familiar case of multifocal fibrosclerosis with retroperitoneal fibrosis, mediastinal fibrosis, sclerosing cholangitis, Riedel’s thyroiditis, and pseudotumor of the orbit, which is now regarded as the synonym of IgG4-RD.
Hamano et al. [9] reported increased serum levels of IgG4 in Japanese patients with AIP, an epoch-making discovery in the history of IgG4-RD. Thereafter, many studies of AIP have been reported, mainly by Japanese investigators. The histopathological findings of LPSP are characterized by the periductal localization of predominantly CD4 positive T-cells, IgG4-positive plasma cells, storiform fibrosis with acinar cell atrophy frequently resulting in stenosis of the main pancreatic duct, and obliterative fibrosis [10]. About 60–80 percent of patients with AIP show obstructive jaundice with sclerosing cholangitis (IgG4-related sclerosing cholangitis; IgG4-SC) and other organ involvement (OOI), in which cholangiographic features are similar to those of primary sclerosing cholangitis (PSC), pancreatic cancer, and cholangiocarcinoma. The steroid responses and the prognoses of sclerosing cholangitis associated with AIP differ from patients with PSC, which suggests different pathological conditions. In 2003, Kamisawa et al. [12] suggested that AIP is a systemic sclerosing disease. This was based on findings that the pancreas and other involved organs have fibrosis with abundant infiltration of IgG4-positive plasma cells. This is similar to the concept of multifocal fibrosclerosis proposed by Comings et al. [39]. Further histological and clinical profiling of patients with “AIP” reveals two distinct subtypes, type 1 and type 2 [40, 41]. Type 1 AIP is classified as a pancreatic manifestation of IgG4-RD, and is probably a systemic disease with an abnormal immunological process. Type 2 AIP is thought to be a specific pancreatic disease with granulocytic epithelial lesion (GEL) [42, 43] and occasional coexistence with ulcerative colitis [41, 44].
Conversely, most patients with MD show elevated serum levels of IgG4, negative anti-SS-A/Ro or anti-SS-B/La antibodies, infiltration of IgG4-positive plasma cells into the glands, and recovery of secretion with steroid treatment. The patients with MD often show steroid responsive OOIs such as AIP, sclerosing cholangitis, retroperitoneal fibrosis, enlarged celiac and hilar lymph nodes, chronic thyroiditis, or interstitial nephritis [1–5, 10]. MD has been considered to be completely different from Sjögren’s syndrome because of this, and because of its responsiveness to steroid treatment [1–5, 10].
In addition to the original concept of multifocal idiopathic fibrosclerosis, recent studies led us to develop a novel concept of a systemic disease such as IgG4-related systemic sclerosing disease [1], systemic IgG4-related plasmacytic syndrome (SIPS) [2], or IgG4-positive multiorgan lymphoproliferative syndrome (IgG4-MOLPS) [3], all of which may refer to the same conditions. Based on these findings, the members of the Japanese Research Committees for “Systemic IgG4-related Sclerosing Disease” (chaired by Professor Okazaki) and “IgG4-MOLPS” (chaired by Professor Umehara), both of which were supported by the Research for Intractable Disease Program from the Ministry of Health, Labor and Welfare of Japan, have agreed that the comprehensive term “IgG4-related disease IgG4-RD)” includes these conditions at a minimum, although pathogenesis and pathophysiology remain unclear [4, 5]. The first International Symposium on IgG4-RD held in Boston (chaired by Professor Stone of Massachusetts General Hospital) endorsed the Japanese concept and proposed nomenclatures and pathological criteria for individual organ lesions [6, 7] (Tables 1, 2, 3).
Table 1.
History of IgG4-related disease
| Year | Authors | References | Evidences/contents |
|---|---|---|---|
| 1892 | Mikulicz et al. | [34] | Mikulicz’s disease (Z. Chir. Fesrschr) |
| 1961 | Sarles et al. | [36] | Hyper-gammaglobulinemia in CP (Am J Dig Di) |
| 1967 | Comings et al. | [39] | Familial multifocal fibrosclerosis. (Ann Intern Med) |
| 1972 | Küttner | [35] | Küttner tumor (Acta Otolaryngol) |
| 1991 | Kawaguchi et al. | [38] | Lymphoplasmacytic sclerosing pancreatitis(Human Pathol) |
| 1995 | Yoshida et al. | [37] | Autoimmune pancreatitis (Dig Dis Sci) |
| 2001 | Hamano et al. | [9] | High IgG4 levels in sclerosing pancreatitis (N Eng J Med) |
| 2002 | Japan Pancreas Society | [57] | Clinical diagnostic criteria for AIP 200 (Suizo) |
| 2006 | Okazaki et al. | [56] | Clinical diagnostic criteria for AIP 2006 (J Gastroenterol) |
| 2006 | Chari et al. | [58] | Mayo criteria (Clin Gastroenterol Hepatol) |
| 2006 | Kamisawa et al. | [12] | IgG4-related sclerosing disease (J Gastroenterol) |
| 2006 | Yamamoto et al. | [2] | IgG4-related plasmacytic disease (Mod Rheumatol) |
| 2008 | Masaki et al. | [3] | IgG4-multiorgan lymphoproliferative syndrome (MOLPS) (Ann Rheum Dis) |
| 2011 | Shimosegawa et al. | [41] | International Consensus Diagnostic Criteria (ICDC) for AIP (Pancreas) |
| 2012 | Umehara, Okazaki, et al. | [4, 5] | Concept and comprehensive Diagnostic Criteria for IgG4-related disease (Mod Rheumatol) |
| 2012 | Deshpande et al. | [6] | International Pathological Consensus for IgG4-RD (Mod Pathol) |
| 2012 | Stone et al. | [7] | Nomenclatures of individual organ manifestation of IgG4-RD (Arthritis Rheum) |
Table 2.
Preferred nomenclature for individual organ system manifestations of IgG4-related disease (from [7], with permission)
| Organ system/tissue | Preferred name |
|---|---|
| Pancreas | Type 1 autoimmune pancreatitis (IgG4-related pancreatitis) |
| Eye | IgG4-related ophthalmic disease is the general term for the peri-ocular manifestations of this disease. There are several subsets, outlined below. |
| Lacrimal glands | IgG4-related dacryoadenitis |
| Orbital soft tissue (orbital inflammatory pseudotumor) | IgG4-related orbital inflammation |
| Extra-ocular muscle disease | IgG4-related orbital myositis |
| Orbit with involvement of multiple anatomic structures | IgG4-related pan-orbital inflammation (includes lacrimal gland disease, extra-ocular muscle involvement, and other potential intra-orbital complications) |
| Salivary glands (parotid and submandibular glands) | IgG4-related sialadenitis or, more specifically, IgG4-related parotitis or IgG4-related submandibular gland disease |
| Pachymeninges | IgG4-related pachymeningitis |
| Hypophysis | IgG4-related hypophysitis |
| Thyroid (Riedel’s thyroiditis) | IgG4-related thyroid disease |
| Aorta | IgG4-related aortitis/peri-aortitis |
| Arteries | IgG4-related periarteritis |
| Mediastinum | IgG4-related mediastinitis |
| Retroperitoneum | IgG4-related retroperitoneal fibrosis |
| Mesentery | IgG4-related mesenteritis |
| Skin | IgG4-related skin disease |
| Lymph node | IgG4-related lymphadenopathy |
| Bile ducts | IgG4-related sclerosing cholangitis |
| Gallbladder | IgG4-related cholecystitis |
| Liver | IgG4-related hepatopathy (refers to liver involvement that is distinct from biliary tract involvement) |
| Lung | IgG4-related lung disease |
| Pleura | IgG4-related pleuritis |
| Pericardium | IgG4-related pericarditis |
| Kidney | IgG4-related kidney disease. The specific renal pattern should be termed IgG4-related tubulointerstitial nephritis and membranous glomerulonephritis secondary to IgG4-RD. Involvement of the renal pelvis should be termed IgG4-related renal pyelitis. |
| Breast | IgG4-related mastitis |
| Prostate | IgG4-related prostatitis |
Table 3.
The three major histopathological features associated with IgG4-RD and the minimal criteria in a new organ/site in the international pathological consensus (from [6])
| The three major histopathological features associated with IgG4-RD |
| 1. Dense lymphoplasmacytic infiltrate |
| 2. Fibrosis, arranged at least focally in a storiform pattern |
| 3. Obliterative phlebitis |
| Other histopathological features associated with IgG4-RD are: |
| 1. Phlebitis without obliteration of the lumen |
| 2. Increased numbers of eosinophils |
| Minimal Criteria for IgG4-RD in a new organ/Site |
| (1) characteristic histopathological findings with an elevated IgG4t plasma cells and IgG4-to-IgG ratio |
| (2) high serum IgG4 concentrations |
| (3) effective response to glucocorticoid therapy |
| (4) reports of other organ involvement that is consistent with IgG4-related disease |
Current concepts of IgG4-RD
General concept of IgG4-RD
Patients with IgG4-RD show diffuse or focal organ enlargement and mass-forming or nodular/thickened lesions in various organs, either synchronously or metachronously. This is due to the prominent infiltration of lymphocytes and plasmacytes with fibrosis [4]. The causes of the disease are still not clear; however, some abnormal immunological mechanisms are involved. The organs known to be affected include the pancreas, biliary duct, lacrimal/salivary glands, retroperitoneum, central nervous system, thyroid gland, lungs, liver, gastrointestinal tracts, kidneys, prostate gland, and lymph nodes [21–41]. IgG4-RD mainly affects middle-aged to elderly men, and clinical symptoms vary depending on the organ in which the lesions are located. Many cases are treated effectively by steroid therapy [1–10]; however, the prognosis is not clear. Some patients develop serious complications such as obstructive jaundice due to hepatic, gallbladder, or pancreatic lesions; hydronephrosis due to retroperitoneal fibrosis; or respiratory symptoms due to pulmonary lesions [1–10, 25, 26, 29, 45–47]. Although the infiltration of IgG4-positive cells and increased serum levels of IgG4 are characteristic in IgG4-RD, the severity of fibrosis seems to be different among the individual organs involved. These conditions are quite similar to multifocal idiopathic fibrosclerosis [39]. Storiform fibrosis and obliterative phlebitis are characteristic in pancreatic and biliary tract lesions, but the degree varies depending on the individual organs. For example, very seldom do lesions appear in the lachrymal/salivary gland or lymph node. The previous nomenclature of “IgG4-related sclerosing disease” is mainly based on the fibrous swollen organs, whereas those of “IgG4-SIPS” and “IgG4+ MOLPS” have been based on lymphoplasmacytic proliferation and swollen lymph nodes without fibrosis.
Although most patients have multiorgan lesions synchronously or metachronously, about 10–20 percent of the patients do not have confirmed OOI. Therefore, it is unclear whether the pathogenetic mechanism is same among individual organs or not.
Concept of IgG4-RD in the hepato-bilio-pancreatic system
Type 1 AIP (IgG4-related pancreatitis)
Recent studies have suggested that AIP manifests as two distinct subtypes, type 1 and type 2 [40, 41]. In type 1 AIP, the histologic description is called lymphoplasmacytic sclerosing pancreatitis (LPSP) and the pancreatic histopathology shows the following characteristic features: (i) abundant infiltration of plasma cells (greater than 10 IgG4+ cells per high power field (hpf) and an IgG4 to IgG ratio greater than 40 percent) and lymphocytes; (ii) peculiar storiform or swirling fibrosis; and (iii) perivenular infiltration with lymphocytes and plasma cells often leading to obliterative phlebitis.
Type 2 AIP was proposed by American and European pathologists based on histological examination of the resected pancreases from patients with chronic non-alcoholic pancreatitis. These pathologists reported another histopathological pattern, which they named idiopathic duct-centric pancreatitis (IDCP) or AIP with granulocytic epithelial lesion (GEL) [41–43]. The most characteristic feature of type 2 AIP is the granulocytic epithelial lesion (GEL), which often presents with destruction and obliteration of the pancreatic duct. Type 2 AIP includes swelling of the pancreas, but no or very few IgG4-positive plasma cells. Type 2 AIP clinical features show a distinctly different profile associated with no serum IgG4, IgG elevation, presence of autoantibodies, or other organ involvement.
IgG4-related sclerosing cholangitis (IgG4-SC)
IgG4-SC is a characteristic type of sclerosing cholangitis with dense infiltration of IgG4-positive plasma cells and extensive fibrosis in the bile duct wall [48]. Circular and symmetrical thickening of the bile duct wall is observed not only in the stenotic areas but also in the areas without stenosis that appear normal in the cholangiogram [49]. IgG4-SC related OOI such as IgG4-related dacryoadenitis or sialadenitis or IgG4-related retroperitoneal fibrosis is frequently associated with type 1 AIP [50–54] The differential diagnosis of IgG4-SC from PSC and cholangiocarcinoma is very important. It is also necessary to rule out secondary sclerosing cholangitis caused by diseases with obvious pathogenesis.
Nomenclatures of individual organ manifestation of IgG4-RD
Because multiorgan involvements may occur in IgG4-RD as described above, IgG4-RD includes a wide variety of diseases, including MD, AIP, hypophysitis, Riedel thyroiditis, interstitial pneumonitis, interstitial nephritis, prostatitis, lymphadenopathy, retroperitoneal fibrosis, inflammatory aortic aneurysm, and inflammatory pseudotumor [1–10]. In the International Symposium on IgG4-RD, the nomenclature of individual organ manifestations of IgG4-RD were proposed (Table 2) using “IgG4-related” as a modifier, except for the pancreatic manifestation [7]. The pancreatic manifestation of IgG4-RD was termed “type 1 autoimmune pancreatitis (IgG4-related pancreatitis).” The term “type 1 AIP” is now widely accepted among gastroenterologists and pancreatic surgeons. It also serves to discriminate between type 1 and type 2 AIP, which is not a part of the IgG4-RD spectrum. When the pathogenesis of type 2 AIP is clarified, the term “type 1 AIP” might be replaced by “IgG4-related pancreatitis.” In the biliary manifestation, the nomenclature for IgG4-related biliary tract (but not gall bladder) disease includes “sclerosing” to distinguish between the primary and IgG4-related forms of sclerosing cholangitis.
Clinical diagnostic criteria for IgG4-RD
International pathological consensus criteria for IgG4-RD
To diagnose IgG4-RD, histopathological findings are critical, and the international histological consensus criteria (IPC) (Table 3) have been proposed [6]. There are three major histopathological features associated with IgG4-RD: (i) dense lymphoplasmacytic infiltration; (ii) fibrosis, arranged at least focally in a storiform pattern; and (iii) obliterative phlebitis. Other minor histopathological features associated with IgG4-RD are phlebitis without obliteration of the lumen and increased numbers of eosinophils. IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders [55]. Therefore, different numbers of IgG4-positive cells among individual organ lesions were required for diagnosing IgG4-RD (Fig. 1). For examples, more than 100 IgG4-positive cells are required in the lacrimal and salivary gland and lymph node, and more than 200 cells in the skin. On the other hand, in the liver, bile duct and pancreas, more than 10 cells by biopsy and 50 cells by surgical specimen are needed. In order to diagnose involvement of a new organ or site, it has been recommended that at least three, but ideally four, of the following criteria for IgG4-RD be met: (i) characteristic histopathological findings of an elevated concentration of IgG4+ plasma cells and elevated IgG4 to IgG ratio; (ii) high serum IgG4 concentrations; (iii) effective response to glucocorticoid therapy; and (iv) other organ involvement that is consistent with IgG4-RD. Appropriate histopathologic findings are essential, but not sufficient, to establish a new manifestation or site of IgG4-RD, so a combination of clinical manifestations, including serum IgG4 levels and histological findings, are necessary.
Fig. 1.

Histologic diagnostic schema of IgG4-related disease (reproduced from [6])
Japanese comprehensive diagnostic criteria for IgG4-RD
The Japanese comprehensive diagnostic criteria for IgG4-RD [5] (Table 4) are based on two major characteristics of IgG4-RD: increased serum concentrations of IgG4 and infiltration of IgG4+ cells into the affected organ. The cutoff value for serum IgG4 concentration, 135 mg/dl, is the same as that in AIP [9]. Although tissue biopsies are difficult to obtain from some organs, including the pancreas, retroperitoneum, and ocular cavity, histopathological examination is required. Pathological criteria should be rigorous because IgG4+ plasma cell infiltration has been reported in various diseases and clinical conditions, such as rheumatoid synovitis, inflammatory oral and skin lesions, and carcinomas with a peritumoral inflammatory response [55]. Histopathological findings of marked IgG4+ cell infiltration (concentration of greater than 10 cells per hpg) and an IgG4/IgG cell ratio of greater than 40 percent are diagnostic of IgG4-RD.
Table 4.
Comprehensive clinical diagnostic criteria for IgG4-RD (from [5], with permission)
| 1. Clinical examination showing characteristic diffuse/localized swelling or masses in single or multiple organs |
| 2. Hematological examination shows elevated serum IgG4 concentrations(135 mg/dl) |
| 3. Histopathologic examination shows: |
| (1) Marked lymphocyte and plasmacyte infiltration and fibrosis. |
| (2) Infiltration of IgG4+ plasma cells: ratio of IgG4+/IgG+ cells >40 % and >10 IgG4+ plasma cells/HPF |
| Definite: 1) + 2) + 3) |
| Probable: 1) + 3) |
| Possible: 1) + 2) |
| However, it is important to differentiate IgG4-RD from malignant tumors of each organ (e.g., cancer, lymphoma) and similar diseases (e.g. Sjögren’s syndrome, primary sclerosing cholangitis, Castleman’s disease, secondary retroperitoneal fibrosis, Wegener’s granulomatosis, sarcoidosis, Churg–Strauss syndrome) by additional histopathological examination |
| Even when patients cannot be diagnosed using the CCD criteria, they may be diagnosed using organ-specific diagnostic criteria for IgG4RD. |
Diagnostic criteria for individual organ manifestation of IgG4-RD in hepato-bilio-pancreatic organs
Diagnostic criteria for type1 AIP
International consensus of diagnostic criteria (ICDC) for type1 AIP
The ICDC for AIP [41] were developed based on previous criteria, including JPS (2002, 2006) [56, 57], HISORt (2006, 2009) [58, 59] Korean (2007) [60], Asian (2008) [61], Mannheim (2009) [62] and Italian (2003, 2009) [63], and first enabled us to make an independent clinical diagnosis of type 1 or type 2 AIP. The diagnosis of type 1 AIP by ICDC requires a combination of five primary cardinal features (Tables 5,6): (i) imaging features of a) pancreatic parenchyma (on CT/MRI) and b) pancreatic duct (ERCP or MRCP); (ii) serology (IgG4); (iii) other organ involvement; (iv) histopathology of the pancreas; and (v) response to steroid therapy. Each criterion, except for steroid responsiveness, is classified as either level 1 or level 2 collateral criteria. Level 1 collateral is highly suggestive of AIP. Patients with obstructive jaundice and a diffusely enlarged pancreas (especially with a capsule-like rim) without pancreatic ductal dilatation/cutoff or pancreatic low-density mass on CT/MRI are highly likely to have AIP. However, subjects with typical findings of pancreatic cancer (e.g., low-density mass on contrast-enhanced CT, pancreatic ductal dilatation/cutoff with or without pancreatic atrophy) should be considered as having pancreatic cancer. Subjects without features typical of AIP or pancreatic cancer should first be investigated for pancreatic cancer. AIP should be considered only after negative work-up of malignancy. Response to steroids can confirm a strong suspicion of AIP. However, a steroid trial as a means to diagnose AIP is to be used sparingly and should not be used as a substitute for a thorough search for an etiology.
Table 5.
Level 1 and Level 2 criteria for type 1 AIP in international consensus of diagnostic criteria (ICDC) (from [41], with permission)
| Criterion | Level 1 | Level 2 | ||
|---|---|---|---|---|
| P | Parenchymal imaging | Typical: | Indeterminate (including Atypicala): | |
| Diffuse enlargement with delayed enhancement (sometimes associated with rim like enhancement) | Segmental/focal enlargement with delayed enhancement | |||
| D | Ductal imaging (ERP) | Long (>1/3 length of the mpd) or multiple strictures without marked upstream dilatation | Segmental/focal narrowing without marked upstream dilatation (duct size <5 mm) | |
| S | Serology | IgG4 >2 X upper limit of normal value | IgG4 1-2 X upper limit of normal value | |
| OOI | Other organ involvement | a or b | a or b | |
| a. Histology of extrapancreatic organs | a. Histology of extrapancreatic organs including endoscopic biopsies of bile ductb: | |||
| any three of the following | both of the following | |||
| i. Marked lymphoplasmacytic infiltration with fibrosis and without granulocytic infiltration | i. Marked lymphoplasmacytic infiltration without granulocytic infiltration | |||
| ii. Striform fibrosis | ii. Abundant (>10 cells/hpf) IgG4 positive cells | |||
| iii. Obliterative phlebitis | ||||
| iv. Abundant (>10 cells/hpf) IgG4 positive cells | ||||
| b. Typical radiological evidence | b. Physical or radiological evidence | |||
| at least one | at least one | |||
| i. Segmental/multiple proximal (hilar/intra hepatic) or proximal and distal bile duct stricture | i. Symmetrically enlarged salivary/lacrimal glands | |||
| ii. Retroperitoneal fibrosis | ii. Radiologic evidence of renal involvement described in association with AIP | |||
| H | histology of the pancreas | LPSP (core biopsy/resection) At least 3 of the following | LPSP (core biopsy) Any 2 of the following | |
| i. Periductal lymphoplasmacytic infiltrate without granulocytic infiltration | i. Periductal lymphoplasmacytic infiltrate without granulocytic infiltration | |||
| ii. Obliterative phlebitis | ii. Obliterative phlebitis | |||
| iii. Storiform fibrosis | iii. Storiform fibrosis | |||
| iv. Abundant (>10 cells/hpf) IgG4 positive cells | iv. Abundant (>10 cells/hpf) IgG4 positive cells | |||
| Diagnostic steroid trial | ||||
| Response to steroid (Rt)c | Rapid (≤2 weeks) radiologically demonstrable resolution or marked improvement in pancreatic/extra-pancreatic Manifestations | |||
aAtypical: some AIP cases may show low-density mass, pancreatic ductal dilatation or distal atrophy. Such atypical imaging findings in patients with obstructive jaundice and/or pancreatic mass are highly suggestive of pancreatic cancer. Such patients should be managed as pancreatic cancer unless there is strong collateral evidence for AIP and a thorough work-up for cancer is negative (see algorithm)
bEndoscopic biopsy of duodenal papilla is a useful adjunctive method because ampulla is often involved pathologically in AIP
cDiagnostic steroid trial should be conducted carefully by pancreatologists with caveats (see text) only after negative work-up for cancer including EUS-FNA
Table 6.
Diagnosis of definitive and probable type 1 AIP using international consensus diagnostic criteria (ICDC) (from [41], with permission)
| Diagnosis | Primary basis for diagnosis | Imaging evidence | Collateral evidence |
|---|---|---|---|
| Definitive Type 1 AIP | Histology | Typical/indeterminate | Histologically confirmed LPSP (Level 1 H) |
| Imaging | Typical | Any non-D Level 1/Level 2 | |
| Indeterminate | Two or more from Level 1 (+Level 2 Da) | ||
| Response to steroid | Indeterminate | Level 1 S/OOI + Rt or IBD + Level 1 D + Level 2 S/OOI/H + Rt | |
| Probable Type 1 AIP | Indeterminate | Level 2 S/OOI/H + Rt |
aLevel 2 D is counted as Level 1 in this setting
Clinical diagnostic criteria for AIP by Japan Pancreas Society in 2011 (JPS-2011)
JPS-2011 [64, 65] (Table 7) took basic concepts from both the previous Japanese criteria [56, 57] and the CDC for type 1 AIP [41]. These include ensuring that the criteria are (i) simple for general physicians’ use; (ii) rely on diffuse/segmental/focal classification of pancreatic imaging; (iii) use IgG4 alone as a serological marker; (iv) identify OOIs such as sclerosing cholangitis; sclerosing sialadenitis and retroperitoneal fibrosis; (v) have no classifications of level1/2 in serum IgG4 and OOI; (vi) apply optional steroid trial only after determining non-malignancy using EUS-FNA. Different from the previous Japanese criteria, with JPS-2011, patients are diagnosed as having definitive, probable, or possible AIP by a combination of the criteria described. This is similar to the concept of the ICDC.
Table 7.
Clinical diagnostic criteria for autoimmune pancreatitis in 2011 by Japan Pancreas Society (JPS-2011) (from [64, 65], with permission)
| A. Diagnostic criterion |
|---|
| I. Enlargement of the pancreas: |
| a. Diffuse enlargement |
| b. Segmental/focal enlargement |
| II. ERP (endoscopic retrograde pancreatography) shows irregular narrowing of the main pancreatic duct |
| III. Serological findings |
| Elevated levels of serum IgG4 (≥135 mg/dl) |
| IV. Pathological findings: among i)–iv) listed below, |
| a. Three or more are observed |
| b. Two are observed |
| i) Prominent infiltration and fibrosis of lymphocytes and plasmacytes |
| ii) Ten or more diffuse IgG4-positive plasmacytes per high-power microscope field |
| iii) Storiform fibrosis |
| iv) Obliterative phlebitis |
| V. Other organ involvement (OOI): sclerosing cholangitis, sclerosing dacryoadenitis/sialoadenitis, retroperitoneal fibrosis |
| a. Clinical lesions |
| Extra-pancreatic sclerosing cholangitis, sclerosing dacryoadenitis/sialoadenitis (Mikulicz disease), or retroperitoneal fibrosis can be diagnosed with clinical and image findings. |
| b. Pathological lesions |
| Pathological examination shows characteristic features of sclerosing cholangitis, sclerosing dacryoadenitis/sialoadenitis, or retroperitoneal fibrosis. |
| <Option> Effectiveness of steroid therapy |
| A specialized facility may include in its diagnosis the effectiveness of steroid therapy, once pancreatic or bile duct cancers have been ruled out. When it is difficult to differentiate from malignant conditions, it is desirable to perform cytological examination using an endoscopic ultrasound-guided fine needle aspiration (EUS-FNA). Facile therapeutic diagnosis by steroids should be avoided unless the possibility of malignant tumor has been ruled out by pathological diagnosis. |
| B. Diagnosis |
|---|
| I. Definite diagnosis |
| 1 Diffuse type |
| I a + < III/IVb/V(a/b)> |
| 2 Segmental/focal type |
| I b + II + two or more of <III/IV b/V (a/b)> |
| I b + II + < III/IV b/V (a/b)> + Option |
| 3 Definite diagnosis by histopathological study |
| IV a |
| II. Probable diagnosis |
| Segmental/focal type: I b + II + < III/IV b/V (a/b)> |
| III. Possible diagnosisa |
| Diffuse type: I a + II + Option |
| Segmental/focal type: I b + II + Option |
When a patient with a focal/segmental image of AIP on CT/MRI without ERCP findings fulfill more than one of III, IVb and V(a/b) criteria, he/she can be diagnosed as possible AIP only after the negative workup for malignancy by EUS-FNA, and confirmed as probable one by an optional steroid response
aPossible diagnosis: a case may be possibly type 2, although it is extremely rare in Japan
“+” refers to “and”, and “/” refers to “or”
Although the JPS-2011 is focused on type 1 AIP, some patients with type 2 AIP, which is extremely rare in Japan, may be diagnosed as possible AIP using these criteria. As ERCP is more commonly performed to diagnose AIP or pancreatic cancer than EUS-FNA in Japan, ERCP is essentially required in the diagnosis of the focal/segmental type of AIP. However, to follow the concept of the ICDC as much as possible, the following exceptional case can be deemed acceptable only by an expert: when a patient with a focal/segmental image of AIP on CT/MRI without ERCP findings fulfills more than one of III (serum IgG4), IVb (two of pathological findings) and V(a/b) (OOI), he can be diagnosed as possible AIP only after the negative workup for malignancy by EUS-FNA and AIP is confirmed as probable by an optional steroid response.
Diagnostic criteria for IgG4-related sclerosing cholangitis
Primary sclerosing cholangitis and cholangiocarcinoma must be discriminated from IgG4-SC. The diagnosis of IgG4-SC is based on the combination of the following criteria [66] (Table 8): (i) characteristic biliary imaging findings; (ii) elevation of serum IgG4 concentrations; (iii) coexistence of IgG4-RDs except those of the biliary tract; and (iv) characteristic histopathological features. It is difficult to obtain sufficient biliary tract tissue to determine the characteristic histology of IgG4-SC by biopsy [67]. Similar to the ICDC or JPS-2011 diagnostic criteria for AIP, the effectiveness of steroid therapy is an optional additional diagnostic criterion to confirm an accurate diagnosis of IgG4-SC.
Table 8.
Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012 (from [66], with permission)
| Diagnostic items |
|---|
| (1) Biliary tract imaging reveals diffuse or segmental narrowing of the intrahepatic and/or extrahepatic bile duct associated with the thickening of bile duct wall |
| (2) Hematological examination shows elevated serum IgG4 concentrations (≥135 mg/dl) |
| (3) Coexistence of autoimmune pancreatitis, IgG4-related dacryoadenitis/sialadenitis, or IgG4-related retroperitoneal fibrosis |
| (4) Histopathological examination shows: |
| a. Marked lymphocytic and plasmacyte infiltration and fibrosis |
| b. Infiltration of IgG4-positive plasma cells:[10 IgG4-positive plasma cells/HPF |
| c. Storiform fibrosis |
| d. Obliterative phlebitis |
| <Option> effectiveness of steroid therapy |
| A specialized facility, in which detailed examinations such as endoscopic biliary biopsy and endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) can be administered, may include in its diagnosis the effectiveness of steroid therapy, once pancreatic or biliary cancers have been ruled out. |
| Diagnosis |
| Definite diagnosis |
| (1) + (3) |
| (1) + (2) + (4) a, b |
| (4) a, b, c |
| (4) a, b, d |
| Probable diagnosis |
| (1) + (2) + option |
| Possible diagnosis |
| (1) + (2) |
It is necessary to exclude PSC, malignant diseases such as pancreatic or biliary cancers, and secondary sclerosing cholangitis caused by the diseases with obvious pathogenesis. When it is difficult to differentiate from malignant conditions, a patient must not be treated with facile steroid therapy, but should be referred to a specialized medical facility
Algorithm for diagnosing IgG4-RD in combination with the comprehensive and individual organ diagnostic criteria
A diagnostic algorithm for IgG4-RD [5], using comprehensive diagnostic criteria combined with organ-specific criteria, is shown in Fig. 2. Table 9 shows a comparison of minimal pathological criteria, comprehensive diagnostic criteria, and individual specific criteria in the hepato-bilio-pancreatic system for IgG4-RD. A diagnosis of IgG4-RD is definitive in patients with: (i) organ enlargement, mass or nodular lesions, or organ dysfunction; (ii) a serum IgG4 concentration of 135 mg/dl or higher; and (iii) histopathological findings of greater than 10 IgG4 cells per HPF and an IgG4+/IgG+ cell ratio greater than 40 percent. A diagnosis of IgG4-RD is possible in patients who fulfill criteria (i) and (ii), but with negative results on histopathology or without histopathologic examination, whereas a diagnosis of IgG4-RD is probable in patients with organ involvement (i) and fulfilled histopathologic criteria (iii), but without increased serum IgG4 concentration (ii). Patients with organ symptoms but without satisfying serologic or histopathologic criteria are considered unlikely to have IgG4-RD. For possible or probable cases, organ-specific criteria for IgG4-RD could be applied, such as those for AIP [41, 56, 64, 65], MD [68], and KD [69] associated with IgG4. Patients who fulfill the organ-specific criteria for IgG4-RD have a definite diagnosis of this disease.
Fig. 2.
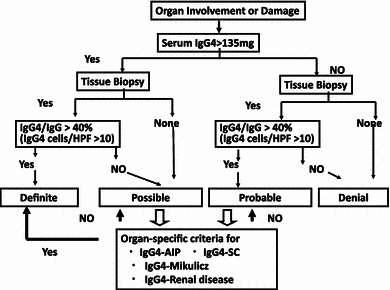
Algorism for Diagnosis of IgG4RD (reproduced from [5], with permission) Diagnostic algorithm performance for comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD) using comprehensive diagnostic criteria combined with organ-specific criteria. A diagnosis of IgG4-RD is definitive in patients with (1) organ enlargement, mass or nodular lesions, or organ dysfunction, (2) a serum IgG4 concentration >135 mg/dl, and (3) histopathological findings of >10 IgG4+ cells/HPF and an IgG4/IgG cell ratio >40 %
Table 9.
Comparison of diagnostic criteria among pathological, comprehensive and individual organ manifestation for IgG4-RD
| Diagnostic criteria (Ref.) | Clinical findings/images | Serology histology (Serum IgG4) | Histology | OOI | Efficacy of Steroid |
|---|---|---|---|---|---|
| Minimal Criteria by the International Pathological Consensus [6] | ND | High | Characteristic histopathological findings with an elevated IgG4t plasma cells and IgG4-to-IgG ratio | Reports of OOI consistent with IgG-RD | Yes |
| Comprehensive Diagnostic Criteria (CDC) for IgG4-RD [5] | Diffuse/localized swelling or masses in single or multiple organs | ≤135 mg/dl | (i) marked lymphocyte and plasmacyte infiltration and fibrosis | ND | No |
| (ii) ratio of IgG4+/IgG+ cells >40 % and >10 IgG4+ plasma cells/HPF | |||||
| ICDC for type1 AIP [41] | (i) parenchymal image on CT/MRI | LPSP | a. Histology of OOI | Yes | |
| (i) Lymphoplasmacyte infiltrate | |||||
| b. Radiological evidence | |||||
| (ii) >10 IgG4+ cells (/hpf) | |||||
| (ii) duct image on ERP | |||||
| (iii) Storiform fibrosis | |||||
| (iv) Obliterative phlebitis | |||||
| Level 1 | Diffuse | >2 × ULN | More than 3 (i-iv) | a. any three (i-iv); b. typical one | |
| Level 2 | Segmental/focal | 1–2 × ULN | Any 2 | a. two (i + ii); b or physical | |
| JPS-2011 [64, 65] | Enlarged pancreas on CT/MRI | ≥135 mg/dl | (i) Lymphoplasmacyte infiltrate | Sclerosing cholangitis, Dacryoadenitis/sialoadenitis | |
| Diffuse enlargement | (ii) >10 IgG4+ cells (/hpf) | ||||
| Segmental/focal enlargement Irregular narrowing of the MPD on ERP | (iii) Storiform fibrosis | Retroperitoneal fibrosis | |||
| (iv) Obliterative phlebitis | (a) Clinical lesions | ||||
| (b) Pathological lesions | |||||
| Clinical Diagnostic Criteria for IgG4-related sclerosing cholangitis 2012 [66] | (i) Narrowing of the intrahepatic/extrahepatic bile duct (Diffuse/Segmental) | ≥135 mg/dl | (i) Lymphoplasmacyte infiltrate | AIP | Yes |
| Dacryoadenitis/sialoadenitis Retroperitoneal fibrosis | |||||
| (ii) >10 IgG4+ cells (/hpf) | |||||
| (ii) Thickening of bile duct wall | (iii) Storiform fibrosis | ||||
| (iv) Obliterative phlebitis | |||||
Conclusion
Current concepts and clinical diagnostic criteria for IgG4-RD and individual organ manifestations are mentioned. The Comprehensive Diagnostic Criteria for IgG4-RD have been proposed for the general and specific criteria for individual organs. It is recommended that in cases of probable or possible IgG4-RD diagnosed by the CDC, organ specific diagnostic criteria should be concurrently used according to the diagnostic algorithm for IgG4-RD in Japan. When the precise mechanism in the development of IgG4-RD is clarified, establishment of the clinical diagnostic criteria for IgG4-RD should be needed in the near future.
Acknowledgments
This study was partly supported by a Grant-in-Aid for Scientific Research from the Ministry of Culture and Science of Japan (23591017), and a Grant-in-Aid for “Research for Intractable Disease” Program from the Ministry of Health, Labor and Welfare of Japan.
Conflict of interest
The authors declare that they have no conflict of interest.
Abbreviations
- AIP
Autoimmune pancreatitis
- CDC
Comprehensive diagnostic criteria
- GEL
Granulocytic epithelial lesion
- ICDC
International consensus diagnostic criteria
- IDCP
Idiopathic duct-centric pancreatitis
- IgG4-RD
IgG4-related disease
- IgG4-SC
IgG4-related sclerosing cholangitis
- IPC
The international pathologic consensus criteria
- JPS
Japan Pancreas Society
- LPSP
Lymphoplasmacytic sclerosing pancreatitis
- MD
Mikulicz’s disease
- MOLPS
Multiorgan lymphoproliferative disease
- OOI
Other organ involvement
- SjS
Sjögren’s syndrome
- PSC
Primary sclerosing cholangitis
- RF
Rheumatoid factor
- SIPS
Systemic IgG4-related plasmacytic syndrome
References
- 1.Okamoto T, Kamisawa A. Autoimmune pancreatitis: proposal of IgG4-related sclerosing disease. J Gastroenterol. 2006;41:613–625. doi: 10.1007/s00535-006-1851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamamoto M, Takahashi H, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, Shinomura Y, Imai K. A new conceptualization for Mikulicz’s disease as an IgG4-related plasmacytic disease. Mod Rheumatol. 2006;16:335–340. doi: 10.1007/s10165-006-0518-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masaki Y, Dong L, Kurose N, Kitagawa K, Morikawa Y, Yamamoto M, et al. Proposal for a new clinical entity, IgG4-positive multi-organ lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis. 2009;68:1310–1315. doi: 10.1136/ard.2008.089169. [DOI] [PubMed] [Google Scholar]
- 4.Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22(1):1–14. doi: 10.1007/s10165-011-0508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22(1):21–30. doi: 10.1007/s10165-011-0571-z. [DOI] [PubMed] [Google Scholar]
- 6.Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–1192. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 7.Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthr Rheum. 2012;64(10):3061–3067. doi: 10.1002/art.34593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okazaki K, Uchida K, Koyabu M, Miyoshi H, Takaoka M. Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. J Gastroenterol. 2011;46(3):277–288. doi: 10.1007/s00535-011-0386-x. [DOI] [PubMed] [Google Scholar]
- 9.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 10.Okazaki K, Kawa S, Kamisawa T, Ito T, Inui K, Irie H, et al. Japanese clinical guidelines for autoimmune pancreatitis. Pancreas. 2009;38:849–866. doi: 10.1097/MPA.0b013e3181b9ee1c. [DOI] [PubMed] [Google Scholar]
- 11.Nakazawa T, Ohara H, Yamada T, Ando H, Sano H, Kajino S, et al. Atypical primary sclerosing cholangitis cases associated with unusual pancreatitis. Hepatogastroenterology. 2001;48:625–630. [PubMed] [Google Scholar]
- 12.Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982–4. [DOI] [PubMed]
- 13.Kishimoto M, Okimura Y, Kimura K, et al. Multifocal fibrosclerosis as a possible cause of panhypopituitarism with central diabetes insipidus. Endocr J. 2000;47(3):335–342. doi: 10.1507/endocrj.47.335. [DOI] [PubMed] [Google Scholar]
- 14.Shimatsu A, Oki Y, Fujiswa I, Sano T. Pituitary and stalk lesions (Infundibulo-hypophysitis) associated with immunoglobulin G4-related systemic disease: an emerging clinical entity. Endocr J. 2009;56(9):1033–1041. doi: 10.1507/endocrj.K09E-277. [DOI] [PubMed] [Google Scholar]
- 15.Komatsu K, Hamano H, Ochi Y, Takayama M, Muraki T, Yoshizawa K, et al. High prevalence of hypothyroidism in patients with autoimmune pancreatitis. Dig Dis Sci. 2005;50:1052–1057. doi: 10.1007/s10620-005-2703-9. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Nishihara E, Hirokawa M, Taniguchi E, Miyauchi A, Kakudo K. Distinct clinical, serological, and sonographic characteristics of Hashimoto’s thyroiditis based with and without IgG4-positive plasma cells. J Clin Endocrinol Metab. 2010;95:1309–1317. doi: 10.1210/jc.2009-1794. [DOI] [PubMed] [Google Scholar]
- 17.Zen Y, Inoue D, Kitao A, Onodera M, Abo H, Miyayama S, et al. IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol. 2009;33(12):1886–1893. doi: 10.1097/PAS.0b013e3181bd535b. [DOI] [PubMed] [Google Scholar]
- 18.Matsui S, Taki H, Shinoda K, Suzuki K, Hayashi R, Tobe K, et al. Respiratory involvement in IgG4-related Mikulicz’s disease. Mod Rheumatol. 2012;22(1):31–39. doi: 10.1007/s10165-011-0504-x. [DOI] [PubMed] [Google Scholar]
- 19.Umemura T, Zen Y, Hamano H, Kawa S, Nakanuma Y, Kiyosawa K. Immunoglobin G4-hepatopathy: association of immunoglobin G4-bearing plasma cells in liver with autoimmune pancreatitis. Hepatology. 2007;46(2):463–471. doi: 10.1002/hep.21700. [DOI] [PubMed] [Google Scholar]
- 20.Umemura T, Zen Y, Hamano H, Joshita S, Ichijo T, Yoshizawa K, et al. Clinical significance of immunoglobulin G4-associated autoimmune hepatitis. J Gastroenterol. 2011;46(Suppl 1):48–55. doi: 10.1007/s00535-010-0323-4. [DOI] [PubMed] [Google Scholar]
- 21.Lopes J, et al. Autoimmune esophagitis: IgG4-related tumors of the esophagus. J Gastrointest Surg. 2010; 14:1031–34. [DOI] [PubMed]
- 22.Uehara T, Hamano H, Kawa S, Sano K, Oki K, Kobayashi Y, et al. Chronic gastritis in the setting of autoimmune pancreatitis. Am J Surg Pathol. 2010;34(9):1241–1249. doi: 10.1097/PAS.0b013e3181ec07ee. [DOI] [PubMed] [Google Scholar]
- 23.Ravi K, Chari ST, Vege SS, Sandborn WJ, Smyrk TC, Loftus EV., Jr Inflammatory bowel disease in the setting of autoimmune pancreatitis. Inflamm Bowel Dis. 2009;15(9):1326–1330. doi: 10.1002/ibd.20898. [DOI] [PubMed] [Google Scholar]
- 24.Ueno K, Watanabe T, Kawata Y, Gotoh T, Tsuji Y, Ida H, et al. IgG4-related autoimmune pancreatitis involving the colonic mucosa. Eur J Gastroenterol Hepatol. 2008;20(11):1118–1121. doi: 10.1097/MEG.0b013e3282f82970. [DOI] [PubMed] [Google Scholar]
- 25.Uchiyama-Tanaka Y, Mori Y, Kimura T, Watanabe S, Nozaki Y, Fujita K, et al. Acute tubulointerstitial nephritis associated with autoimmune-related pancreatitis. Am J Kidney Dis. 2004;43:e18–e25. doi: 10.1053/j.ajkd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant. 2004;19:474–476. doi: 10.1093/ndt/gfg477. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimura Y, Takeda S, Ieki Y, Takazakura E, Koizumi H, Takagawa K. IgG4-associated prostatitis complicating autoimmune pancreatitis. Intern Med. 2006;45:897–901. doi: 10.2169/internalmedicine.45.1752. [DOI] [PubMed] [Google Scholar]
- 28.Nishimori I, Kohsaki T, Onishi S, Shuin T, Kohsaki S, Ogawa Y, et al. IgG4-related autoimmune prostatitis: two cases with or without autoimmune pancreatitis. Intern Med. 2007;46(24):1983–1989. doi: 10.2169/internalmedicine.46.0452. [DOI] [PubMed] [Google Scholar]
- 29.Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359:1403–1404. doi: 10.1016/S0140-6736(02)08359-9. [DOI] [PubMed] [Google Scholar]
- 30.Stone JH, Khosroshahi A, Hilgenberg A, Spooner A, Isselbacher EM, Stone JR. IgG4-related systemic disease and lymphoplasmacytic aortitis. Arthritis Rheum. 2009;60(10):3139–3145. doi: 10.1002/art.24798. [DOI] [PubMed] [Google Scholar]
- 31.Saegusa H, Momose M, Kawa S, Hamano H, Ochi Y, Takayama M, et al. Hilar and pancreatic gallium-67 accumulation is characteristic feature of autoimmune pancreatitis. Pancreas. 2003;27:20–25. doi: 10.1097/00006676-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Sato Y, Takeuchi M, Takata K, Ohno K, Iwaki N, Orita Y, Goto N, et al. Clinicopathologic analysis of IgG4-related skin disease. Mod Pathol. doi:10.1038/modpathol.2012. [DOI] [PubMed]
- 33.Cheuk W, Chan AC, Lam WL, Chow SM, Crowley P, Lloydd R, et al. IgG4-related sclerosing mastitis: description of a new member of the IgG4-related sclerosing diseases. Am J Surg Pathol. 2009;33(7):1058–1064. doi: 10.1097/PAS.0b013e3181998cbe. [DOI] [PubMed] [Google Scholar]
- 34.Mikulicz J. Über eine eigenartige symmetrishe Erkrankung der Tränen und Mundspeicheldrüsen. Stuttgart: Beitr z Chir Fesrschr f Theodor Billroth; 1892. pp. 610–630. [Google Scholar]
- 35.Küttner H. Ü ber entzündiche Tumoren der submaaxilla¨ren Speicheldru¨se. Beitr Klin Chir. 1896;15:815–834. [Google Scholar]
- 36.Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas–an autonomous pancreatic disease? Am J Dig Dis. 1961;6:688–698. doi: 10.1007/BF02232341. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40:1561–1568. doi: 10.1007/BF02285209. [DOI] [PubMed] [Google Scholar]
- 38.Kawaguchi K, Koike M, Tsuruta K, Okamoto A, Tabata I, Fujita N. Lymphoplasmacytic sclerosing pancreatitis with cholangitis: a variant of primary sclerosing cholangitis extensively involving pancreas. Hum Pathol. 1991;22:387–395. doi: 10.1016/0046-8177(91)90087-6. [DOI] [PubMed] [Google Scholar]
- 39.Comings DE, Skubi KB, Van Eyes J, Motulsky AG. Familial multifocal fibrosclerosis. Ann Intern Med. 1967;66:884–892. doi: 10.7326/0003-4819-66-5-884. [DOI] [PubMed] [Google Scholar]
- 40.Chari ST, Kloeppel G, Zhang L, Notohara K, Lerch MM, Shimosegawa T, et al. Histopathologic and clinical subtypes of autoimmune pancreatitis: the Honolulu consensus document. Pancreas. 2010;39:549–554. doi: 10.1097/MPA.0b013e3181e4d9e5. [DOI] [PubMed] [Google Scholar]
- 41.Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, et al. International consensus diagnostic criteria for autoimmune pancreatitis: Guidelines of the International Association of Pancreatology. Pancreas. 2011;40(3):352–358. doi: 10.1097/MPA.0b013e3182142fd2. [DOI] [PubMed] [Google Scholar]
- 42.Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol. 2003;27:1119–1127. doi: 10.1097/00000478-200308000-00009. [DOI] [PubMed] [Google Scholar]
- 43.Zamboni G, Luttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch. 2004;445:552–563. doi: 10.1007/s00428-004-1140-z. [DOI] [PubMed] [Google Scholar]
- 44.Klöppel G, Detlefsen S, Chari ST, Longnecker DS, Zamboni G. Autoimmune pancreatitis: the clinicopathological characteristics of the subtype with granulocytic epithelial lesions. J Gastroenterol. 2010;45:787–793. doi: 10.1007/s00535-010-0265-x. [DOI] [PubMed] [Google Scholar]
- 45.Okazaki K, Kawa S, Kamisawa T, Shimosegawa T, Tanaka M, et al. Japanese consensus guidelines for management of autoimmune pancreatitis: I. Concept and diagnosis of autoimmune pancreatitis. J Gastroenterol. 2010;45:249–265. doi: 10.1007/s00535-009-0184-x. [DOI] [PubMed] [Google Scholar]
- 46.Kawa S, Okazaki K, Kamisawa T, Shimosegawa T, Tanaka M, et al. Japanese consensus guidelines for management of autoimmune pancreatitis: II. Extrapancreatic lesions, differential diagnosis. J Gastroenterol. 2010;45:355–369. doi: 10.1007/s00535-009-0197-5. [DOI] [PubMed] [Google Scholar]
- 47.Kamisawa T, Okazaki K, Kawa S, et al. Japanese consensus guidelines for management of autoimmune pancreatitis: III. Treatment and prognosis of AIP. J Gastroenterol. 2010;45:471–477. doi: 10.1007/s00535-010-0221-9. [DOI] [PubMed] [Google Scholar]
- 48.Zen Y, Harada K, Sasaki M, Sato Y, Tsuneyama K, Haratake J, et al. IgG4-related sclerosing cholangitis with and without hepatic inflammatory pseudotumor, and sclerosing pancreatitis associated sclerosing cholangitis: do they belong to a spectrum of sclerosing pancreatitis? Am J Surg Pathol. 2004;28:1193–1203. doi: 10.1097/01.pas.0000136449.37936.6c. [DOI] [PubMed] [Google Scholar]
- 49.Naitoh I, Nakazawa T, Ohara H, Andoh T, Hayashi K, Tanaka H, et al. Endoscopic transpapillary intraductal ultrasonography and biopsy in the diagnosis of IgG4-related sclerosing cholangitis. J Gastroenterol. 2009;44:1147–1155. doi: 10.1007/s00535-009-0108-9. [DOI] [PubMed] [Google Scholar]
- 50.Nakazawa T, Ohara H, Yamada T, Ando H, Sano H, Kajino S, et al. Atypical primary sclerosing cholangitis cases associated with unusual pancreatitis. Hepatogastroenterology. 2001;48:625–630. [PubMed] [Google Scholar]
- 51.Nakazawa T, Ohara H, Sano H, Ando T, Aoki S, Kobayashi S, et al. Clinical differences between primary sclerosing cholangitis and sclerosing cholangitis with autoimmune pancreatitis. Pancreas. 2005;30:20–25. [PubMed] [Google Scholar]
- 52.Nishino T, Toki F, Oyama H, Oi I, Kobayashi M, Takasaki K, et al. Biliary tract involvement in autoimmune pancreatitis. Pancreas. 2005;30:76–82. [PubMed] [Google Scholar]
- 53.Hirano K, Tada M, Isayama H, Yagioka H, Sasaki T, Kogure H, et al. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut. 2007;56:1719–1724. doi: 10.1136/gut.2006.115246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–715. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 55.Strehl JD, Hartmann A, Agaimy A. Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol. 2011;64(3):237–243. doi: 10.1136/jcp.2010.085613. [DOI] [PubMed] [Google Scholar]
- 56.Okazaki K, Kawa S, Kamisawa T, et al. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol. 2006;41:626–631. doi: 10.1007/s00535-006-1868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Members of the Criteria Committee for Autoimmune Pancreatitis of the Japan Pancreas Society. Diagnostic criteria for autoimmune pancreatitis by the Japan Pancreas Society (2002). J Jpn Pancreas Soc (Suizou). 2002;17:585–7.
- 58.Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006;4:1010–1016. doi: 10.1016/j.cgh.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 59.Chari ST, Takahashi N, Levy MJ, et al. A diagnostic strategy to distinguish autoimmune pancreatitis from pancreatic cancer. Clin Gastroenterol Hepatol. 2009;7:1097–1103. doi: 10.1016/j.cgh.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 60.Kwon S, Kim M-H, Choi EK. The diagnostic criteria for autoimmune chronic pancreatitis: it is time to make a consensus. Pancreas. 2007;34:279–286. doi: 10.1097/MPA.0b013e31802eff5f. [DOI] [PubMed] [Google Scholar]
- 61.Otsuki M, Chung JB, Okazaki K, et al. Asian diagnostic criteria for autoimmune pancreatitis: consensus of the Japan-Korea Symposium on Autoimmune Pancreatitis. J Gastroenterol. 2008;43:403–408. doi: 10.1007/s00535-008-2205-6. [DOI] [PubMed] [Google Scholar]
- 62.Schneider A, Löhr JM. Autoimmune pancreatitis. Internist (Berl) 2009;50:318–330. doi: 10.1007/s00108-008-2262-1. [DOI] [PubMed] [Google Scholar]
- 63.Pearson RK, Longnecker DS, Chari ST, et al. Controversies in clinical pancreatology: autoimmune pancreatitis: does it exist? Pancreas. 2003;27:1–13. doi: 10.1097/00006676-200307000-00001. [DOI] [PubMed] [Google Scholar]
- 64.The Japan Pancreas Society, the Ministry of Health and Welfare Investigation Research Team for Intractable Pancreatic Disease. Clinical Diagnostic for Autoimmune Pancreatitis 2011 (Proposal) (in Japanese with English abstract). J Jpn Pancreas (Suizo). 2012;27:17–25.
- 65.Shimosegawa T. The working Group Members of the Japan Pancreas Society and the Research Committee for Intractable Pancreatic Disease by the Ministry of Labor, Health and Welfare of Japan. The Amendment of the Clinical Diagnostic Criteria in Japan (JPS2011) in Response to the Proposal of the International Consensus of Diagnostic Criteria (ICDC) for Autoimmune Pancreatitis. Pancreas. 2012;41(8):1341–2. [DOI] [PubMed]
- 66.Ohara H, Okazaki K, Tsubouchi H, Inui K, Kawa S, Kamisawa T, et al. Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012. J Hepatobiliary Pancreat Sci. 2012;19(5):536–542. doi: 10.1007/s00534-012-0521-y. [DOI] [PubMed] [Google Scholar]
- 67.Kawakami H, Zen Y, Kuwatani M, Eto K, Haba S, Yamato H, et al. IgG4-related sclerosing cholangitis and autoimmune pancreatitis: histological assessment of biopsies from Vateampulla and the bile duct. J Gastroenterol Hepatol. 2010;25:648–655. doi: 10.1111/j.1440-1746.2010.06346.x. [DOI] [PubMed] [Google Scholar]
- 68.Masaki Y, Sugai S, Umehara H. IgG4-related diseases including Mikulicz’s disease and sclerosing pancreatitis: diagnostic insights. J Rheum. 2010;37:1380–1385. doi: 10.3899/jrheum.091153. [DOI] [PubMed] [Google Scholar]
- 69.Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15(5):615–626. doi: 10.1007/s10157-011-0521-2. [DOI] [PubMed] [Google Scholar]