

Abstract
Antidepressants are among the most commonly prescribed drugs, and a range of medications are available. However, treatment response to a particular drug varies greatly between patients, with only 30% of patients responding well to the first treatment administered. Given evidence that antidepressant treatment response is a heritable trait, together with technological advances in genetic research, three recently published genome-wide investigations into antidepressant responses have examined the determinants of variability in treatment outcomes between depressed patients. Here, we review these studies within the context of wider research efforts to identify treatment response predictors. Some interesting genes have been implicated, but no variants have yet been robustly and reliably linked to response. This may suggest that genetic effect sizes are smaller than originally anticipated. Candidate gene approaches in these samples have lent support to the involvement of serotonergic, glutamatergic and stress-response systems in treatment response, although corroborative evidence from genome-wide analyses indicates these results should be interpreted cautiously. Closer examination of antidepressant response, considering it as a complex trait, has indicated that multiple genes of small effect are likely to be involved. Furthermore, there is some evidence that genetic influence on response to treatment may vary between patients with different symptom profiles or environmental exposures. This has implications for the translation of pharmacogenetic findings into clinical practice: genotypic information from multiple loci and data on non-genetic factors are likely to be needed to tailor antidepressant treatment to the individual patient.
Keywords: Antidepressants, genome-wide analysis, personalized treatment, pharmacogenetics, pharmacogenomics, treatment response
The genetics of antidepressant response
Depression is a serious and prevalent psychiatric disorder, and, while there are a range of treatment options available, there is a high degree of variability between patients in terms of their response to a particular treatment. Genes are likely to play an important role in this variability, and with the rapid pace of technological development in the field of genetics there is a growing interest in using pharmacogenetic approaches to identify predictors of antidepressant response.
This review will focus on the three large genome-wide analyses of antidepressant response that have recently been published, and consider the findings within the context of wider research efforts to identify treatment response predictors. While genetic effect sizes appear to be smaller than originally anticipated, analyses considering possible interactions between both genetic and environmental factors, as well as methods that attempt to address the symptomatic heterogeneity of depression, may point the way to fruitful new research avenues for identifying clinically valuable predictors of treatment response.
Depressive disorders and diagnosis
Depression is a common and disabling illness with a lifetime prevalence of up to 17% [1]. The World Health Organization projects that by 2020 depression will be the second leading contributor to the global burden of disease [2]. The disorder is characterized by low mood, loss of interest and reduced energy. Depression is also associated with cognitive symptoms such as reduced concentration, low self-esteem and suicidal ideations, as well as somatic symptoms such as early morning wakening, and loss of appetite and libido. There is a relatively high degree of symptomatic heterogeneity between depressed patients, with some showing 'atypical' features such as increased sleep and appetite.
Both the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV) [3], and the International Classification of Diseases, 10th revision (ICD-10) [4], give classification criteria for depression (Table 1). The disorder is considered a single diagnostic entity, and the separation of depression into 'neurotic' and 'endogenous' subtypes has fallen out of favor. However, other additional specifiers can be used within both classification systems to more precisely describe patients. To establish if a patient fulfils the criteria for depression as defined in DSM-IV or ICD-10, the majority of research studies use structured or semistructured diagnostic interviews such as the Schedules for Clinical Assessment in Neuropsychiatry [5] or the Composite International Diagnostic Interview [6]. These methods attempt to achieve both diagnostic validity and reliability.
Table 1.
Symptoms and classification of depression
| Depressive symptoms | DSM-IV classification of depressive episode | ICD-10 classification of depressive episode |
|---|---|---|
| (1) Depressed mood for at least 2 weeks | Five or more symptoms, including (1) or (2) | Mild: four or more symptoms, including two of (1), (2) or (3)Moderate: six or more symptoms, including two of (1), (2) or (3)Severe: eight or more symptoms, including (1), (2) and (3) |
| (2) Loss of interest and enjoyment | ||
| (3) Increased fatigability | ||
| (4) Loss of confidence/self-esteema | ||
| (5) Self-reproach/guilt | ||
| (6) Suicidal thoughts or intent | ||
| (7) Reduced concentration/indecisiveness | ||
| (8) Agitation | ||
| (9) Sleep disturbance | ||
| (10) Altered appetite | ||
| Course | Single episode or recurrent | Single episode or recurrent |
| Additional specifiers | With/without psychotic featuresb | With/without psychotic featuresb (severe depression only) |
| With/without catatonic features | With/without somatic symptoms | |
| With/without atypical features | ||
| With/without postpartum onset |
Nevertheless, definitions of depression should not be considered absolute or immutable; there is continued debate over the best way to understand and define the illness. Indeed there is an ongoing research effort to confirm whether depression is best considered as a homogeneous clinical entity, given the variability seen between patients in terms of symptoms, course of illness and treatment response.
In order to measure symptoms and establish treatment response over time, numerous assessment tools are available, including clinician-rated scales such as the Hamilton Depression Rating Scale (HAM-D) [7], as well as self-report alternatives such as the Beck Depression Inventory (BDI) [8]. However, there are differences in the items included in each of these scale, as well as variability in the weightings given to certain symptoms, reflecting the lack of consensus regarding the core features of depression.
Antidepressant treatment options
There are various treatment options for depressed patients, including pharmacotherapy, psychological treatments and, in more severe cases, electroconvulsive therapy. To date, however, the majority of work looking at genetic predictors of response has focused on pharmacological treatments. The main classes of commonly used antidepressant medications are tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors (SNRIs) (Table 2).
Table 2.
Commonly used antidepressant medications
| Antidepressant class | Examples | Mode of action | Adverse effects |
|---|---|---|---|
| Tricyclic antidepressants (TCAs) | Nortriptyline, amitriptyline, imipramine, dothiepin | Block norepinephrine and serotonin reuptake transporters; also antagonize serotoninergic, histaminergic, adrenergic and cholinergic receptors | Common side effects include dry mouth and constipation; cardiovascular issues can occur; fatal in overdose |
| Selective serotonin reuptake inhibitors (SSRIs) | Fluoxetine, sertraline, paroxetine, citalopram, escitalopram | Block serotonin reuptake transporters; more selective than TCAs | Common side effects include nausea, sexual dysfunction; lower cardiotoxicity than TCAs (therefore safer in overdose) |
| Serotonin-norepinephrine reuptake inhibitors (SNRIs) | Venlafaxine, duloxetine | Block norepinephrine and serotonin reuptake transporters; more selective than TCAs | Common side effects similar to SSRIs; also associated with increased blood pressure |
TCAs act by blocking both norepinephrine and serotonin transporters. This inhibits reuptake of these two neurotransmitters into the presynaptic neuron, so increasing available levels in the synaptic cleft to bind to postsynaptic receptors. TCAs also act as antagonists at serotonergic, histaminergic, cholinergic and adrenergic receptors. However, these actions are associated with commonly reported side effects (such as dry mouth and constipation). Furthermore, in some cases TCAs can cause cardiovascular issues and be fatal in overdose.
Given these adverse effects, SSRIs were developed to target the serotonin transporter much more selectively. SSRIs are widely used in depression, and have replaced TCAs as the first line of treatment for depression due to their more limited side-effect profile and improved safety. Nevertheless, TCAs remain useful for patients who do not respond to newer antidepressants. The dual-action SNRIs have similar selectivity and side-effect profiles to SSRIs, but inhibit the reuptake of both serotonin and norepinephrine.
The antidepressants described above have comparable average efficacy [9]. However, individuals vary widely in their response to treatments, with only an estimated 30% of patients responding sufficiently to their first treatment [10,11]. Antidepressants have a delayed onset of action, and it can take up to 8 weeks to assess treatment response [11] and, prior to commencing treatment, it is unclear which drug will produce an optimal response for a particular patient. Given the time and costs required to identify the most effective medication, together with the negative impact of multiple treatment steps (which are associated with increased risk of relapse [11]), there is considerable clinical value in identifying predictors of treatment response.
The role of genetics in determining antidepressant response
While it is logistically difficult to collect the samples required to investigate whether antidepressant treatment response is a heritable trait, family studies looking at response to antidepressants indicate that variability in response to these medications is likely to be at least partially genetic in nature [12,13]. Given this evidence, a growing number of studies are investigating which genes might determine antidepressant treatment response. This research has the dual aims of better understanding how these drugs exert their therapeutic action, and enabling individualized treatment recommendations.
Pharmacogenomic approaches
One key recent development within the rapidly evolving area of genetic research is the ability to perform genome-wide tests of association, where variation across the entire genome is explored in relation to a particular phenotype (in this case, antidepressant treatment response). Genotyping is performed at 500,000 to upwards of 1 million single nucleotide polymorphisms (SNPs), as made possible by commercially available microarray technology (for example: Affymetrix, South San Francisco, CA, USA; Illumina Inc., San Diego, CA, USA). Genomic approaches allow researchers to explore genetic variation in a hypothesis-free fashion, with no requirement to preselect genes of interest. If previously unsuspected genes are found to be associated with variability in treatment response through these systematic analyses, 'pharmacogenomics' has the potential to push forward scientific understanding of how antidepressants work. To date there have been three key genome-wide association studies (GWASs) of antidepressant treatment response [14-16], as summarized in Figure 1.
Figure 1.
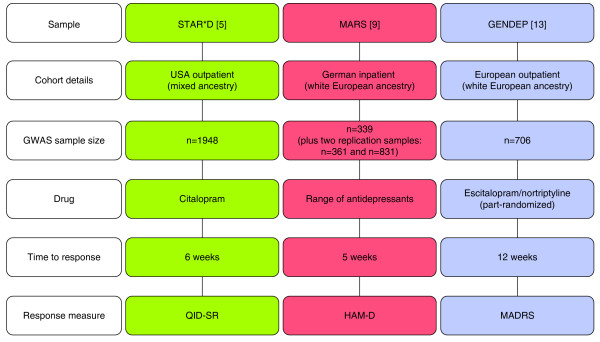
Summary of the study designs for the three genome-wide association studies of antidepressant treatment response published to date. The details of each GWAS are included, with population ancestry, study size, antidepressant treatment used, treatment response time and outcome measure [5,9,13]. The differences between studies may play some role in the lack of overlap in results between studies, although a more major factor is likely to be limitations in statistical power to detect smaller genetic effects. GWAS, genome-wide association study; HAM-D, Hamilton Depression Rating Scale; MADRS, Montgomery-Asberg Depression Rating scale; QID-SR, Quick Inventory of Depressive Symptomatology Self-Report version.
Sequenced Treatment Alternatives to Relieve Depression
The US-based Sequenced Treatment Alternatives to Relieve Depression (STAR*D) project [17] enrolled a total of 4,041 treatment-seeking adults. All patients included in the study had a diagnosis of non-psychotic major depressive disorder (as confirmed by DSM-IV criteria), scoring greater than 14 on the HAM-D (that is, suffered from moderate to severe depression). Among the sample, there were high rates of chronic or recurrent depression. STAR*D had a multistep design; all patients received citalopram (an SSRI) in the first treatment level, then those without sufficient symptomatic benefit moved onto further treatment levels where they received either alternative or augmented treatment options.
STAR*D is the largest and longest study to ever be conducted evaluating antidepressant treatments. It was found that with first-level treatment with citalopram, approximately 30% of patients achieved remission. While the study was not expressly designed for pharmacogenetic purposes, DNA was collected from 1,953 patients, allowing a GWAS of treatment outcomes [15].
Looking at those patients who were enrolled on the trial for at least 6 weeks, the investigators elected to explore two outcome phenotypes: response and remission to citalopram treatment. Treatment response was defined as a greater than 50% reduction in scores on the Quick Inventory of Depressive Symptomatology Self-Report version (QIDS-SR) [18] at the patient's final visit, giving 883 responders and 608 non-responders. Remission was defined as scoring 5 or less on QIDS-SR at follow-up, and patients achieving remission (n = 743) were compared against those classified as non-responders. Genotyping was performed on either the Affymetrix 500K or 5.0 Human SNP arrays, and 430,198 SNPs were used in the genome-wide analysis. The study was designed to be representative of the ancestral diversity within the US population and so stringent statistical controls for population stratification were required.
Three SNPs were associated with treatment response at a suggested level of significance (P <1 × 10-5), and these same SNPs were also among the top 25 hits associated with the remission phenotype (however, it should be noted that remitters represent a subgroup of responding patients). Rs6966038, which is located near the ubiquitin protein ligase E3C gene (UBE3C; involved in protein ubiquitination to promote degradation [19]), was associated with response at P = 4.65 × 10-7 and remission at P = 3.63 × 10-7. Rs6127921 is closest in location to the bone morphogenic protein 7 gene (BMP7; linked to bone development), and was associated with response at P = 3.45 × 10-6 and remission at P = 1.07 × 10-6. Finally, rs809736 is an intronic SNP in the RAR-related orphan receptor alpha gene (RORA; a nuclear receptor [20]) and was associated with response at P = 8.19 × 10-6 and remission at P = 7.64 × 10-5. There was no evidence that treatment side effects might confound the above findings.
Munich Antidepressant Response Signatures
The Munich Antidepressant Response Signatures (MARS) study is a German pharmacogenomic project, with genome-wide data available for 339 inpatients with either major depression or bipolar disorder (11.2%) [14]. The majority of patients were German (85.1%), and all reported white European ancestry. The study was naturalistic in design; patients were treated with a range of antidepressants. Depression symptoms were measured using the HAM-D [7] and three outcome phenotypes were considered: early partial response (HAM-D score improvement greater than 25% after 2 weeks), response (greater than 50% improvement in scores after 5 weeks) and remission (a score of <10 on the HAM-D after 5 weeks or before hospital discharge). Genotyping was performed using the Sentrix Human-1 and HumanHap300 arrays. While MARS was a smaller study than STAR*D, the genome-wide analysis not only used a replication sample of 361 German inpatients, but also a subsample of 832 white outpatients from the STAR*D study to further validate findings.
The most significant association (P = 7.6 × 10-7) was seen between early partial response and rs6989467, a SNP in the 5' region of the cadherin 17 gene (CDH17; encoding a calcium-dependent membrane-associated glycoprotein). Looking across all three phenotypes, the SNP most consistently associated with treatment outcome was rs1502174 (P = 8.5 × 10-5), located downstream of EPHB1 (encoding ephrin type-B receptor 1 protein, a transmembrane protein involved in developmental processes in the nervous system).
Genome Based Therapeutic Drugs for Depression
The Genome Based Therapeutic Drugs for Depression (GENDEP) study is a European, multicenter, 12-week study with two active pharmacological treatment arms (for further details see [21]). A total of 811 outpatients with moderate to severe depression received one of two antidepressants, nortriptyline (a TCA) or escitalopram (an SSRI), in a partially randomized design. The primary outcome measure was the clinician-rated Montgomery-Asberg Depression Rating scale (MADRS) [22], on which each patient was measured weekly. For the pharmacogenomic analysis [16], treatment response was defined using percentage change in the MADRS from baseline to week 12 of the study and genome-wide data were available (using the Illumnia Human610 Quad chip) for 706 patients, 394 of whom were taking escitalopram and 312 nortriptyline. In a whole-sample analysis, associations at a suggestive level of significance were seen in intergenic regions on chromosome 1 (rs2136093, rs6701608, rs2136094) and chromosome 10 (rs16920624, rs11598854, rs7081156). However, when drug-specific analyses were performed, a genome-wide significant association with nortriptyline response was seen with an intronic SNP (rs 2500535) contained within the UST gene. UST encodes uronyl-2-sulphotransferase, an enzyme with an important link to neurotrophic mechanisms [23]. This SNP also reached suggested levels of significance when testing gene-drug interactions (that is, the SNP differentially predicted treatment outcome with escitalopram and nortriptyline). The best performing SNP within the escitalopram-specific analysis was rs1126757 (P = 2.83 × 10-6), a synonymous SNP within the coding region of the IL11 (interleukin 11) gene. Interleukins are neuropoietic cytokines involved in inflammatory responses, and inflammation processes have been associated with depression [24].
Across these three pharmacogenomic studies, only one genome-wide significant finding was observed (the relationship between UST and response to nortriptyline in GENDEP), and, as can been seen from Table 3, there is no overlap between the three samples in the observed suggestive genetic signals. This may be due to differences in sample characteristics (inpatients versus outpatients, variation in cohort ancestry), the antidepressant treatment received (medication taken, length of time receiving drug), or analysis approach (treatment outcome definitions used). Alternatively, this could simply reflect the role of chance; even with true genetic associations, when looking at P-values for over 500,000 markers, the 'top hits' of a genome-wide study can be highly unstable. An additional consideration when assessing pharmacogenomic research is that sample sizes are still very small in comparison with cohorts collected in the field of disease genetics. Studies looking at drug-response phenotypes are limited by the greatly increased cost of sample collection (for example, due to the requirement of following patients over a period of time to assess treatment response) and so the cohorts collected to date may not be sufficiently large to detect the genetic effects that influence treatment response at a genome-wide level of significance.
Table 3.
Genome-wide association studies of antidepressant treatment response
| Sample | GWAS sample size (N) | Microarray used | Primary outcome measure | Response phenotype definition | SNPs reaching suggestive levels of significance (nearest gene) | |
|---|---|---|---|---|---|---|
| STAR*D [5] | 1,948 | Affymetrix 500K/5.0 human SNP arrays | QID-SR | Response: >50% reduction in score | rs6966038 (UBE3C), rs6127921 (BMP7), rs809736 (RORA) | |
| Remission: score <5 | ||||||
| MARS [9] | 339 (two replication samples, N=361 and N=831) | Sentrix Human-1 and HumanHap300 BeadChip | HAM-D | Early partial response: >25% reduction in score after 2 weeks of treatment | rs6989467 (CDH17)a, rs1502174 (EPHB1) | |
| Response: >50% reduction in score | ||||||
| Remission: score <10 | ||||||
| GENDEP [13] | 706 | Illumina 610 array | MADRS | Percentage change from baseline to week 12 | Whole-sample analysis | rs2136093, rs6701608 and rs2136094 (intergenic, on chromosome 1)rs16920624, rs11598854 and rs7081156 (intergenic, on chromosome 10) |
| Escitalopram-specific analysis | rs1126757 (IL11) | |||||
| Nortriptyline-specific analysis | rs2500535 (UST) | |||||
| Gene-by-drug analysis | rs2500535 (UST) | |||||
aOnly associated with early partial response. GWAS, genome-wide association study; HAM-D, Hamilton Depression Rating Scale; MADRS, Montgomery-Asberg Depression Rating scale; QID-SR, Quick Inventory of Depressive Symptomatology Self-Report version; SNP, single nucleotide polymorphism.
Candidate gene approaches
By testing for genetic association at over 500,000 SNPs, genome-wide analyses must compensate for the very large number of hypotheses tested with an appropriate adjustment of the significance thresholds used (generally set at P < 5 × 10-8) [25]. Aiming to avoid this multiple hypothesis testing burden, candidate gene studies preselect appropriate genes of interest. Although this hypothesis-driven method cannot reveal novel treatment pathways, this approach is better powered to detect genes with smaller effect sizes. There is a lot of published literature on candidate genes in antidepressant response. However, candidate gene studies are vulnerable to publication bias, where the same few 'favorite' genes have been repeatedly tested in many small cohorts, with only the more interesting (generally significant) results being published. Therefore, when interpreting results, key considerations are replication and sample size (as emphasized by Horstmann and Binder [26]). Given this, the focus is placed on replicated candidate gene findings emerging from large cohort studies (Table 4). We describe findings for two genes involved in serotonergic pathways (SLC6A4 and HTR2A), one gene linked to the hypothalamic-pituitary-adrenal axis (FKBP5) and one glutamatergic receptor gene (GRIK4).
Table 4.
Candidate gene studies of antidepressant treatment response undertaken in large cohorts
| Gene | Study | Drug | Sample size | Significant association |
|---|---|---|---|---|
| SLC6A4 (solute carrier family 6 (neurotransmitter transporter, serotonin), member 4) | STAR*D [1] | Citalopram | 1,775 | No (but see [2]) |
| GENDEP [3] | Escitalopram | 450 | 5HTTLPR, rs2020933 | |
| Nortriptyline | 345 | No | ||
| GenPod [4] | Citalopram/reboxetine | 520 | No | |
| HTR2A (5-hydroxytryptamine (serotonin) receptor 2A, G-protein-coupled) | STAR*D [5] | Citalopram | 1,297 | rs7997012 (also see [6]) |
| MARS [7] | Various | 451 (plus replication sample) | rs7997012: reverse allelic association to [5] | |
| GENDEP [3] | Escitalopram | 422 | rs931623, rs2224721 | |
| FKBP5 (FK506 binding protein 5) | STAR*D [8] | Citalopram | 1,809 | rs4713916 |
| MARS [9] | Various | 294 (plus replication sample) | rs1360780, rs3800373, rs4713916 | |
| GENDEP [3] | Escitalopram or nortriptyline | 733 | No | |
| GRIK4 (glutamate receptor, ionotropic, kainate 4) | STAR*D [6] | Citalopram | 1,816 | rs1954787 |
| MARS [10] | Various | 300 | No |
SLC6A4
The serotonin transporter is a key target for many antidepressants (including SSRIs, SNRIs and TCAs), and so the gene encoding this protein (SLC6A4) represents a prominent candidate gene for pharmacogenetics. In the promoter region of this gene there is a repeat polymorphism comprising a 44 bp deletion [27], known as 5HTTLPR (serotonin transport-linked polymorphic region). This variant was first associated with treatment response by Smeraldi et al. [28], a finding supported by a meta-analysis in 2007 [29]. However, when 5HTTLPR was examined within large cohort samples, the results were mixed. Within the GENDEP sample, the polymorphism did indeed predict treatment outcome, but only for those patients on escitalopram [30]. Sex-specific analysis suggested that this effect was concentrated in males. Furthermore, an additional variant (rs2020933) within the SLC6A4 gene also demonstrated a significant effect on treatment outcome.
However, other large studies report conflicting results. The Genetic and Clinical Predictors of Treatment Response in Depression (GenPod) project is a pharmacogenetic study based in the UK, where genotypic and outcome data are available for 520 patients, randomized to receive citalopram or reboxetine [31]. Reports from GenPod as well as those from STAR*D [32] found no association between 5HTTLPR and treatment outcome. Inconsistent findings could result from ethnic variability [33] or differences in outcome response definitions, but in the recent meta-analysis by Taylor et al. [34], including both GENDEP and STAR*D data (although not GenPod), a weak effect of 5HTTLPR genotype on remission did not reach significance once potential publication bias was accounted for.
HTR2A
Another serotonergic gene that has been implicated in antidepressant response is HTR2A. This encodes the serotonin receptor 2A, which is downregulated by antidepressant treatment in parallel with clinical improvement [35]. A significant association between a marker in the HTR2A gene and treatment outcomes was observed when looking at 68 candidate genes (768 SNPs) in the STAR*D cohort (n = 1,297) [36]. A significant association was seen between rs7997012 and treatment response (the association with remission was not significant), and other markers within this gene also reached suggested levels of significance. Given differences in allele frequencies, the authors suggested that HTR2A may play a role in the racial differences in treatment response found in their mixed-ancestry sample. The top two SNPs implicated in STAR*D were also followed up in the MARS cohort [37]. In a combined sample of 637 inpatients (as in the genome-wide analysis, a second independent German sample was used in addition to the 451 patients recruited as part of the MARS project) a significant association was seen between rs7997012 and both remission and response phenotypes. In that report, however, the allele association was the inverse of that observed in STAR*D; in MARS the AA genotype was associated with impaired treatment response whereas in STAR*D the G allele was associated with impaired treatment response.
When HTR2A was examined in the GENDEP cohort, as part of an analysis looking at 10 different candidate genes (n = 760), once again an association was seen with treatment outcomes, but there was no replication of the specific allelic relationship seen in STAR*D [38]. In an escitalopram-specific analysis, no association was observed with the rs7997012 SNP. Instead, two intronic markers in the HTR2A gene (rs931623 and rs2224721, which are in linkage disequilibrium) were both significantly associated with response to the SSRI. Therefore, while several studies indicated a relationship between genetic variation in the HTR2A gene and treatment response, there has been no replicated evidence that specific variants confer increased likelihood of favorable outcomes. Further attempts at replication in large cohorts are needed to clarify this issue, and it may also be valuable to perform sequencing in this region in an attempt to identify the causal variant that might underlie these findings. Interestingly, Murphy et al. [39] have implicated genetic variation in HTR2A in side effects to the SSRI paroxetine. Thus, the potentially confounding effects of treatment discontinuation should also be considered.
FKBP5
In addition to serotonergic pathways, the hypothalamic-pituitary-adrenal (HPA) axis has also been implicated in antidepressant treatment response [40]. When looking across eight genes in the HPA axis in the MARS sample [41], significant associations were observed between treatment outcomes and three SNPs within the glucocorticoid-receptor-associated co-chaperone gene FKBP5 (rs1360780, rs3800373 and rs4713916). Follow-up work demonstrated that these SNPs were linked to increased intracellular expression of FKBP5, which leads to regulation of the HPA axis via adaptive changes in the glucocorticoid receptor. This evidence supports the proposal that effective antidepressant treatment may involve modulation of the HPA axis. Examination of these three SNPs in STAR*D replicated the association between rs4713916 and remission [42], and it was noted that the linkage disequilibrium patterns observed in the mixed-ancestry sample of STAR*D could suggest that the causal variant underlying this relationship may lie in the promoter region of the gene. However, the association with FKBP5 was not replicated in GENDEP [38]. As part of a ten-candidate gene analysis, the HPA axis genes FKBP5 and NR3C1 (which encodes the glucocorticoid receptor) were both examined. In a whole-sample analysis, no associations between treatment response and any of the markers tested in the FKBP5 gene (including the rs1360780 SNP implicated by Binder et al. [41]) were identified, although two SNPs within NR3C1 were associated at suggested levels of significance (rs852977 and rs10482633).
GRIK4
The final gene implicated in these large cohort studies of antidepressant treatment response is GRIK4, a gene encoding glutamatergic receptor, kainate 4. Paddock et al. [43] examined the 68 candidate genes previously considered in the STAR*D sample [36], but were able to incorporate genetic data from a second wave of genotyping, taking the sample size from 1,297 to 1,816. The authors not only replicated the HTR2A signal outlined above [36], but also reported evidence implicating rs1954787 within GRIK4 in this enlarged sample. The glutamatergic system has previously been considered within treatment response pathways [44] and this association could lend weight to this hypothesis, but, as highlighted, further replication is crucial. It was also observed that the combined effect of GRIK4 and HTR2A on treatment outcomes was more robust than when each gene was considered separately.
Despite the evidence above linking candidate genes and treatment outcome, if these findings were true, it would be expected that there would be a consistent pattern of associations seen across GWASs. Nevertheless, in both the STAR*D and GENDEP genome-wide analyses [15,16], a priori candidate genes did not show associations with treatment outcomes above the levels expected by chance (although the STAR*D analysis does draw attention to 'nominally significant' associations with several SNPs of interest in the HTR2A gene). Although meta-analyses are needed, the results so far indicate that the current genes of interest do not exert a sizable direct effect on antidepressant treatment response.
Gene-gene interactions
Alternatively, the observation of combined effects of GRIK4 and HTR2A in the STAR*D sample may indicate that genetic variation has a more complex relationship with treatment outcomes. Horstmann et al. [45] used data from MARS to consider whether three of the genes highlighted in these large cohort studies (HTR2A, GRIK4 and FKBP5) may contribute to treatment response variability, in an additive or interactive fashion (Figure 2). While the specific SNPs in HTR2A and GRIK4 implicated in the STAR*D sample did not replicate in MARS, when looking across all the variants analyzed in each gene, significant associations with treatment response were found. Exploring the most predictive SNPs, the best-fitting model of genetic moderation of treatment response included a main effect of GRIK4 genotype, and two interaction terms, between GRIK4 and FKBP5, and GRIK4 and HTR2A. This three-SNP interaction model accounted for 13.1% of the variation in treatment response, a substantial improvement over simpler, main effect models. This supports the proposal that antidepressant treatment response phenotypes are influenced by multiple genes, and that these genes are likely to have both independent and interactive (or epistatic) effects. This idea was also investigated in the MARS sample where a multilocus analysis was used to score patients as possessing high or low numbers of response alleles, based on the top 310 most informative SNPs in their GWAS analysis [14].
Figure 2.
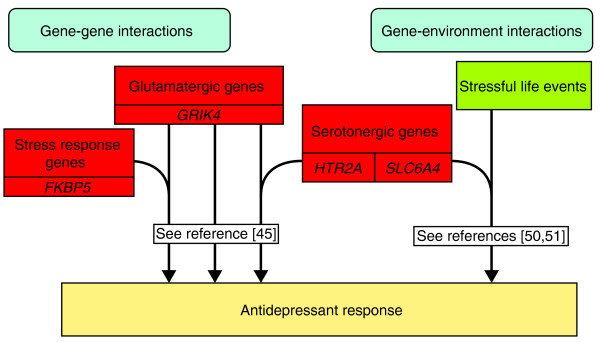
Examples of reported interactions between genetic and non-genetic factors in determining antidepressant treatment response. Work looking at the relationship between potential response predictors indicates that antidepressant treatment response is determined by multiple interacting factors. For example, genetic variation in GRIK4 (glutamate receptor, ionotropic, kainate 4) has been reported to have both a main effect and interactive effects with FKBP5 (FK506 binding protein 5) and HTR2A (5-hydroxytryptamine (serotonin) receptor 2A, G-protein-coupled) in its influence on response outcomes [45]. Similarly, an association between the occurrence of stressful life events and response may be moderated by genetic variation in SLC6A4 (solute carrier family 6 (neurotransmitter transporter, serotonin), member 4) [50,51].
It has been calculated that STAR*D and GENDEP are powered to detect single SNP main effects on general treatment response that are clinically significant in size [46]. However, it now seems likely that treatment response is a complex trait that is not determined by a single genetic variant, but results from multiple interacting genetic effects. In which case, the failure of pharmacogenetic and pharmacogenomic approaches to identify any replicated genetic variants associated with clinically meaningful effects on treatment response may be because studies are underpowered to detect these effects.
Gene-environment interactions in predicting treatment response
Effects at one gene locus may not only be moderated by other genetic variants, but may also be dependent on environmental exposures. Environments such as stressful life events (SLEs) have been associated with depression [47], and investigations into this relationship have indicated that 5HTTLPR moderates the effect of SLEs on the emergence of depressive symptoms [48,49]. Mandelli et al. [50] reported initial evidence that this same interaction may also predict antidepressant treatment response, in a sample of 159 mood-disorder patients treated with an SSRI. This interaction between SLEs and 5HTTLPR was also replicated in GENDEP, although only among patients taking escitalopram, not among those taking nortriptyline [51] (Figure 2). This could suggest that the gene-environment interaction is drug-specific, and may be why no statistically significant interactions were found with 5HTTLPR in a sample of 290 patients who were receiving a range of different antidepressants [52]. Thus, it is likely that the accurate prediction of antidepressant treatment response will require the consideration of multiple factors, both genetic and environmental, and the interaction between them.
Symptomatic heterogeneity in depression
Beyond SLEs, other non-genetic factors that may influence antidepressant response, such as symptomatic differences between patients, have also been explored as potential indicators of antidepressant response. This reflects the ongoing debate as to whether depression is a single, homogeneous clinical entity. Three commonly considered subgroups, defined on their symptomatic presentation, are: melancholic depression (akin to 'endogenous' depression, with pervasive anhedonia, sleep and appetite disturbance plus diurnal variation of mood); atypical depression (with features including mood reactivity, hyperphagia and hypersomnia); and anxious depression (reflecting the high rates of anxious symptoms and co-morbid anxiety disorders seen in depression [53,54]). In the STAR*D sample, differences in response to citalopram seen between melancholic and non-melancholic and between atypical and non-atypical depression were accounted for by demographic and clinical factors at baseline [55,56], but anxious depression was linked to poorer treatment response [57]. However, this pattern with anxious depression is not consistently replicated [58,59]. Studies comparing the efficacy of different antidepressant drugs in these subtypes have also been inconsistent in their findings [60-63].
As noted by Uher et al. [46], it is important to consider not only whether associations between depression subtype and treatment response can be replicated, but also whether the observed effect sizes are sufficiently large to be of clinical value.
Interestingly, more robust predictors of treatment response were identified when the symptomatic variation in depression was explored in terms of dimensional traits (rather than the above categorical subtypes). Using detailed symptomatic data available in GENDEP, six dimensional facets of depressive symptoms were described: mood, anxiety, pessimism, interest activity (describing symptoms of indecisiveness and reduced activity), sleep and appetite [64]. The interest-activity facet was the most robust predictor of response in GENDEP; symptoms of low interest and decreased activity predicted significantly worse treatment outcomes [65]. Importantly, the association between interest-activity symptoms and treatment outcome was also replicated in the STAR*D sample.
Differential response prediction
The predictive value of the interest-activity facet is substantial enough to be of clinical importance (one additional remission could be accurately predicted for every three or four patients where interest activity was measured), and obtaining the required information is relatively inexpensive. Thus, interest-activity scores appear to be valuable in translational terms. Nonetheless, low levels of interest and activity predicted poorer outcomes for patients, regardless of which antidepressant was used. As Simon and Perlis [66] highlight, the greatest clinical utility is obtained from factors that differentially predict response to alternative treatments, as these may be used to guide treatment recommendations. Consequently, assessment of whether other treatment options might be effective in patients displaying loss of interest and decreased activity would be of translational value. Therefore, in addition to between-drug comparisons, work in 'therapygenetics' [67] (where predictors of response to talking therapies are considered) points to a potentially fruitful direction for exploring differential predictors of treatment response. If studies allowing direction comparisons between psychological and pharmacological treatments could be undertaken, response predictors with high translational value might be revealed. For example, there is preliminary evidence that the 5HTTLPR variant linked with poorer antidepressant response is associated with better response to cognitive behavioral therapy [67].
The observed heterogeneity in treatment response associated with symptomatic differences also points to interesting new avenues for pharmacogenetic research, as it may indicate that genetic moderation of response to antidepressants could be symptom-specific. For example, among patients taking nortriptyline in GENDEP, genetic variation in the beta-3 subunit of the G-protein complex (GNB3) was associated specifically with differences in improvement of neurovegetative symptoms (that is, symptoms relating to sleep and appetite), but was not associated with improvement in other symptom dimensions (such as observed mood or cognitive symptoms) [68]. Therefore, consideration of the symptomatic diversity encompassed by the diagnostic category of depression (possibly through dimensional rather than categorical models) may allow the identification of genetic effects on specific components of treatment response that are not otherwise seen.
Towards an understanding of mechanisms underlying antidepressant treatments
Genetic research into antidepressant response not only aims to identify outcome predictors, but also provides valuable insight into the physiological processes underlying successful treatment of depression. The lack of strong genetic signals in either genome-wide or candidate gene studies indicates that antidepressant treatment response is not a phenotype determined by a small number of highly influential genes. Instead, the genetic evidence suggests that antidepressant treatment response is best considered a complex phenotype, determined by multiple genes of small effect. While candidate gene findings require further replication and confirmation, the evidence suggests that these genes lie in multiple, interacting biological systems.
The serotonergic system, as the canonical target for antidepressants, is a key candidate for investigators, and has been implicated with associations observed between treatment response and variation in SLC6A4 [30] and HTR2A [36-38], although effect sizes are small when considered alone. The effects of successful antidepressant treatment on the HPA axis suggest that this system also plays an important role in the physiological processes that lead to remission [69]. This proposal is supported by the observed association between FKBP5 (which acts as a regulator within this system) and treatment response [41,42]. Indeed, there has been a large research effort in attempting to develop novel antidepressants targeting the HPA system [70], although this is yet to achieve success [71]. Similarly, evidence of glutamatergic involvement in both depression and treatment response [72] has led to investigations of ketamine as another potential new treatment for depression [73], and the association between variability in the glutamatergic gene GRIK4 and treatment response strengthens the hypothesis that this system also plays an important role in antidepressant action.
Arguably, what is most interesting about the genetic findings is that they provide preliminary indications that, rather than purely additive effects of each of the different pathways contributing to treatment response, the serotonergic, glutamatergic and HPA systems may have interactive effects. This might indicate a physiological interaction, as experimental observations in humans seem to suggest (for example, [74]).
The non-genetic predictors of response outlined here also may give an insight into the mechanisms underlying antidepressant response. For example, the finding that interest-activity symptoms (of low interest, reduced activity, indecisiveness and lack of enjoyment) predict poorer treatment response [65] could be interpreted to suggest that the pathophysiological processes producing these symptoms are, to some extent, distinct, and less well targeted by antidepressants.
Nevertheless, the identification of treatment response predictors is only the first step in understanding the actions of antidepressants; further exploration of the biological impact of these genetic variants and the mechanism by which suggested predictors may be associated with treatment response is needed, as well as a greater understanding of how these predictors may interact.
Conclusions and future directions
Genome-wide analyses of antidepressant treatment response have proved disappointing to date, with no strong signals emerging from the three studies conducted thus far. Given the small genetic effect sizes observed in candidate gene studies, it seems that existing studies are likely to be underpowered on a genome-wide scale, and strong conclusions cannot be drawn without increasing sample sizes and undertaking formal meta-analyses.
Nonetheless, there is some evidence from candidate gene approaches within these large samples indicating that genetic variability in the serotonergic, glutamatergic and stress-response pathways may influence antidepressant treatment response. These associations remain small in effect size; no genes of interest have yet been reliably linked with clinically significant effects. This suggests that, rather than single gene effects, there may instead be multiple genetic influences that determine treatment outcomes in combination with environmental factors. The interactions between each of these various elements must be considered if we wish to better provide treatment recommendations that are optimized for each individual.
Indeed, the best-performing predictor identified in the three large GWAS samples has been the symptom dimension of interest activity, and there is some evidence for symptom-specific improvements. This indicates that the future for pharmacogenomic studies lies not only in larger samples, but also in the collection of detailed and accurate patient data, to allow the identification of any subgroup-specific relationships.
The translational value of pharmacogenetic work with antidepressants has yet to be fully realized; indeed, for real clinical utility, studies must focus on comparing different treatments for depression in order to identify differential predictors of response. Furthermore, it is likely that translation into clinics will only be achieved by combining information from multiple response predictors. Nonetheless, the candidate genes identified to date provide an insight into the mechanisms of antidepressant action, and may enable the development of novel treatments for depression.
Abbreviations
BDI: Beck Depression Inventory; bp: base pair; DSM-IV: Diagnostic and Statistical Manual of Mental Disorders: 4th edition; GENDEP: Genome Based Therapeutic Drugs for Depression; GenPod: Genetic and Clinical Predictors of Treatment response in Depression; GWAS: genome-wide association study; HAM-D: Hamilton Depression Rating Scale; HPA: hypothalamic-pituitary-adrenal; ICD-10: International Classification of Diseases: 10th revision; MADRS: Montgomery-Asberg Depression Rating scale; MARS: Munich Antidepressant Response Signatures; QIDS-SR: Quick Inventory of Depressive Symptomatology: self-report version; SLE: stressful life event; SNP: single nucleotide polymorphism; SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; STAR*D: Sequenced Treatment Alternatives to Relieve Depression; TCA: tricyclic antidepressant; 5HTTLPR: serotonin transport-linked polymorphic region.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Karen Hodgson, Email: karen.hodgson@kcl.ac.uk.
Shaista Jeelani Mufti, Email: shayamufti@gmail.com.
Rudolf Uher, Email: Rudolf.Uher@cdha.nshealth.ca.
Peter McGuffin, Email: peter.mcguffin@kcl.ac.uk.
Acknowledgements
Karen Hodgson is supported by an MRC PhD scholarship. This work was in part supported by the South London and Maudsley NHS Foundation Trust NIHR Biomedical Research Centre.
References
- Blazer DG, Kessler RC, McGonagle KA, Swartz MS. The prevalence and distribution of major depression in a national community sample: the National Comorbidity Survey. Am J Psychiatry. 1994;4:979–986. doi: 10.1176/ajp.151.7.979. [DOI] [PubMed] [Google Scholar]
- Lopez AD, Murray CC. The global burden of disease, 1990-2020. Nat Med. 1998;4:1241–1243. doi: 10.1038/3218. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- World Health Organization. ICD-10 Classifications of Mental and Behavioural Disorder: Clinical Descriptions and Diagnostic Guidelines. Geneva: WHO; 1992. [Google Scholar]
- Wing JK, Sartorius N, Üstün TB. Diagnosis and Clinical Measurement in Psychiatry: a Reference Manual for SCAN. Cambridge: Cambridge University Press; 1998. [Google Scholar]
- Robins LN, Wing J, Wittchen HU, Helzer JE, Babor TF, Burke J, Farmer A, Jablenski A, Pickens R, Regier DA, Sartorius N, Towle LH. The Composite International Diagnostic Interview. An epidemiologic instrument suitable for use in conjunction with different diagnostic systems and in different cultures. Arch Gen Psychiatry. 1988;4:1069–1077. doi: 10.1001/archpsyc.1988.01800360017003. [DOI] [PubMed] [Google Scholar]
- Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol. 1967;4:278–296. doi: 10.1111/j.2044-8260.1967.tb00530.x. [DOI] [PubMed] [Google Scholar]
- Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- Arroll B, Elley CR, Fishman T, Goodyear-Smith FA, Kenealy T, Blashki G, Kerse N, Macgillivray S. Antidepressants versus placebo for depression in primary care. Cochrane Database Syst Rev. 2009. p. CD007954. [DOI] [PMC free article] [PubMed]
- Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry. 2001;4:234–241. doi: 10.1192/bjp.178.3.234. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;4:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- Franchini L, Serretti A, Gasperini M, Smeraldi E. Familial concordance of fluvoxamine response as a tool for differentiating mood disorder pedigrees. J Psychiatr Res. 1998;4:255–259. doi: 10.1016/S0022-3956(98)00004-1. [DOI] [PubMed] [Google Scholar]
- O'Reilly RL, Bogue L, Singh SM. Pharmacogenetic response to antidepressants in a multicase family with affective disorder. Biol Psychiatry. 1994;4:467–471. doi: 10.1016/0006-3223(94)90642-4. [DOI] [PubMed] [Google Scholar]
- Ising M, Lucae S, Binder EB, Bettecken T, Uhr M, Ripke S, Kohli MA, Hennings JM, Horstmann S, Kloiber S, Menke A, Bondy B, Rupprecht R, Domschke K, Baune BT, Arolt V, Rush AJ, Holsboer F, Muller-Myhsok B. A Genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch Gen Psychiatry. 2009;4:966–975. doi: 10.1001/archgenpsychiatry.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GD, Reinalda MS, Slager SL, McGrath PJ, Hamilton SP. A genomewide association study of citalopram response in major depressive disorder. Biol Psychiatry. 2010;4:133–138. doi: 10.1016/j.biopsych.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uher R, Perroud N, Ng MY, Hauser J, Henigsberg N, Maier W, Mors O, Placentino A, Rietschel M, Souery D, Zagar T, Czerski PM, Jerman B, Larsen ER, Schulze TG, Zobel A, Cohen-Woods S, Pirlo K, Butler AW, Muglia P, Barnes MR, Lathrop M, Farmer A, Breen G, Aitchison KJ, Craig I, Lewis CM, McGuffin P. Genome-wide pharmacogenetics of antidepressant response in the GENDEP project. Am J Psychiatry. 2010;4:555–564. doi: 10.1176/appi.ajp.2009.09070932. [DOI] [PubMed] [Google Scholar]
- Rush AJ. STAR*D: what have we learned?. Am J Psychiatry. 2007;4:201–204. doi: 10.1176/appi.ajp.164.2.201. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, Markowitz JC, Ninan PT, Kornstein S, Manber R, Thase ME, Kocsis JH, Keller MB. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 2003;4:573–583. doi: 10.1016/S0006-3223(02)01866-8. [DOI] [PubMed] [Google Scholar]
- Urso ML, Chen YW, Scrimgeour AG, Lee PC, Lee KF, Clarkson PM. Alterations in mRNA expression and protein products following spinal cord injury in humans. J Physiol. 2007;4:877–892. doi: 10.1113/jphysiol.2006.118042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere V, Beatty B, Squire J, Copeland NG, Jenkins NA. The orphan nuclear receptor ROR alpha (RORA) maps to a conserved region of homology on human chromosome 15q21-q22 and mouse chromosome 9. Genomics. 1995;4:596–598. doi: 10.1006/geno.1995.1197. [DOI] [PubMed] [Google Scholar]
- Uher R, Maier W, Hauser J, Marusic A, Schmael C, Mors O, Henigsberg N, Souery D, Placentino A, Rietschel M, Zobel A, Dmitrzak-Weglarz M, Petrovic A, Jorgensen L, Kalember P, Giovannini C, Barreto M, Elkin A, Landau S, Farmer A, Aitchison KJ, McGuffin P. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry. 2009;4:252–259. doi: 10.1192/bjp.bp.108.057554. [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;4:382–389. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- Akita K, von Holst A, Furukawa Y, Mikami T, Sugahara K, Faissner A. Expression of multiple chondroitin/dermatan sulfotransferases in the neurogenic regions of the embryonic and adult central nervous system implies that complex chondroitin sulfates have a role in neural stem cell maintenance. Stem Cells. 2008;4:798–809. doi: 10.1634/stemcells.2007-0448. [DOI] [PubMed] [Google Scholar]
- Raison CL, Miller AH. Is depression an inflammatory disorder?. Curr Psychiatry Rep. 2011;4:467–475. doi: 10.1007/s11920-011-0232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pe'er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;4:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther. 2009;4:57–73. doi: 10.1016/j.pharmthera.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Heils A, Teufel A, Petri S, Stober G, Riederer P, Bengel D, Lesch KP. Allelic variation of human serotonin transporter gene expression. J Neurochem. 1996;4:2621–2624. doi: 10.1046/j.1471-4159.1996.66062621.x. [DOI] [PubMed] [Google Scholar]
- Smeraldi E, Zanardi R, Benedetti F, Di Bella D, Perez J, Catalano M. Polymorphism within the promoter of the serotonin transporter gene and antidepressant efficacy of fluvoxamine. Mol Psychiatry. 1998;4:508–511. doi: 10.1038/sj.mp.4000425. [DOI] [PubMed] [Google Scholar]
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry. 2007;4:247–257. doi: 10.1038/sj.mp.4001926. [DOI] [PubMed] [Google Scholar]
- Huezo-Diaz P, Uher R, Smith R, Rietschel M, Henigsberg N, Marusic A, Mors O, Maier W, Hauser J, Souery D, Placentino A, Zobel A, Larsen ER, Czerski PM, Gupta B, Hoda F, Perroud N, Farmer A, Craig I, Aitchison KJ, McGuffin P. Moderation of antidepressant response by the serotonin transporter gene. Br J Psychiatry. 2009;4:30–38. doi: 10.1192/bjp.bp.108.062521. [DOI] [PubMed] [Google Scholar]
- Lewis G, Mulligan J, Wiles N, Cowen P, Craddock N, Ikeda M, Grozeva D, Mason V, Nutt D, Sharp D, Tallon D, Thomas L, O'Donovan MC, Peters TJ. Polymorphism of the 5-HT transporter and response to antidepressants: randomised controlled trial. Br J Psychiatry. 2011;4:464–471. doi: 10.1192/bjp.bp.110.082727. [DOI] [PubMed] [Google Scholar]
- Hu X-Z, Rush AJ, Charney D, Wilson AF, Sorant AJM, Papanicolaou GJ, Fava M, Trivedi MH, Wisniewski SR, Laje G, Paddock S, McMahon FJ, Manji H, Lipsky RH. Association between a functional serotonin transporter promoter polymorphism and citalopram treatment in adult outpatients with major depression. Arch Gen Psychiatry. 2007;4:783–792. doi: 10.1001/archpsyc.64.7.783. [DOI] [PubMed] [Google Scholar]
- Mrazek DA, Rush AJ, Biernacka JM, O'Kane DJ, Cunningham JM, Wieben ED, Schaid DJ, Drews MS, Courson VL, Snyder KA, Black JL, Weinshilboum RM. SLC6A4 variation and citalopram response. Am J Med Genet B Neuropsychiatr Genet. 2009;4:341–351. doi: 10.1002/ajmg.b.30816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Sen S, Bhagwagar Z. Antidepressant response and the serotonin transporter gene-linked polymorphic region. Biol Psychiatry. 2010;4:536–543. doi: 10.1016/j.biopsych.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC. Neuroimaging studies of mood disorders. Biol Psychiatry. 2000;4:813–829. doi: 10.1016/S0006-3223(00)01020-9. [DOI] [PubMed] [Google Scholar]
- McMahon FJ, Buervenich S, Charney D, Lipsky R, Rush AJ, Wilson AF, Sorant AJ, Papanicolaou GJ, Laje G, Fava M, Trivedi MH, Wisniewski SR, Manji H. Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. Am J Hum Genet. 2006;4:804–814. doi: 10.1086/503820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucae S, Ising M, Horstmann S, Baune BT, Arolt V, Muller-Myhsok B, Holsboer F, Domschke K. HTR2A gene variation is involved in antidepressant treatment response. Eur Neuropsychopharmacol. 2010;4:65–68. doi: 10.1016/j.euroneuro.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Uher R, Huezo-Diaz P, Perroud N, Smith R, Rietschel M, Mors O, Hauser J, Maier W, Kozel D, Henigsberg N, Barreto M, Placentino A, Dernovsek MZ, Schulze TG, Kalember P, Zobel A, Czerski PM, Larsen ER, Souery D, Giovannini C, Gray JM, Lewis CM, Farmer A, Aitchison KJ, McGuffin P, Craig I. Genetic predictors of response to antidepressants in the GENDEP project. Pharmacogenomics J. 2009;4:225–233. doi: 10.1038/tpj.2009.12. [DOI] [PubMed] [Google Scholar]
- Murphy GM Jr, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry. 2003;4:1830–1835. doi: 10.1176/appi.ajp.160.10.1830. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Owens MJ. Treatment of mood disorders. Nat Neurosci. 2002. pp. 1068–1070. [DOI] [PubMed]
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B, Papiol S, Seaman S, Lucae S, Kohli MA, Nickel T, Kunzel HE, Fuchs B, Majer M, Pfennig A, Kern N, Brunner J, Modell S, Baghai T, Deiml T, Zill P, Bondy B, Rupprecht R, Messer T, Kohnlein O, Dabitz H, Bruckl T, Muller N, Pfister H, Lieb R. et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;4:1319–1325. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- Lekman M, Laje G, Charney D, Rush AJ, Wilson AF, Sorant AJ, Lipsky R, Wisniewski SR, Manji H, McMahon FJ, Paddock S. The FKBP5-gene in depression and treatment response - an association study in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Cohort. Biol Psychiatry. 2008;4:1103–1110. doi: 10.1016/j.biopsych.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddock S, Laje G, Charney D, Rush AJ, Wilson AF, Sorant AJ, Lipsky R, Wisniewski SR, Manji H, McMahon FJ. Association of GRIK4 with outcome of antidepressant treatment in the STAR*D cohort. Am J Psychiatry. 2007;4:1181–1188. doi: 10.1176/appi.ajp.2007.06111790. [DOI] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;4:351–354. doi: 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Lucae S, Menke A, Hennings JM, Ising M, Roeske D, Muller-Myhsok B, Holsboer F, Binder EB. Polymorphisms in GRIK4, HTR2A, and FKBP5 show interactive effects in predicting remission to antidepressant treatment. Neuropsychopharmacology. 2010;4:727–740. doi: 10.1038/npp.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uher R, Tansey KE, Malki K, Perlis RH. Biomarkers predicting treatment outcome in depression: what is clinically significant?. Pharmacogenomics. 2012;4:233–240. doi: 10.2217/pgs.11.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. 1997;4:191–214. doi: 10.1146/annurev.psych.48.1.191. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;4:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Uher R, McGuffin P. The moderation by the serotonin transporter gene of environmental adversity in the etiology of depression: 2009 update. Mol Psychiatry. 2010;4:18–22. doi: 10.1038/mp.2009.123. [DOI] [PubMed] [Google Scholar]
- Mandelli L, Marino E, Pirovano A, Calati R, Zanardi R, Colombo C, Serretti A. Interaction between SERTPR and stressful life events on response to antidepressant treatment. Eur Neuropsychopharmacol. 2009;4:64–67. doi: 10.1016/j.euroneuro.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Keers R, Uher R, Huezo-Diaz P, Smith R, Jaffee S, Rietschel M, Henigsberg N, Kozel D, Mors O, Maier W, Zobel A, Hauser J, Souery D, Placentino A, Larsen ER, Dmitrzak-Weglarz M, Gupta B, Hoda F, Craig I, McGuffin P, Farmer AE, Aitchison KJ. Interaction between serotonin transporter gene variants and life events predicts response to antidepressants in the GENDEP project. Pharmacogenomics J. 2011;4:138–145. doi: 10.1038/tpj.2010.14. [DOI] [PubMed] [Google Scholar]
- Bukh JD, Bock C, Vinberg M, Werge T, Gether U, Kessing LV. No interactions between genetic polymorphisms and stressful life events on outcome of antidepressant treatment. Eur Neuropsychopharmacol. 2010;4:327–335. doi: 10.1016/j.euroneuro.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Zimmerman M, Chelminski I, McDermut W. Major depressive disorder and axis I diagnostic comorbidity. J Clin Psychiatry. 2002;4:187–193. doi: 10.4088/JCP.v63n0303. [DOI] [PubMed] [Google Scholar]
- Fawcett J, Kravitz HM. Anxiety syndromes and their relationship to depressive illness. J Clin Psychiatry. 1983;4:8–11. [PubMed] [Google Scholar]
- McGrath PJ, Khan AY, Trivedi MH, Stewart JW, Morris DW, Wisniewski SR, Miyahara S, Nierenberg AA, Fava M, Rush AJ. Response to a selective serotonin reuptake inhibitor (citalopram) in major depressive disorder with melancholic features: a STAR*D report. J Clin Psychiatry. 2008;4:1847–1855. doi: 10.4088/JCP.v69n1201. [DOI] [PubMed] [Google Scholar]
- Stewart JW, McGrath PJ, Fava M, Wisniewski SR, Zisook S, Cook I, Nierenberg AA, Trivedi MH, Balasubramani GK, Warden D, Lesser I, John Rush A. Do atypical features affect outcome in depressed outpatients treated with citalopram?. Int J Neuropsychopharmacol. 2010;4:15–30. doi: 10.1017/S1461145709000182. [DOI] [PubMed] [Google Scholar]
- Fava M, Rush AJ, Alpert JE, Balasubramani GK, Wisniewski SR, Carmin CN, Biggs MM, Zisook S, Leuchter A, Howland R, Warden D, Trivedi MH. Difference in treatment outcome in outpatients with anxious versus nonanxious depression: a STAR*D report. Am J Psychiatry. 2008;4:342–351. doi: 10.1176/appi.ajp.2007.06111868. [DOI] [PubMed] [Google Scholar]
- Nelson JC. Anxiety does not predict response to duloxetine in major depression: results of a pooled analysis of individual patient data from 11 placebo-controlled trials. Depress Anxiety. 2010;4:12–18. doi: 10.1002/da.20632. [DOI] [PubMed] [Google Scholar]
- Nelson JC, Delucchi K, Schneider LS. Anxiety does not predict response to antidepressant treatment in late life depression: results of a meta-analysis. Int J Geriatr Psychiatry. 2009;4:539–544. doi: 10.1002/gps.2233. [DOI] [PubMed] [Google Scholar]
- Joyce PR, Mulder RT, McKenzie JM, Luty SE, Cloninger CR. Atypical depression, atypical temperament and a differential antidepressant response to fluoxetine and nortriptyline. Depress Anxiety. 2004;4:180–186. doi: 10.1002/da.20001. [DOI] [PubMed] [Google Scholar]
- McGrath PJ, Stewart JW, Janal MN, Petkova E, Quitkin FM, Klein DF. A placebo-controlled study of fluoxetine versus imipramine in the acute treatment of atypical depression. Am J Psychiatry. 2000;4:344–350. doi: 10.1176/appi.ajp.157.3.344. [DOI] [PubMed] [Google Scholar]
- Brown WA. Treatment response in melancholia. Acta Psychiatr Scand Suppl. 2007. pp. 125–129. [DOI] [PubMed]
- Uher R, Dernovsek MZ, Mors O, Hauser J, Souery D, Zobel A, Maier W, Henigsberg N, Kalember P, Rietschel M, Placentino A, Mendlewicz J, Aitchison KJ, McGuffin P, Farmer A. Melancholic, atypical and anxious depression subtypes and outcome of treatment with escitalopram and nortriptyline. J Affect Disord. 2011;4:112–120. doi: 10.1016/j.jad.2011.02.014. [DOI] [PubMed] [Google Scholar]
- Uher R, Farmer A, Maier W, Rietschel M, Hauser J, Marusic A, Mors O, Elkin A, Williamson RJ, Schmael C, Henigsberg N, Perez J, Mendlewicz J, Janzing JG, Zobel A, Skibinska M, Kozel D, Stamp AS, Bajs M, Placentino A, Barreto M, McGuffin P, Aitchison KJ. Measuring depression: comparison and integration of three scales in the GENDEP study. Psychol Med. 2008;4:289–300. doi: 10.1017/S0033291707001730. [DOI] [PubMed] [Google Scholar]
- Uher R, Perlis RH, Henigsberg N, Zobel A, Rietschel M, Mors O, Hauser J, Dernovsek MZ, Souery D, Bajs M, Maier W, Aitchison KJ, Farmer A, McGuffin P. Depression symptom dimensions as predictors of antidepressant treatment outcome: replicable evidence for interest-activity symptoms. Psychol Med. 2011. pp. 1–14. [DOI] [PMC free article] [PubMed]
- Simon GE, Perlis RH. Personalized medicine for depression: can we match patients with treatments?. Am J Psychiatry. 2010;4:1445–1455. doi: 10.1176/appi.ajp.2010.09111680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley TC, Hudson JL, Creswell C, Tropeano M, Lester KJ, Cooper P, Farmer A, Lewis CM, Lyneham HJ, Rapee RM, Uher R, Zavos HMS, Collier DA. Therapygenetics: the 5HTTLPR and response to psychological therapy. Mol Psychiatry. 2012;4:236–237. doi: 10.1038/mp.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keers R, Bonvicini C, Scassellati C, Uher R, Placentino A, Giovannini C, Rietschel M, Henigsberg N, Kozel D, Mors O, Maier W, Hauser J, Souery D, Mendlewicz J, Schmal C, Zobel A, Larsen ER, Szczepankiewicz A, Kovacic Z, Elkin A, Craig I, McGuffin P, Farmer AE, Aitchison KJ, Gennarelli M. Variation in GNB3 predicts response and adverse reactions to antidepressants. J Psychopharmacol. 2011;4:867–874. doi: 10.1177/0269881110376683. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Binder EB. Glucocorticoids as predictors of treatment response in depression. Harv Rev Psychiatry. 2011;4:125–143. doi: 10.3109/10673229.2011.586550. [DOI] [PubMed] [Google Scholar]
- McQuade R, Young AH. Future therapeutic targets in mood disorders: the glucocorticoid receptor. Br J Psychiatry. 2000;4:390–395. doi: 10.1192/bjp.177.5.390. [DOI] [PubMed] [Google Scholar]
- Cowen PJ. Not fade away: the HPA axis and depression. Psychol Med. 2010;4:1–4. doi: 10.1017/S0033291709005558. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;4:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW. Ketamine as a novel antidepressant: from synapse to behavior. Clin Pharmacol Ther. 2012;4:303–309. doi: 10.1038/clpt.2011.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter R, Gallagher P, Watson S, Young A. Corticosteroid-serotonin interactions in depression: a review of the human evidence. Psychopharmacology. 2004;4:1–17. doi: 10.1007/s00213-004-1774-1. [DOI] [PubMed] [Google Scholar]