

Abstract
Despite improvements in disease management, multiple myeloma (MM) remains incurable. Conventional treatment methods are unsatisfactory, leading to a pattern of regression and remission, and ultimately failure. This pattern suggests that one of the possible strategies for improving outcomes is continuous therapy to maintain suppression of the surviving tumor cells. Optimal management of MM requires potent agents and modalities with direct tumoricidal activity, which can also provide continuous suppression of the residual tumor to prevent disease relapse. Immunomodulatory agents exert immunomodulatory and tumoricidal effects, and cause disruption of stromal cell support from the bone marrow microenvironment. Therefore continuous therapy with immumomodulatory agents may be able to provide both tumor reduction and tumor suppression, enabling physicians to consider the possibility of incorporating continuous therapy into the treatment paradigm of patients with MM.
Keywords: Multiple myeloma, Cell biology, Bone marrow microenvironment, Mode of action, Immunomodulatory drugs, Proteosome inhibitors
1. Introduction
Multiple myeloma (MM) is characterized by the accumulation of clonal plasma cells in the bone marrow, the presence of monoclonal immunoglobulin (Ig) in the serum or urine, osteolytic bone lesions, renal disease, and immunodeficiency. It is principally a disease of older patients, with a median age at diagnosis of 65–70 years. The first stage in the development of MM is the emergence of asymptomatic monoclonal gammopathy of undetermined significance (MGUS). In some of these patients, this progresses to smoldering MM and ultimately to symptomatic MM, with an annual risk of around 1% for patients with MGUS [1]. The reasons why MGUS progresses to MM in only a small proportion of patients are unclear, and both genetic and environmental factors have been implicated [2]. Progression to MM is associated with a series of complex genetic events in MM cells, as well as changes in the bone marrow microenvironment, including increased angiogenesis, suppression of the immune response, increased bone resorption, and the establishment of aberrant signaling-loops involving cytokines and growth factors associated with the clinical features of MM and its resistance to treatment [3].
Despite the improvements in overall survival associated with the use of conventional high-dose chemotherapy and autologous stem-cell transplantation (HDT-ASCT), median overall survival remained at around 33 months until the introduction of the novel anti-myeloma agents, thalidomide, lenalidomide, and bortezomib [4]. For patients diagnosed since 2000, the use of the novel agents has improved survival times significantly, particularly for younger patients [5,6]. The median survival time for patients under 65 years of age treated with novel agents is 56 months [6]. The course of MM treated with conventional chemotherapy, such as single-agent alkylating drugs, corticosteroids or combination chemotherapy involving novel anti-myeloma agents is characterized by a pattern of remission and relapse, with a decreasing duration of response and increasing number of salvage regimens (Fig. 1). This reflects the development of drug resistance, which eventually results in refractory disease (Fig. 1) [7]. This pattern suggests the presence of residual disease after treatment, even following an apparently complete response. Therefore, the incurable nature of MM necessitates treatment with agents and modalities that not only provide direct tumoricidal effects to reduce tumor burden, but also suppress residual disease with continuous use.
Fig. 1.
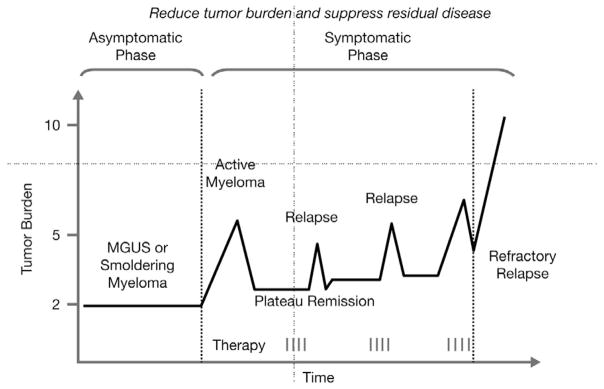
Characteristic pattern of remission and relapse following conventional chemotherapy in multiple myeloma. MGUS, monoclonal gammopathy of undetermined significance.
The development of novel anti-MM agents relies on an understanding of the biology of MM and the multiple factors involved in its pathogenesis and response to treatment. As well as genetic aberrations in essential growth- and tumor-suppressor genes, there is increasing evidence that interactions between tumor cells and their bone marrow microenvironment play a pivotal role in the development, maintenance, and progression of MM, and thus, in the development of drug resistance. This knowledge has improved treatment options leading to the approval of drugs such as thalidomide, bortezomib, and lenalidomide, which not only target malignant cells directly, but also their supporting bone marrow microenvironment. In addition to their tumoricidal effects, immunomodulatory agents also act on the immune system, potentially helping to overcome MM-associated immunodeficiency and enhancing anti-MM immune activity.
This article aims to give an overview of the biology of MM, focusing on the pivotal role of the bone marrow microenvironment and its relevance to tumor survival and proliferation. It will also discuss how new agents have the potential to modify MM biology, offering the prospect of a shift in treatment paradigm to a focus on sustaining disease control with long-term treatment, which may transform myeloma into a chronic disease.
2. The biology of MM
The bone marrow of patients with MM contains malignant cells that have the morphology of mature plasma cells or plasmablasts. However, the origin of MM cells and their developmental relationship to non-malignant counterpart cells remains obscure. The vast majority of MM cells appear to be mature, quiescent, and terminally differentiated; therefore, they do not have long-term proliferative potential. This raises questions about which cells in MM patients are clonogenic and capable of proliferation, and at what stage these cells develop. It is possible that clonogenic growth may be restricted to a minor, specialized population of cells that is distinct from the differentiated/mature cells that comprise the bulk of disease, such as post-germinal center memory B cells [8], and that these cells may be similar to the cancer stem cells seen in other malignancies [9].
Identification of the stage at which tumor growth develops in MM has been an important research aim. During normal differentiation from stem cells to plasma cells, immature B lymphocytes first differentiate in the bone marrow, where they undergo immunoglobulin heavy-chain (IgH) VDJ gene rearrangement, resulting in the expression of surface IgM (Fig. 2). The B cells then migrate as naïve B lymphocytes to the secondary lymphoid tissue where antigen stimulation leads to their proliferation. During this period, somatic hypermutation in the IgH and light-chain genes gives rise to the selection of B-cell clones expressing high-affinity Igs. At this stage the cells may either leave the secondary lymphoid organs and circulate as memory B cells or differentiate into post-follicular plasmablasts following a switch in Ig class from IgM to IgG, IgA, IgD, or IgE. Plasmablasts migrate back to the bone marrow to undergo terminal differentiation into plasma cells. During this process, B cells express a range of surface markers used to assess their developmental stage (Fig. 2). Cell-surface markers and IgH chain gene sequences in MM cells define the nature of the malignant cells, and their analysis has revealed both similarities and differences between normal plasma cells and MM cells. Both cell types typically express CD138 (syndecan-1), which is considered to be a universal marker of both cell types; they also express CD38, although expression levels have been found to vary between cell types [10,11]. MM cells also show extensive somatic hypermutations of rearranged Ig genes and almost exclusively express Ig isotypes other than IgM, indicating a mature, post-follicular B-cell origin.
Fig. 2.

B-cell maturation and cell-surface marker expression. * Or other Ig isotype. Ig, immunoglobulin; VDJ, variable diversity, and joining.
MM cells express not only markers associated with plasma cells, but also markers associated with natural killer (NK) cells (CD56/NCAM), T cells (CD28); and occasionally also the pan-B-cell marker, CD20 [11]. Interestingly, cell-surface markers usually found on B-cells during the early stages of differentiation, such as CD10 and CD19, have been observed on subpopulations of MM cells [12–14]. This provides evidence for the existence of an early-lineage precursor for MM cells, which is further supported by the identification of unique MM idiotypic determinants in pre-B-cell populations [15] and mRNA expression of cell-surface proteins characteristic of myeloid, erythroid, and platelet lineages in MM cells [16]. Several studies have described varying frequencies of clonal cells expressing B-cell characteristics rather than plasma cell characteristics in the bone marrow and peripheral blood of patients with MM [17,18]. Such studies have suggested the presence of clonogenic stem cells in MM arising from a post-germinal center compartment [18,19], possibly equivalent to memory B cells [9]. The exact phenotype of the clonogenic cells in MM remains to be definitively established, and their role in the pathogenesis of disease is controversial [9]. The results of a recent study revealed the presence of a stem cell phenotype [20], and those of other studies have suggested that such clonogenic cells may be resistant to chemotherapy and that they may persist following treatment [14,21,22], making them a particularly interesting target for MM therapy.
Whatever the cell of origin, the majority of MM cases are characterized by complex chromosomal abnormalities (Table 1) [23–30]. Karyotype analysis has demonstrated the presence of two major cytogenetic categories: hyperdiploid MM, which includes numerous chromosomal trisomies and is associated with a low prevalence of IgH translocations; and non-hyperdiploid MM, which encompasses hypodiploid, pseudodiploid, and near-tetraploid MM and is associated with a high level of IgH rearrangements [23]. Some of the most frequent and early genetic events involve the IgH gene locus on 14q32, which is commonly part of a translocation [24]. Interphase fluorescence in situ hybridization (FISH) analysis has shown that these translocations are present in about half of patients with MGUS and up to 75% of patients with MM [24,29,31]. The most common of these translocations leads to dysregulation of oncogenes at translocation partner regions (Table 1) [23,24]. Frequent translocations involving the IgH gene locus and 14q32 are t(11;14)(q13;q32), t(4;14)(p16;q32), and t(14;16)(q32;q23) [23–27, 30], some of which have been associated with poor survival [28,32]. In terms of prognosis, deletion of 17p13, involving the tumor suppressor gene p53, is the most important cytogenetic factor; it is associated with worse treatment outcomes (Table 1) [23,24]. However, 13q deletion, which has been traditionally considered an adverse prognostic factor, is associated with poor prognosis only if other cytogenetic abnormalities, such as t(4;14) and deletion of 17p13, are present [28]. Although several genetic mutations seen in MM patients have been linked to disease progression, clinical findings, and response to therapy, it is important to note that the behavior of MM cells at the biological and clinical level is also crucially influenced by interactions between tumor cells and the bone marrow microenvironment [33].
Table 1.
| Chromosomal aberrations | Protein affected | Biological consequence | Frequency | Effect on treatment efficacy |
|---|---|---|---|---|
| t(11;14)(q13;q32) [23,24] | Cyclin D1 | Over-expression; cell cycle dysregulation | 15% | |
| t(4;14)(p16;q32) [24–27] | FGFR3 [23] or MMSET | Over-expression and activation; MM cell proliferation/apoptosis prevention MMSET probably linked to crucial transforming event | 25% | Poor survival [28,29] |
| t(14;16)(q32;q23) [30] | c-MAF | Over-expression; involvement in IL-4 regulation | 5% | Poor survival [28,29] |
| del 17p13 [23] | p53 | Cell-cycle dysregulation/apoptosis | Rare in newly diagnosed MM; frequency increasing in relapsed patients; not present in MGUS | Short survival, aggressive disease, short response duration following ASCT [23] |
ASCT, autologous stem-cell transplantation; del, deletion; FGFR3, fibroblast growth factor receptor 3; IL, interleukin; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; MMSET, multiple myeloma SET; t, translocation.
3. The role of the bone marrow microenvironment in MM pathogenesis
MM has become the prototypical tumor model for characterizing the interaction between tumor cells and their local milieu [34,35]. The bone marrow microenvironment refers not only to bone marrow stromal cells (BMSCs), but also to the non-cellular component composed of extracellular matrix (ECM) proteins such as collagen, fibronectin and laminin, and the extracellular fluid containing cytokines and growth factors. The bone marrow microenvironment supports normal hematopoiesis and these support mechanisms are harnessed by MM cells. The provision of various cytokines, growth factors, and receptors to MM cells increases their replicative capacity and confers resistance to pro-apoptotic signals, including those induced by conventional chemotherapy drugs [33]. The key processes include: direct MM cell–BMSC interactions and MM cell interactions with other components of the bone marrow microenvironment; indirect effects of cytokines produced by BMSCs or MM cells following such interactions; and the resulting activation of proliferative and anti-apoptotic signaling pathways (Fig. 3) [36]. The list of adhesion molecules and cytokines implicated in the pathophysiology of MM is extensive and expanding. The signaling cascades induced by these factors affect not only the proliferation and survival of tumor cells, but also other key aspects of MM pathology, including the development of osteolytic lesions and angiogenesis. A full discussion of the molecules and signaling pathways implicated in MM pathophysiology is beyond the scope of this article; however, some of the most important points are discussed below.
Fig. 3.
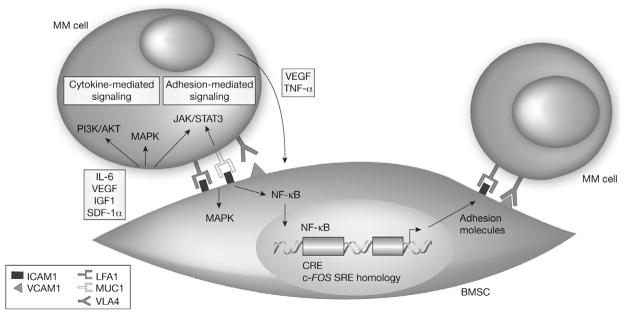
Multiple myeloma (MM) cell–bone marrow stromal cell (BMSC) interaction. Reproduced with permission from [36] © 2002, Rights Managed by Nature Publishing Group. AKT, protein kinase B; CRE, cyclic AMP-responsive element; ICAM1; intercellular adhesion molecule 1; IGF1, insulin-like growth factor-1; IL-6, interleukin-6; JAK, janus kinase; LFA1, lymphocyte function-associated antigen 1; MAPK, mitogen-activated protein kinase; muc1, mucin 1; NF-κB, nuclear factor κB; PI3K, phosphatidylinositol 3-kinase; SDF-1α, stromal-cell-derived factor-1α; SRE, serum response element; STAT3, signal transducer and activator of transcription 3; TNF-α, tumor necrosis factor-α; VCAM1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor; VLA4, very-late antigen 4.
Cell adhesion molecules (CAMs) such as CD44 (H-CAM), CD56 (N-CAM), members of the CD49 integrin family (including very-late antigen VLA-4 and VLA-5), lymphocyte function-associated antigen-1, syndecan-1, and selectin have been shown to mediate the interaction of tumor cells with, and their adherence to, ECM proteins and BMSCs [37]. These molecules play crucial roles in MM pathogenesis, including in the homing and localization of MM cells to the bone marrow [37]. Binding of MM cells to BMSCs induces the activation of p42/44 mitogen-activated protein kinase, and nuclear factor κB (NF-κB), resulting in increased expression of adhesion molecules on both MM cells and BMSCs, which in turn leads to increased production of cytokines, in particular interleukin (IL)-6 [38–40]. Adhesion molecules may also play a direct role in the response of MM to therapy; for example, MM cells resistant to melphalan and doxorubicin typically overexpress VLA-4. Adherence of VLA-4 to ECM proteins, such as fibronectin, induces CAM-mediated drug resistance, including up-regulation of p27 in tumor cells [41]. Concomitant exposure of MM cells to IL-6 and adhesion to fibronectin has been shown to result in an increase in STAT3 phosphorylation, nuclear translocation, and DNA binding, leading to transactivation of genes involved in proliferation, differentiation, and survival [42]. Another adhesion molecule, selectin, has been shown to modulate the interactions between MM cells and surrounding stromal cells; indeed, selectin inhibitors and the proteasome inhibitor bortezomib have been shown to reduce tumor burden [43].
Of the cytokines involved in MM pathogenesis, IL-6 is a major growth and survival factor for MM cells that is predominantly produced by BMSCs. Adhesion of MM cells to BMSCs up-regulates IL-6 secretion via NF-κB-dependent transcription [38,39]. IL-6 acts via its receptor (CD126) to activate signal transduction pathways (JAK/STAT3 and PI3K/Akt), inducing proliferation and preventing apoptosis, and thereby contributing to drug resistance [24,33]. IL-6 is known to mediate both MM cell proliferation and inhibition of Fas-induced apoptosis [44,45]. Additionally, it enhances production of vascular endothelial growth factor (VEGF) [44–46] and it may also play a role in the differentiation of osteoclasts [47].
Tumor necrosis factor (TNF)-α is a potent mediator of inflammation and bone resorption [48]. It has been shown to enhance MM-cell survival, trigger proliferation, and promote cell migration [49,50]. Levels of the pro-inflammatory chemokines mediated by TNF-α are higher in patients with MM than in patients with MGUS, suggesting that TNF-α levels increase with disease severity [51]. In MM, TNF-α also mediates the up-regulation of adhesion molecules on MM cells and BMSCs, which in turn results in activation of the NF-κB pathway [48]. High serum levels of TNF-α in MM patients are associated with osteolytic lesions, because TNF-α induces osteoclast differentiation [51]. B-cell activating factor (BAFF), a member of the TNF superfamily of proteins, is crucial for the maintenance and homeostasis of normal B-cell development, and has been shown to confer a survival advantage on MM cells [52–54]. MM cell adhesion to BMSCs augments BAFF production via NF-κB activation, and BAFF itself strengthens this adhesion [52].
Other relevant cytokines include insulin-like growth factor-1, a growth and survival factor in MM cells [55] that induces sustained activation of proliferative/anti-apoptotic signaling cascades (e.g., PI3K/Akt; IKK/NF-κB) [56]. VEGF is a pro-angiogenic molecule expression which is up-regulated by MM cell–BMSC adhesion, and is associated with tumor cell migration, growth, and survival [44,45]. VEGF also stimulates microvascular endothelial cells and BMSCs to increase IL-6 secretion, thus contributing further to MM cell proliferation and survival. IL-17 is a cytokine that, in addition to exerting an effect on cell survival [57], has also recently been identified as a key mediator of bone disease in myeloma [58]. Interestingly, the extent of lytic bone disease appears to be largely mediated by IL-17 produced by T cells, independent of the tumor burden, underscoring the crucial interplay of the immune system with the tumor microenvironment in the pathogenesis of MM [58].
It is clear that there is a complex web of autocrine and paracrine interactions between components of the bone marrow microenvironment and MM cells. However, other factors also contribute to the many aspects of MM pathophysiology, including the effects of the malignant cells on the immune system.
4. Immunodeficiency in MM
Immune dysfunction is an important feature of MM and is associated with an increased incidence of infections, which are a major cause of morbidity and mortality in myeloma patients. Importantly, immunodeficiency impacts disease progression and resistance to chemotherapy. Several factors produced as a result of MM cell–BMSC interactions also alter the functions of the host immune effector cells, thus interfering with immune surveillance and preventing immune-mediated tumor rejection [59].
MM patients show a pattern of global immunosuppression, with significantly reduced numbers of NK, B, and memory T cells, and low levels of non-myeloma Igs [60]. CD4+ and CD8+ T cells from MM patients show multiple abnormalities, particularly in more advanced stages, including reduced expression of cell-surface markers associated with T-cell signaling (such as CD28 and CD152), aberrant signal transduction, and impaired activation-induced cytokine production [61]. Defects have also been demonstrated in NK T cells [62] and antigen-presenting cells [63], as well as during B-cell differentiation and antibody responses [64,65]. Cytokine production resulting from MM cell–BMSC interactions is thought to play a role in immune dysfunction; for example, transforming growth factor-β has several effects in MM, including suppression of normal B-cell function [66].
Increasing knowledge about the interactions among cells, signaling proteins, and the bone marrow microenvironment has been important in providing potential targets for new therapies. Therapies that affect both the tumor cells and their bone marrow microenvironment, and that enable the patient’s own immune system to mount a more effective anti-MM response, are required.
5. Mode of action of immunomodulatory agents
Thalidomide, a synthetic derivative of glutamic acid, has been found to have a range of properties including anti-inflammatory effects via inhibition of TNF-α [67], inhibition of angiogenesis [68], and immunomodulatory properties, including enhancement of T cell- and NK cell-mediated immunity [69,70]. These properties stimulated interest in thalidomide as an anti-cancer drug, particularly for the treatment of MM. Thalidomide has been relatively successful in improving survival in patients with MM, initially as monotherapy and later in combination with dexa-methasone. However, it is associated with a range of toxicities in addition to its known teratogenic effect, including polyneuropathy, somnolence and, particularly when administered in combination with dexamethasone or chemotherapy, venous thromboembolism.
Immunomodulatory agents, synthetic analogs of thalidomide, were developed by Celgene Corporation to have increased potency with less toxicity compared with the parent compound [71]. Lenalidomide and pomalidomide are more potent inhibitors of TNF-α in vivo than thalidomide [72], and are more potent T-cell co-stimulators [73]. To date, clinical data on this class of compounds have come predominantly from studies on lenalidomide (phases I-IV), with pomalidomide currently in phase II–III development [73–78]. Lenalidomide is administered orally, has a favorable safety profile and has been shown to be highly effective in treating MM [79]. It is currently approved for use with dexamethasone in patients with MM who have received at least one prior therapy. Although their exact mode of action in MM remains unknown, studies suggest that immunomodulatory agents have a combination of anti-myeloma actions including direct tumoricidal effects, disruption of stromal cell support from the bone marrow microenvironment, and a number of immunomodulatory effects including anti-proliferative, apoptotic, anti-inflammatory, and anti-angiogenic effects. Recently, expression of cereblon (CRBN), a thalidomide-binding protein and teratogenic target [80] has been shown to be an essential requirement for immunomodulatory activity [81], and to be an important molecular target of lenalidomide and pomalidomide [82]. A positive association has been identified between high levels of CRBN expression and a good clinical response to treatment with lenalidomide and dexamethasone [83]. The multiple effects of immunomodulatory agents have been cited as the likely reason for the breadth of activity of this class of drugs [84].
6. Direct tumoricidal effects and modulation of the tumor microenvironment
Immunomodulatory agents have been shown to have several direct and indirect effects on MM cells, via both direct tumoricidal effects and modulation of the bone marrow microenvironment, including the prevention of angiogenesis and osteoclastogenesis.
Lenalidomide down-regulates expression of the MM cell survival factor interferon regulatory factor-4 [85–87]. Conversely, it induces the expression of cyclin-dependent kinase inhibitors, including p21, p27, and p15, and the early response transcription factors Egr1, Egr2, and Egr3, which are implicated in the regulation of tumor suppressor and cell-cycle regulatory genes [85,88,89]. Lenalidomide has also been shown to activate caspases, directly triggering tumor cell death [89,90], with the activation of caspases 3, 8, and 9 by lenalidomide being synergistically enhanced by dexamethasone [89,90].
Lenalidomide and pomalidomide also act by disrupting the stromal support within the bone marrow that is needed for the production of a range of cytokines including VEGF, IL-6, and TNF-α [72,91,92]. By inhibiting TNF-α expression, and thereby, reducing the expression of adhesion molecules on both MM cells and BMSCs [93], immunomodulatory agents have been shown to reduce levels of IL-6 induced by MM–BMSC interactions [44,91]. The down-regulation of adhesion molecules also has implications for signaling pathways. Indeed, lenalidomide has been shown to down-regulate NF-κB in vitro [92], resulting in reduced expression of anti-apoptotic proteins [93].
Angiogenesis in MM has been associated with active disease and the adhesion-molecule-mediated interactions between MM cells and the microvasculature have been implicated in the ability of a tumor to disseminate [94,95]. The anti-angiogenic effects of immunomodulatory agents are likely to be due to anti-migratory mechanisms mediated via modulation of chemotactic factors such as TNFα, VEGF, and basic fibroblast growth factor rather than direct inhibition of endothelial cell proliferation [92,96,97]. Lenalidomide has also been shown to inhibit growth factor-induced phosphorylation of Akt, a key signaling step in the Akt pathway involved in malignant transformation, chemoresistance, and invasiveness, by inducing cell survival, growth, migration, and angiogenesis [97,98]. Immunomodulatory agents have also been shown to directly inhibit osteoclast maturation, associated with a reduction in osteoclast expression of cathespin K, markers of osteoclast differentiation [99], and markers of bone metabolism [100].
7. Immunomodulatory properties
In vitro, immunomodulatory agents have been shown to augment both the adaptive and innate immune systems via enhancement of T-cell and NK-cell immune responses, both of which are reduced in MM patients [69,89,101–103]. Immunomodulatory agents induce cell-surface expression of positive co-stimulatory molecules on T cells, including CD28, which is down-regulated in MM. T-cell co-stimulation by immunomodulatory agents via the B7-CD28 pathway is associated with up-regulation of cytokines, such as IL-2 and interferon (IFN)-γ, which mediate T-cell activation, proliferation, and anti-tumor immune responses [73].
Treatment with immunomodulatory agents has been shown to diminish the expression of suppressor of cytokine signaling (SOCS)1, a negative regulator of IL-2 and IFN-γ signaling, in immune effector cells (CD4+, CD8+ cells, NKT cells, and NK cells) from both the peripheral blood and bone marrow of MM patients [104]. SOCS1 also negatively regulates IL-6 signaling, and is silenced by hypermethylation in approximately 75% of MM patients [105]. Interestingly, immunomodulatory agents were found to demethylate the SOCS1 gene; this not only abrogated IL-6 expression, but also enhanced the susceptibility of MM cell lines to cytotoxic T-lymphocyte killing [104].
The increase in production of immunostimulatory cytokines by immunomodulatory agents also results in augmentation of other aspects of the immune response, including NK-cell function and dendritic cell activity [69,103,106,107]. For example, immunomodulatory agents have been shown to augment NK-cell proliferation and activity in the presence of IL-2, resulting in NK cell-mediated lysis of MM cells [69]. The use of lenalidomide as salvage therapy after ASCT for MM resulted in increased levels of activated NK cells [108]. Lenalidomide has also been shown to enhance anti-tumor antibody-dependent cellular cytotoxicity as a result of increased IL-2 production by T cells [107,109]. This effect is reflected in patients responding to treatment with thalidomide, who showed increases in both the numbers of NK cells, and in IL-2 and IFN-γ secretion [69]. Taken together, these data suggest that immunomodulatory compounds exert a positive regulatory function, enhancing the anti-MM immune response [93,104].
There is also evidence that immunomodulatory agents regulate humoral immune responses in MM. The positive co-stimulatory surface marker ICOS and its ligand, which are up-regulated by immunomodulatory agents [104], regulate T-cell-mediated immune responses by controlling T-cell/B-cell interactions. This is achieved by stimulating the production of cytokines, such as IL-4 and IL-10, which play crucial roles in B-cell growth, maturation, and isotype switching [110]. Lenalidomide-based therapy was found to improve the humoral immune response in a significant proportion of responding patients [111]. In addition, MM patients treated with the polyvalent pneumococcal vaccine, in combination with lenalidomide, showed significantly greater B-cell and T-cell responses to the vaccine, underscoring its potential role as an immunostimulatory vaccine adjuvant [112].
Given the complexity of the immune response in vivo, the ultimate immunomodulatory effects of immunomodulatory agents in patients are probably considerably more complex and dynamic than current evidence indicates, and are also dependent on the individual’s immune status and cytokine profile in the bone marrow.
8. Mode of action of proteasome inhibitors
Proteasome inhibitors act by targeting intracellular protein turnover via inhibition of the ubiquitin–proteasome pathway [113]. The proteasome inhibitor bortezomib, a specific inhibitor of the 26S proteasome, was the first of its class to enter clinical trials for the treatment of MM. Bortezomib is administered by injection and is indicated for the treatment of patients who have already received at least one prior therapy and undergone, or are unsuitable for, bone marrow transplantation, or those patients with previously untreated MM who are ineligible for high-dose chemotherapy with bone marrow transplantation. The second generation proteasome inhibitor carfilzomib is currently undergoing phase III trials in patients with relapsed/refractory MM in the ASPIRE and FOCUS trials, as well as in newly diagnosed (CYCLONE) and elderly (NCT01279694) MM patients. Carfilzomib is selective and structurally distinct from bortezomib, and shows more sustained proteasome inhibition as its effect is mechanistically irreversible [114] it is being assessed in a head-to-head phase III trial versus bortezomib (ENDEAVOR study). As such, the majority of data on proteasome inhibitors in MM have been derived from studies on bortezomib.
9. Direct tumoricidal effects and modulation of the tumor microenvironment
The therapeutic effects of bortezomib probably result from a combination of direct toxicity and its effects on the bone marrow microenvironment [3,115]. Proteasome inhibition results in cytoplasmic accumulation of IκB, which blocks the nuclear translocation and transcriptional activity of NF-κB. As discussed previously, inhibition of NF-κB results in a decrease in expression of a range of adhesion molecules and cytokines such as IL-6, which is involved in the growth and survival of MM cells. In vitro, proteasome inhibitors have been shown to inhibit growth, induce apoptosis, and overcome drug resistance in MM cells [116,117]. The exact means by which “non-specific” proteasome inhibition results in selective killing of tumor cells is unclear [118]. Inhibition of the NF-κB pathway and associated down-regulation of inhibitors of apoptosis such as Bcl-2, A1, cIAP-2, and XIAP, promotion of endoplasmic reticulum stress-induced apoptosis, induction of p53-dependent apoptosis, and cell cycle disruption, have all been implicated in the selective killing of tumor cells by bortezomib [115,116,118,119]. In keeping with this, bortezomib has been shown to directly inhibit proliferation and induce apoptosis in MM cell lines and tumor cells from MM patients [36]. Bortezomib has also been shown to overcome chemoresistance following conventional chemotherapy in vitro via inhibition of NF-κB [117,120], and by lowering the apoptotic threshold of resistant cells via down-regulation of effectors involved in the cellular response to genotoxic stress induced by DNA-damaging chemotoxic agents [120].
Bortezomib has both direct and indirect effects on endothelial cells, inhibiting the proliferation of MM patient-derived endothelial cells and human umbilical vein endothelial cells, and inhibiting angiogenesis in vitro. It inhibits VEGF and IL-6 production dose-dependently, and down-regulates expression of angiopoietin-1 and -2 [121]. In addition, bortezomib has been shown to inhibit osteoclast differentiation and bone resorption activity in vitro via inhibition of p38, AP-1, and NF-κB [122], although clinical observations suggest that response to treatment is associated with osteoblast activation in patients with MM [123,124]. Inhibition of the ubiquitin–proteasome pathway also reportedly modulates osteoblast differentiation through up-regulation of the expression of bone morphogenetic protein-2 [125].
10. Effects on the immune system
Proteasome inhibition sensitizes tumor cells to NK cell-mediated lysis through TNF-related apoptosis-inducing ligand and/or Fas/Fas ligand pathways [126,127]. However, bortezomib has also been shown to disrupt NK-mediated immunity via induction of NK-cell apoptosis and suppression of NKp46 receptor-mediated cytotoxicity [128,129]. In addition, proteasome inhibitors induce apoptosis in activated and proliferating human T cells [130], interfere with the dendritic cell function [131] and antigen presentation [132], and suppress essential immune functions of CD4+ T cells [133], all of which contribute to an overall immunosuppressive action. Clinically, the immunomodulatory effects of bortezomib result in an increased incidence of herpes zoster reactivation during treatment [134]. The long-term effects of this action in patients with MM-induced immunosuppression are unclear.
11. Summary
In summary, MM is an incurable disease in which relapse is characterized by re-growth of residual tumor and immune suppression with a complex biology that affects many aspects of the disease and its response to treatment. As such, this disease requires effective long-term treatment strategies [135,136]. Conventional treatment, even with the addition of novel anti-myeloma agents, remains unsatisfactory. The disease is characterized by a pattern of regression and remission, and ultimately failure, indicating the presence of residual disease or resistant cells, even in patients who initially show a complete clinical response to treatment. This pattern suggests that continuous therapy may be required to maintain suppression of surviving tumor cells, which confound conventional therapy (Fig. 4), although unanswered questions remain regarding the long-term safety of continuous treatment with these agents.
Fig. 4.
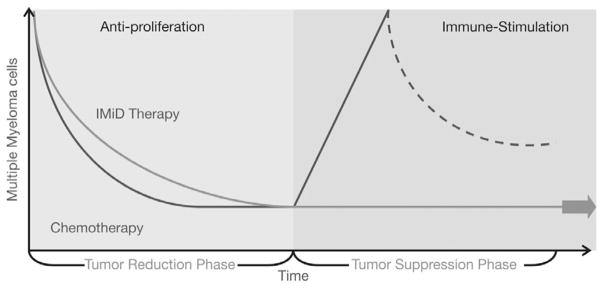
Continuous therapy with IMiD® immunomodulatory agents versus conventional chemotherapy.
Studies of the biology of MM have highlighted the need for agents to target not only the tumor cells themselves, but also to disrupt their supportive microenvironment in the bone marrow. Immunomodulatory agents and proteasome inhibitors act directly on malignant clones and also on their environment through inhibition of stromal cell support. This may explain the high clinical response rates associated with treatment regimens involving these agents. In addition to their ability to directly induce tumor cell death and to interfere with tumor cell–microenvironment interactions, immunomodulatory agents also offer the potential to enhance anti-tumor immune responses in MM as a consequence of the immunostimulatory effects they exert on both cellular and humoral immunity. The ability to modify the biology of MM using such new therapies raises the question of whether a change in treatment paradigm towards continuous therapy, providing both tumor reduction and tumor suppression, is warranted. Future studies are needed to test such paradigms.
Acknowledgments
We thank Shanthi Jayawardena, PhD, and Eva Polk, PhD (Excerpta Medica), for writing assistance in the preparation of the manuscript. Editorial support was funded by Celgene Corporation. The authors were fully responsible for all content and editorial decisions for this manuscript.
Footnotes
Conflict of interest statement
Dr. Borrello, MD, obtains research support and serves as a consultant to Celgene Corporation.
References
- 1.Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564–9. doi: 10.1056/NEJMoa01133202. [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA, Kumar S. The significance of monoclonal gammopathy of undetermined significance. Haematologica. 2009;94(12):1641–4. doi: 10.3324/haematol.2009.013961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351(18):1860–73. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 4.Kyle RA, Gertz MA, Witzig TE, Lust JA, Lacy MQ, Dispenzieri A, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21–33. doi: 10.4065/78.1.21. [DOI] [PubMed] [Google Scholar]
- 5.Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–20. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turesson I, Velez R, Kristinsson SY, Landgren O. Patterns of improved survival in patients with multiple myeloma in the twenty-first century: a population-based study. J Clin Oncol. 2010;28(5):830–4. doi: 10.1200/JCO.2009.25.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar SV, Therneau TM, Gertz MA, Lacy MQ, Dispenzieri A, Rajkumar SV, et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin Proc. 2004;79(7):867–74. doi: 10.4065/79.7.867. [DOI] [PubMed] [Google Scholar]
- 8.Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103(6):2332–6. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huff CA, Matsui W. Multiple myeloma cancer stem cells. J Clin Oncol. 2008;26(17):2895–900. doi: 10.1200/JCO.2007.15.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood. 1984;63(6):1424–33. [PubMed] [Google Scholar]
- 11.Bataille R, Jégo G, Robillard N, Barillé-Nion S, Harousseau JL, Moreau P, et al. The phenotype of normal, reactive and malignant plasma cells. Identification of “many and multiple myelomas” and of new targets for myeloma therapy. Haematologica. 2006;91(9):1234–40. [PubMed] [Google Scholar]
- 12.Grogan TM, Durie BG, Lomen C, Spier C, Wirt DP, Nagle R, et al. Delineation of a novel pre-B cell component in plasma cell myeloma: immunochemical, immunophenotypic, genotypic, cytologic, cell culture, and kinetic features. Blood. 1987;70(4):932–42. [PubMed] [Google Scholar]
- 13.Cao J, Vescio RA, Rettig MB, Hong CH, Kim A, Lee JC, et al. A CD10-positive subset of malignant cells is identified in multiple myeloma using PCR with patient-specific immunoglobulin gene primers. Leukemia. 1995;9(11):1948–53. [PubMed] [Google Scholar]
- 14.Kiel K, Cremer FW, Rottenburger C, Kallmeyer C, Ehrbrecht E, Atzberger A, et al. Analysis of circulating tumor cells in patients with multiple myeloma during the course of high-dose therapy with peripheral blood stem cell transplantation. Bone Marrow Transplant. 1999;23(10):1019–27. doi: 10.1038/sj.bmt.1701767. [DOI] [PubMed] [Google Scholar]
- 15.Kubagawa H, Vogler LB, Capra JD, Conrad ME, Lawton AR, Cooper MD. Studies on the clonal origin of multiple myeloma. Use of individually specific (idiotype) antibodies to trace the oncogenic event to its earliest point of expression in B-cell differentiation. J Exp Med. 1979;150(4):792–807. doi: 10.1084/jem.150.4.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epstein J, Xiao HQ, He XY. Markers of multiple hematopoietic-cell lineages in multiple myeloma. N Engl J Med. 1990;322(10):664–8. doi: 10.1056/NEJM199003083221005. [DOI] [PubMed] [Google Scholar]
- 17.Billadeau D, Ahmann G, Greipp P, Van Ness B. The bone marrow of multiple myeloma patients contains B cell populations at different stages of differentiation that are clonally related to the malignant plasma cell. J Exp Med. 1993;178(3):1023–31. doi: 10.1084/jem.178.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakkus MH, Van Riet I, Van Camp B, Thielemans K. Evidence that the clonogenic cell in multiple myeloma originates from a pre-switched but somatically mutated B cell. Br J Haematol. 1994;87(1):68–74. doi: 10.1111/j.1365-2141.1994.tb04872.x. [DOI] [PubMed] [Google Scholar]
- 19.Vescio RA, Cao J, Hong CH, Lee JC, Wu CH, Der Danielian M, et al. Myeloma Ig heavy chain V region sequences reveal prior antigenic selection and marked somatic mutation but no intraclonal diversity. J Immunol. 1995;155(5):2487–97. [PubMed] [Google Scholar]
- 20.Svachova H, Pour L, Sana J, Kovarova L, Muthu Raja KR, Hajek R. Stem cell marker nestin is expressed in plasma cells of multiple myeloma patients. Leuk Res. 2011;35(8):1008–13. doi: 10.1016/j.leukres.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Pilarski LM, Belch AR. Circulating monoclonal B cells expressing P glycoprotein may be a reservoir of multidrug-resistant disease in multiple myeloma. Blood. 1994;83(3):724–36. [PubMed] [Google Scholar]
- 22.Rottenburger C, Kiel K, Bösing T, Cremer FW, Moldenhauer G, Ho AD, et al. Clonotypic CD20+ and CD19+ B cells in peripheral blood of patients with multiple myeloma post high-dose therapy and peripheral blood stem cell transplantation. Br J Haematol. 1999;106(2):545–52. doi: 10.1046/j.1365-2141.1999.01548.x. [DOI] [PubMed] [Google Scholar]
- 23.Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, et al. International Myeloma Working Group. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210–21. doi: 10.1038/leu.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seidl S, Kaufmann H, Drach J. New insights into the pathophysiology of multiple myeloma. Lancet Oncol. 2003;4(9):557–64. doi: 10.1016/s1470-2045(03)01195-1. [DOI] [PubMed] [Google Scholar]
- 25.Chesi M, Nardini E, Brents LA, Schröck E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32. 3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16(3):260–4. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plowright EE, Li Z, Bergsagel PL, Chesi M, Barber DL, Branch DR, et al. Ectopic expression of fibroblast growth factor receptor 3 promotes myeloma cell proliferation and prevents apoptosis. Blood. 2000;95(3):992–8. [PubMed] [Google Scholar]
- 27.Martinez-Garcia E, Popovic R, Min D-J, Sweet SM, Thomas PM, Zamdborg L, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4; 14) multiple myeloma cells. Blood. 2011;117(1):211–20. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007;109(8):3489–95. doi: 10.1182/blood-2006-08-040410. [DOI] [PubMed] [Google Scholar]
- 29.Fonseca R, Bailey RJ, Ahmann GJ, Rajkumar SV, Hoyer JD, Lust JA, et al. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood. 2002;100(4):1417–24. [PubMed] [Google Scholar]
- 30.Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, et al. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood. 1998;91(12):4457–63. [PubMed] [Google Scholar]
- 31.Avet-Loiseau H, Brigaudeau C, Morineau N, Talmant P, Laï JL, Daviet A, et al. High incidence of cryptic translocations involving the Ig heavy chain gene in multiple myeloma, as shown by fluorescence in situ hybridization. Genes Chromosomes Cancer. 1999;24(1):9–15. doi: 10.1002/(sici)1098-2264(199901)24:1<9::aid-gcc2>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 32.Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569–75. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- 33.Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PG, Anderson KC. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: interplay of growth factors, their receptors and stromal interactions. Eur J Cancer. 2006;42(11):1564–73. doi: 10.1016/j.ejca.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 34.Mitsiades CS, Mitsiades NS, Richardson PG, Munshi NC, Anderson KC. Multiple myeloma: a prototypic disease model for the characterization and therapeutic targeting of interactions between tumor cells and their local microenvironment. J Cell Biochem. 2007;101(4):950–68. doi: 10.1002/jcb.21213. [DOI] [PubMed] [Google Scholar]
- 35.Mitsiades CS, McMillin DW, Klippel S, Hideshima T, Chauhan D, Richardson PG, et al. The role of the bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol Oncol Clin North Am. 2007;21(6):1007–34. vii–viii. doi: 10.1016/j.hoc.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Hideshima T, Anderson KC. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat Rev Cancer. 2002;2(12):927–37. doi: 10.1038/nrc952. [DOI] [PubMed] [Google Scholar]
- 37.Teoh G, Anderson KC. Interaction of tumor and host cells with adhesion and extracellular matrix molecules in the development of multiple myeloma. Hematol Oncol Clin North Am. 1997;11(1):27–42. doi: 10.1016/s0889-8588(05)70413-5. [DOI] [PubMed] [Google Scholar]
- 38.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87(3):1104–12. [PubMed] [Google Scholar]
- 39.Uchiyama H, Barut BA, Mohrbacher AF, Chauhan D, Anderson KC. Adhesion of human myeloma-derived cell lines to bone marrow stromal cells stimulates interleukin-6 secretion. Blood. 1993;82(12):3712–20. [PubMed] [Google Scholar]
- 40.Hideshima T, Akiyama M, Hayashi T, Richardson P, Schlossman R, Chauhan D, et al. Targeting p38 MAPK inhibits multiple myeloma cell growth in the bone marrow milieu. Blood. 2003;101(2):703–5. doi: 10.1182/blood-2002-06-1874. [DOI] [PubMed] [Google Scholar]
- 41.St Croix B, Florenes VA, Rak JW, Flanagan M, Bhattacharya N, Slingerland JM, et al. Impact of the cyclin-dependent kinase inhibitor p27Kip1 on resistance of tumor cells to anticancer agents. Nat Med. 1996;2(11):1204–10. doi: 10.1038/nm1196-1204. [DOI] [PubMed] [Google Scholar]
- 42.Shain KH, Yarde DN, Meads MB, Huang M, Jove R, Hazlehurst LA, et al. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009;69(3):1009–15. doi: 10.1158/0008-5472.CAN-08-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azab AK, Quang P, Azab F, Magnani J, Patton J, Smith T, et al. Selectin inhibition disrupts multiple myeloma cells interaction with the bone marrow microenvironment and sensitizes them to therapy. Blood. 2010;116:Abstract 453. [Google Scholar]
- 44.Gupta D, Treon SP, Shima Y, Hideshima T, Podar K, Tai YT, et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15(12):1950–61. doi: 10.1038/sj.leu.2402295. [DOI] [PubMed] [Google Scholar]
- 45.Dankbar B, Padró T, Leo R, Feldmann B, Kropff M, Mesters RM, et al. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000;95(8):2630–6. [PubMed] [Google Scholar]
- 46.Chauhan D, Kharbanda S, Ogata A, Urashima M, Teoh G, Robertson M, et al. Interleukin-6 inhibits Fas-induced apoptosis and stress-activated protein kinase activation in multiple myeloma cells. Blood. 1997;89(1):227–34. [PubMed] [Google Scholar]
- 47.Löwik CW, van der Pluijm G, Bloys H, Hoekman K, Bijvoet OL, Aarden LA, et al. Parathyroid hormone (PTH) and PTH-like protein (PLP) stimulate interleukin-6 production by osteogenic cells: a possible role of interleukin-6 in osteoclastogenesis. Biochem Biophys Res Commun. 1989;162(3):1546–52. doi: 10.1016/0006-291x(89)90851-6. [DOI] [PubMed] [Google Scholar]
- 48.Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene. 2001;20(33):4519–27. doi: 10.1038/sj.onc.1204623. [DOI] [PubMed] [Google Scholar]
- 49.Jourdan M, Tarte K, Legouffe E, Brochier J, Rossi JF, Klein B. Tumor necrosis factor is a survival and proliferation factor for human myeloma cells. Eur Cytokine Netw. 1999;10(1):65–70. [PMC free article] [PubMed] [Google Scholar]
- 50.Jöhrer K, Janke K, Krugmann J, Fiegl M, Greil R. Transendothelial migration of myeloma cells is increased by tumor necrosis factor (TNF)-alpha via TNF receptor 2 and autocrine up-regulation of MCP-1. Clin Cancer Res. 2004;10(6):1901–10. doi: 10.1158/1078-0432.ccr-1053-03. [DOI] [PubMed] [Google Scholar]
- 51.Giuliani N, Lisignoli G, Colla S, Lazzaretti M, Storti P, Mancini C, et al. CC-Chemokine ligand 20/macrophage inflammatory protein-3α and CC-chemokine receptor 6 are overexpressed in myeloma microenvironment related to osteolytic bone lesions. Cancer Res. 2008;68(16):6840–50. doi: 10.1158/0008-5472.CAN-08-0402. [DOI] [PubMed] [Google Scholar]
- 52.Tai YT, Li XF, Breitkreutz I, Song W, Neri P, Catley L, et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006;66(13):6675–82. doi: 10.1158/0008-5472.CAN-06-0190. [DOI] [PubMed] [Google Scholar]
- 53.Neri P, Kumar S, Fulciniti MT, Vallet S, Chhetri S, Mukherjee S, et al. Neutralizing B-cell activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin Cancer Res. 2007;13(19):5903–9. doi: 10.1158/1078-0432.CCR-07-0753. [DOI] [PubMed] [Google Scholar]
- 54.Moreaux J, Legouffe E, Jourdan E, Quittet P, Rème T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103(8):3148–57. doi: 10.1182/blood-2003-06-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Georgii-Hemming P, Wiklund HJ, Ljunggren O, Nilsson K. Insulin-like growth factor I is a growth and survival factor in human multiple myeloma cell lines. Blood. 1996;88(6):2250–8. [PubMed] [Google Scholar]
- 56.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Akiyama M, et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell. 2004;5(3):221–30. doi: 10.1016/s1535-6108(04)00050-9. [DOI] [PubMed] [Google Scholar]
- 57.Prabhala RH, Pelluru D, Fulciniti M, Prabhala HK, Nanjappa P, Song W, et al. Elevated IL-17 produced by Th17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood. 2010;115(26):5385–92. doi: 10.1182/blood-2009-10-246660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Noonan K, Marchionni L, Anderson J, Pardoll D, Roodman GD, Borrello I. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood. 2010;116(18):3554–63. doi: 10.1182/blood-2010-05-283895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cook G, Campbell JD. Immune regulation in multiple myeloma: the host-tumour conflict. Blood Rev. 1999;13(3):151–62. doi: 10.1054/blre.1999.0111. [DOI] [PubMed] [Google Scholar]
- 60.Schütt P, Brandhorst D, Stellberg W, Poser M, Ebeling P, Müller S, et al. Immune parameters in multiple myeloma patients: influence of treatment and correlation with opportunistic infections. Leuk Lymphoma. 2006;47(8):1570–82. doi: 10.1080/10428190500472503. [DOI] [PubMed] [Google Scholar]
- 61.Mozaffari F, Hansson L, Kiaii S, Ju X, Rossmann ED, Rabbani H, et al. Signalling molecules and cytokine production in T cells of multiple myeloma-increased abnormalities with advancing stage. Br J Haematol. 2004;124(3):315–24. doi: 10.1046/j.1365-2141.2003.04789.x. [DOI] [PubMed] [Google Scholar]
- 62.Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med. 2003;197(12):1667–76. doi: 10.1084/jem.20021650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ratta M, Fagnoni F, Curti A, Vescovini R, Sansoni P, Oliviero B, et al. Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood. 2002;100(1):230–7. doi: 10.1182/blood.v100.1.230. [DOI] [PubMed] [Google Scholar]
- 64.Rawstron AC, Davies FE, Owen RG, English A, Pratt G, Child JA, et al. B-lymphocyte suppression in multiple myeloma is a reversible phenomenon specific to normal B-cell progenitors and plasma cell precursors. Br J Haematol. 1998;100(1):176–83. doi: 10.1046/j.1365-2141.1998.00525.x. [DOI] [PubMed] [Google Scholar]
- 65.Li J, Li M, Wang C, Wang J, Sanchez E, Li ZW, et al. Lack of binding of monocolonal protein produced by multiple meloma (MM) cells to FcγRIIb reduces immunoreceptor tyrosine-based inhibitory motif (ITIM) signaling and provides the rationale for a novel therapeutic approach to MM. Blood. 2010;116:Abstract 455. [Google Scholar]
- 66.Urashima M, Ogata A, Chauhan D, Hatziyanni M, Vidriales MB, Dedera DA, et al. Transforming growth factor-beta1: differential effects on multiple myeloma versus normal B cells. Blood. 1996;87(5):1928–38. [PubMed] [Google Scholar]
- 67.Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med. 1991;173(3):699–703. doi: 10.1084/jem.173.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. 1994;91(9):4082–5. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood. 2001;98(1):210–6. doi: 10.1182/blood.v98.1.210. [DOI] [PubMed] [Google Scholar]
- 70.Haslett PAJ, Corral LG, Albert M, Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med. 1998;187(11):1885–92. doi: 10.1084/jem.187.11.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muller GW, Corral LG, Shire MG, Wang H, Moreira A, Kaplan G, et al. Structural modifications of thalidomide produce analogs with enhanced tumor necrosis factor inhibitory activity. J Med Chem. 1996;39(17):3238–40. doi: 10.1021/jm9603328. [DOI] [PubMed] [Google Scholar]
- 72.Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, et al. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-α. J Immunol. 1999;163(1):380–6. [PubMed] [Google Scholar]
- 73.LeBlanc R, Hideshima T, Catley LP, Shringarpure R, Burger R, Mitsiades N, et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood. 2004;103(5):1787–90. doi: 10.1182/blood-2003-02-0361. [DOI] [PubMed] [Google Scholar]
- 74.Lacy MQ, Hayman SR, Gertz MA, Dispenzieri A, Buadi F, Kumar S, et al. Pomalidomide (CC4047) plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J Clin Oncol. 2009;27(30):5008–14. doi: 10.1200/JCO.2009.23.6802. [DOI] [PubMed] [Google Scholar]
- 75.Lacy MQ, Hayman SR, Gertz MA, Short KD, Dispenzieri A, Kumar S, et al. Pomalidomide (CC4047) plus low dose dexamethasone (Pom/dex) is active and well tolerated in lenalidomide refractory multiple myeloma (MM) Leukemia. 2010;24(11):1934–9. doi: 10.1038/leu.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lacy MQ, Allred JB, Gertz MA, Hayman SR, Short KD, Buadi F, et al. Pomalidomide plus low-dose dexamethasone in myeloma refractory to both bortezomib and lenalidomide: comparison of 2 dosing strategies in dual-refractory disease. Blood. 2011;118(11):2970–5. doi: 10.1182/blood-2011-04-348896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leleu X, Attal M, Arnulf B, Duhamel A, Moreau P, Traulle C, et al. Phase 2 multicenter, randomized open label study of 2 modalities of pomalidomide plus low-dose dexamethasone (POMD) in patients with multiple myeloma (MM), refractory to both lenalidomide and bortezomib (double refractory). IFM 2009-02. Heamatologica. 2011;96(Suppl 1):S72. [Abstract P-148] [Google Scholar]
- 78.Richardson PG, Siegel DS, Vij R, Hofmeister CC, Jagannath S, Chen C, et al. Randomized, open label phase 1/2 study of pomalidomide (POM) alone or in combination with low-dose dexamethasone (LoDex) in patients (pts) with relapsed and refractory multiple myeloma who have received prior treatment that includes lenalidomide (LEN) and bortezomib (BORT): phase 2 results. Blood. 2011;118 [Abstract 634] [Google Scholar]
- 79.Zeldis JB, Knight RD, Jacques C, Tozer A, Bizzari JP. Lenalidomide in multiple myeloma: current role and future directions. Expert Opin Pharmacother. 2010;11(5):829–42. doi: 10.1517/14656561003645611. [DOI] [PubMed] [Google Scholar]
- 80.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–50. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 81.Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118(18):4771–9. doi: 10.1182/blood-2011-05-356063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012 May 3; doi: 10.1038/leu.2012.119. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heintel D, Bolomsky A, Schreder M, Pfeifer S, Zojer N, Jäger U, et al. High expression of the thalidomide-binding protein cereblon (CRBN) is associated with improved clinical response in patients with multiple myeloma treated with lenalidomide and dexamethasone. Blood (ASH Annual Meeting Abstracts) 2011;118:Abstract 2879. [Google Scholar]
- 84.List AF. Lenalidomide – the phoenix rises. N Engl J Med. 2007;357(21):2183–6. doi: 10.1056/NEJMe078203. [DOI] [PubMed] [Google Scholar]
- 85.Lopez-Girona A, Heintel D, Zhang LH, Mendy D, Gaidarova S, Brady H, et al. Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response. Br J Haematol. 2011;154(3):325–36. doi: 10.1111/j.1365-2141.2011.08689.x. [DOI] [PubMed] [Google Scholar]
- 86.Li S, Pal R, Monaghan SA, Schafer P, Ouyang H, Mapara M, et al. IMiD immunomodulatory compounds block C/EBPβ translation through eIF4E down-regulation resulting in inhibition of MM. Blood. 2011;117(19):5157–65. doi: 10.1182/blood-2010-10-314278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang X, Di Liberto M, Jayabalan D, Hussein M, Randolph S, Niesvizky R, et al. Lenalidomide targets myeloma cells preferentially during prolonged early G1 arrest but not synchronization into S phase by selective and reversible inhibition of CDK4/CDK6 through loss of IRF-4. Blood (ASH Annual Meeting Abstracts) 2010;116 [Abstract 449] [Google Scholar]
- 88.Escoubet-Lozach L, Lin IL, Jensen-Pergakes K, Brady HA, Gandhi AK, Schafer PH, et al. Pomalidomide and lenalidomide induce p21 WAF-1 expression in both lymphoma and multiple myeloma through a LSD1-mediated epigenetic mechanism. Cancer Res. 2009;69(18):7347–56. doi: 10.1158/0008-5472.CAN-08-4898. [DOI] [PubMed] [Google Scholar]
- 89.Gandhi AK, Kang J, Capone L, Parton A, Wu L, Zhang LH, et al. Dexamethasone synergizes with lenalidomide to inhibit multiple myeloma tumor growth, but reduces lenalidomide-induced immunomodulation of T and NK cell function. Curr Cancer Drug Targets. 2010;10(2):155–67. doi: 10.2174/156800910791054239. [DOI] [PubMed] [Google Scholar]
- 90.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, et al. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood. 2002;99(12):4525–30. doi: 10.1182/blood.v99.12.4525. [DOI] [PubMed] [Google Scholar]
- 91.Hideshima T, Chauhan D, Shima Y, Raje N, Davies FE, Tai YT, et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood. 2000;96(9):2943–50. [PubMed] [Google Scholar]
- 92.De Luisi A, Ferrucci A, Coluccia AML, Ria R, Moschetta M, de Luca E, et al. Lenalidomide restrains motility and overangiogenic potential of bone marrow endothelial cells in patients with active multiple myeloma. Clin Can Res. 2011;17(7):1935–46. doi: 10.1158/1078-0432.CCR-10-2381. [DOI] [PubMed] [Google Scholar]
- 93.Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, Smyth MJ, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22–32. doi: 10.1038/leu.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, et al. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994;87(3):503–8. doi: 10.1111/j.1365-2141.1994.tb08304.x. [DOI] [PubMed] [Google Scholar]
- 95.Vacca A, Di LM, Ribatti D, Di Stefano R, Gadaleta-Caldarola G, Iodice G, et al. Bone marrow of patients with active multiple myeloma: angiogenesis and plasma cell adhesion molecules LFA-1, VLA-4, LAM-1, and CD44. Am J Hematol. 1995;50(1):9–14. doi: 10.1002/ajh.2830500103. [DOI] [PubMed] [Google Scholar]
- 96.Dredge K, Marriott JB, Macdonald CD, Man HW, Chen R, Muller GW, et al. Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br J Cancer. 2002;87(10):1166–72. doi: 10.1038/sj.bjc.6600607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dredge K, Horsfall R, Robinson SP, Zhang LH, Lu L, Tang Y, et al. Orally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvasc Res. 2005;69(1–2):56–63. doi: 10.1016/j.mvr.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 98.Jakubikova J, Adamia S, Kost-Alimova M, Klippel S, Cervi D, Daley JF, et al. Lenalidomide targets clonogenic side population in multiple myeloma: pathophysiologic and clinical implications. Blood. 2011;117(17):4409–19. doi: 10.1182/blood-2010-02-267344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Breitkreutz I, Raab MS, Vallet S, Hideshima T, Raje N, Mitsiades C, et al. Lenalidomide inhibits osteoclastogenesis, survival factors and bone-remodeling markers in multiple myeloma. Leukemia. 2008;22(10):1925–32. doi: 10.1038/leu.2008.174. [DOI] [PubMed] [Google Scholar]
- 100.Terpos E, Christoulas D, Katodritou E, Kastritis E, Kyrtsonis MC, Papatheodorou A, et al. The effect of lenalidomide and dexamethasone combination on bone remodeling of relapsed/refractory myeloma: final results of two studies of the Greek Myeloma Study Group with 205 patients. Haematologica (EHA Annual Meeting Abstracts) 2011;96(Suppl 2):117. [Abstract 0283] [Google Scholar]
- 101.Schafer PH, Gandhi AK, Loveland MA, Chen RS, Man HW, Schnetkamp PP, et al. Enhancement of cytokine production and AP-1 transcriptional activity in T cells by thalidomide-related immunomodulatory drugs. J Pharmacol Exp Ther. 2003;305(3):1222–32. doi: 10.1124/jpet.102.048496. [DOI] [PubMed] [Google Scholar]
- 102.Haslett PAG, Hanekom WA, Muller G, Kaplan G. Thalidomide and a thalidomide analogue drug costimulate virus-specific CD8+ T cells in vitro. J Infect Dis. 2003;187(6):946–55. doi: 10.1086/368126. [DOI] [PubMed] [Google Scholar]
- 103.Chang DH, Liu N, Klimek V, Hassoun H, Mazumder A, Nimer SD, et al. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications. Blood. 2006;108(2):618–21. doi: 10.1182/blood-2005-10-4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Görgün G, Calabrese E, Soydan E, Hideshima T, Perrone G, Bandi M, et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood. 2010;116(17):3227–37. doi: 10.1182/blood-2010-04-279893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yamamoto M, Nishimoto N, Davydova J, Kishimoto T, Curiel DT. Suppressor of cytokine signaling-1 expression by infectivity-enhanced adenoviral vector inhibits IL-6-dependent proliferation of multiple myeloma cells. Cancer Gene Ther. 2006;13(2):194–202. doi: 10.1038/sj.cgt.7700873. [DOI] [PubMed] [Google Scholar]
- 106.Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, Roth M, Vaughn M, Knight J, et al. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol. 2008;140(1):36–45. doi: 10.1111/j.1365-2141.2007.06841.x. [DOI] [PubMed] [Google Scholar]
- 107.Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res. 2008;14(14):4650–7. doi: 10.1158/1078-0432.CCR-07-4405. [DOI] [PubMed] [Google Scholar]
- 108.Lioznov M, El-Cheikh J, Jr, Hoffmann F, Hildebrandt Y, Ayuk F, Wolschke C, et al. Lenalidomide as salvage therapy after allo-SCT for multiple myeloma is effective and leads to an increase of activated NK (NKp44(+)) and T (HLA-DR(+)) cells. Bone Marrow Transplant. 2010;45(2):349–53. doi: 10.1038/bmt.2009.155. [DOI] [PubMed] [Google Scholar]
- 109.Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol. 2005;128(2):192–203. doi: 10.1111/j.1365-2141.2004.05286.x. [DOI] [PubMed] [Google Scholar]
- 110.Suh WK, Tafuri A, Berg-Brown NN, Shahinian A, Plyte S, Duncan GS, et al. The inducible costimulator plays the major costimulatory role in humoral immune responses in the absence of CD28. J Immunol. 2004;172(10):5917–23. doi: 10.4049/jimmunol.172.10.5917. [DOI] [PubMed] [Google Scholar]
- 111.Baz R, Dimopoulos M, Richardson P, Yu Z, Hussein M, Chanan-Khan A. Lenalaidomide-based thearapy leads to improvement of humoral immune system in relapsed or refractory mutliple myeloma patients who respond to the therapy. Haematologica. 2009;94(Suppl 2):159. [Abstract 0395] [Google Scholar]
- 112.Noonan KA, Rudraraju L, Ferguson A, Emerling A, Pasetti MF, Huff CA, et al. Lenalidomide augments immune responses to Prevnar vaccination in relapsed myeloma patients: implications for cancer and infectious vaccines. Clin Cancer Res. 2012;18(5):1426–34. doi: 10.1158/1078-0432.CCR-11-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Myung J, Kim KB, Crews CM. The ubiquitin-proteasome pathway and proteasome inhibitors. Med Res Rev. 2001;21(4):245–73. doi: 10.1002/med.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteosome. Cancer Res. 2007;67(13):6383–91. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 115.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci USA. 2002;99(22):14374–9. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–6. [PubMed] [Google Scholar]
- 117.Ma MH, Yang HH, Parker K, Manyak S, Friedman JM, Altamirano C, et al. The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin Cancer Res. 2003;9(3):1136–44. [PubMed] [Google Scholar]
- 118.Yang Y, Kitagaki J, Wang H, Hou DX, Perantoni AO. Targeting the ubiquitin-proteasome system for cancer therapy. Cancer Sci. 2009;100(1):24–28. doi: 10.1111/j.1349-7006.2008.01013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Piazza F, Manni S, Brancalion A, Quotti Tubi L, Pavan L, Gurrieri C, et al. Inhibition of protein kinase CK2 enhances the cytotoxic effects of bortezomib on multiple myeloma cells. Hematologica (EHA Annual Meeting Abstracts) 2011;96(Suppl 2):117. [Abstract 0284] [Google Scholar]
- 120.Mitsiades N, Mitsiades CS, Richardson PG, Poulaki V, Tai YT, Chauhan D, et al. The proteasome inhibitor PS–341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101(6):2377–80. doi: 10.1182/blood-2002-06-1768. [DOI] [PubMed] [Google Scholar]
- 121.Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006;66(1):184–91. doi: 10.1158/0008-5472.CAN-05-1195. [DOI] [PubMed] [Google Scholar]
- 122.von Metzler I, Krebbel H, Hecht M, Manz RA, Fleissner C, Mieth M, et al. Bortezomib inhibits human osteoclastogenesis. Leukemia. 2007;21(9):2025–34. doi: 10.1038/sj.leu.2404806. [DOI] [PubMed] [Google Scholar]
- 123.Zangari M, Esseltine D, Lee CK, Barlogie B, Elice F, Burns MJ, et al. Response to bortezomib is associated to osteoblastic activation in patients with multiple myeloma. Br J Haematol. 2005;131(1):71–3. doi: 10.1111/j.1365-2141.2005.05733.x. [DOI] [PubMed] [Google Scholar]
- 124.Lund T, Søe K, Abildgaard N, Garnero P, Pedersen PT, Ormstrup T, et al. First-line treatment with bortezomib rapidly stimulates both osteoblast activity and bone matrix deposition in patients with multiple myeloma, and stimulates osteoblast proliferation and differentiation in vitro. Eur J Haematol. 2010;85(4):290–9. doi: 10.1111/j.1600-0609.2010.01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Garrett IR, Chen D, Gutierrez G, Zhao M, Escobedo A, Rossini G, et al. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J Clin Invest. 2003;111(11):1771–82. doi: 10.1172/JCI16198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, et al. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006;66(14):7317–25. doi: 10.1158/0008-5472.CAN-06-0680. [DOI] [PubMed] [Google Scholar]
- 127.Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180(1):163–70. doi: 10.4049/jimmunol.180.1.163. [DOI] [PubMed] [Google Scholar]
- 128.Wang X, Ottosson A, Ji C, Feng X, Nordenskjöld M, Henter JI, et al. Proteasome inhibition induces apoptosis in primary human natural killer cells and suppresses NKp46-mediated cytotoxicity. Haematologica. 2009;94(4):470–8. doi: 10.3324/haematol.13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Feng X, Yan J, Wang Y, Zierath JR, Nordenskjöld M, Henter JI, et al. Proteosome inhibitor bortezomib disrupts tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression and natural killer (NK) cell killing of TRAIL receptor-positive multiple myeloma cells. Blood (ASH Annual Meeting Abstracts) 2010;116 doi: 10.1016/j.molimm.2010.05.003. [Abstract 5021] [DOI] [PubMed] [Google Scholar]
- 130.Blanco B, Pérez-Simón JA, Sánchez-Abarca LI, Carvajal-Vergara X, Mateos J, Vidriales B, et al. Bortezomib induces selective depletion of alloreactive T lymphocytes and decreases the production of Th1 cytokines. Blood. 2006;107(9):3575–83. doi: 10.1182/blood-2005-05-2118. [DOI] [PubMed] [Google Scholar]
- 131.Nencioni A, Schwarzenberg K, Brauer KM, Schmidt SM, Ballestrero A, Grünebach F, et al. Proteasome inhibitor bortezomib modulates TLR4-induced dendritic cell activation. Blood. 2006;108(2):551–8. doi: 10.1182/blood-2005-08-3494. [DOI] [PubMed] [Google Scholar]
- 132.Basler M, Lauer C, Beck U, Groettrup M. The proteasome inhibitor bortezomib enhances the susceptibility to viral infection. J Immunol. 2009;183(10):6145–50. doi: 10.4049/jimmunol.0901596. [DOI] [PubMed] [Google Scholar]
- 133.Berges C, Haberstock H, Fuchs D, Miltz M, Sadeghi M, Opelz G, et al. Proteasome inhibition suppresses essential immune functions of human CD4+ T cells. Immunology. 2008;124(2):234–46. doi: 10.1111/j.1365-2567.2007.02761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dasanu CA, Alexandrescu DT. Prophylactic antivirals may be helpful in prevention of varicella-zoster virus reactivation in myeloma, but are they safe? J Oncol Pharm Pract. 2010;16(4):266–8. doi: 10.1177/1078155209350374. [DOI] [PubMed] [Google Scholar]
- 135.Attal M, Harousseau JL, Leyvraz S, Doyen C, Hulin C, Benboubker L, et al. Inter-Groupe Francophone du Myélome (IFM) Maintenance therapy with thalidomide improves survival in patients with multiple myeloma. Blood. 2006;108(10):3289–94. doi: 10.1182/blood-2006-05-022962. [DOI] [PubMed] [Google Scholar]
- 136.Spencer A, Prince HM, Roberts AW, Prosser IW, Bradstock KF, Coyle L, et al. Consolidation therapy with low-dose thalidomide and prednisolone prolongs the survival of multiple myeloma patients undergoing a single autologous stem-cell transplantation procedure. J Clin Oncol. 2009;27(11):1788–93. doi: 10.1200/JCO.2008.18.8573. [DOI] [PubMed] [Google Scholar]