

Abstract
Background
New antiplatelet agents that provide greater, more consistent inhibition of the platelet ADP receptor P2Y12 may be used in combination with glycoprotein (GP) IIb‐IIIa antagonists, but their combined effect on platelet function and procoagulant activity is not well studied. Therefore, the objective of this study was to evaluate the independent and complementary effects of P2Y12 and GPIIb‐IIIa inhibition on platelet function and procoagulant activity.
Methods and Results
Healthy donor blood was treated with the active metabolite of prasugrel (R‐138727 5 μmol/L), GPIIb‐IIIa antagonists (abciximab 3 μg/mL or eptifibatide 0.9 μg/mL), and combinations thereof, exposed to physiologically relevant agonists (collagen and ADP) and then evaluated for markers of platelet activation and procoagulant activity. Significant interactions between R‐138727 and GPIIb‐IIIa antagonists were observed. R‐138727 and the GPIIb‐IIIa antagonists had additive inhibitory effects on collagen‐stimulated platelet aggregation and on the collagen plus ADP–stimulated level of activated platelet surface GPIIb‐IIIa. R‐138727 and abciximab each inhibited collagen plus ADP–stimulated platelet phosphatidylserine expression and prothrombin cleavage, and the combination produced greater inhibition than achieved with abciximab alone. In contrast, eptifibatide did not inhibit, but instead enhanced, collagen plus ADP–stimulated prothrombin cleavage. Addition of R‐138727 reduced prothrombin cleavage in eptifibatide‐treated samples, suggesting a novel mechanism for potential benefit from combined prasugrel and eptifibatide treatment.
Conclusions
The complementary effects of abciximab and R‐138727 on platelet activation, aggregation, and procoagulant activity suggest their combined use may, to a greater degree than with either agent alone, reduce thrombus formation in vivo.
Keywords: glycoprotein IIb‐IIIa receptor antagonists, platelets, prasugrel
Introduction
In addition to the most commonly used antiplatelet agent, aspirin, which inhibits platelet cyclooxygenase‐1, two distinct classes of antiplatelet agents with distinct mechanisms of action, glycoprotein (GP) IIb‐IIIa antagonists (eg, abciximab, eptifibatide) and antagonists of the platelet ADP receptor P2Y12 (eg, clopidogrel, prasugrel), are used for the prevention of cardiac ischemic complications in the setting of percutaneous coronary intervention (PCI).1–2 Abciximab is the Fab fragment of the chimeric human–murine monoclonal antibody 7E3, whereas eptifibatide is a cyclic hexapeptide, small‐molecule inhibitor.3 Abciximab and eptifibatide bind to the GPIIb‐IIIa (integrin αIIbβ3) receptor of human platelets, inhibiting the final common pathway of platelet aggregation in response to all agonists. Clopidogrel and prasugrel are thienopyridine prodrugs, each with an active metabolite that irreversibly inhibits the platelet ADP receptor P2Y12 and, thereby, inhibits ADP‐induced platelet activation and aggregation.1
While aspirin plus clopidogrel is the most frequently orally administered antiplatelet combination in PCI,4 novel P2Y12 inhibitors such as ticagrelor and prasugrel are also used in the setting of acute coronary syndrome PCI.5 As GPIIb‐IIIa and P2Y12 antagonists target distinct steps of the thrombus formation process, supplementing aspirin plus P2Y12 antagonist therapy with a GPIIb‐IIIa antagonist enhances platelet inhibition and may have clinical benefits in patients, particularly patients with large thrombus burden or poor response to oral antiplatelet agents.2,4,6 Although combination GPIIb‐IIIa and P2Y12 inhibition on an aspirin background is used in the clinical setting, the combined effect of these inhibitors on platelet responses has not been well studied, particularly in the case of the newer P2Y12 agents.
In in vitro studies, the active metabolite of prasugrel, R‐138727, in the absence or presence of aspirin, inhibits platelet activation as well as platelet procoagulant and proinflammatory responses;7–9 however, the effect of concomitant prasugrel and GPIIb‐IIIa antagonist treatment on these end points remains unknown. Therefore, the objective of this study was to evaluate the independent and complementary effects of P2Y12 and GPIIb‐IIIa inhibition on platelet function. On a background of aspirin, we evaluated the combined effect of in vitro treatment of whole blood with the active metabolite of prasugrel, R‐138727, plus abciximab or eptifibatide on platelet activation, aggregation, and procoagulant activity.
Methods
Materials
ADP was purchased from Bio/Data Corporation, and fibrillar type I collagen was purchased from Chrono‐log. Gly‐Pro‐Arg‐Pro (GPRP) was purchased from Bachem. The fluorogenic activated factor XIII (FXIIIa) substrate was from Zedira. Fluorescein isothiocyanate (FITC)‐conjugated annexin V, FITC‐labeled PAC1 (an IgM monoclonal antibody [mAb] specific for the activated conformation of GPIIb‐IIIa), phycoerythrin (PE)‐Cy5–conjugated CD42b‐specific mAb, and PE‐conjugated CD42a‐ and CD62P‐specific mAbs were from Becton Dickinson. PE‐Cy5–labeled CD14‐specific mAb was from Beckman Coulter. FITC‐conjugated anti–tissue factor mAb (clone VD8) was from American Diagnostica. Purified human coagulation factors Va and Xa (FVa and FXa, respectively) were purchased from Haematologic Technologies. The prothrombin fragment 1.2 (F1.2) ELISA was from Dade Behring.
Blood Collection and Treatment
Institutional review board–approved written informed consent was obtained from all subjects. Two hours after healthy volunteers ingested aspirin 325 mg (Bayer HealthCare), blood was collected into a final concentration of sodium citrate 0.32%, hirudin 25 μg/mL, or d‐Phe‐Pro‐Arg‐chloromethylketone 300 μmol/L (PPACK; EMD Biosciences). Platelet counts for all donors were within the normal range of 150 to 400×103/μL. R‐138727 (provided by Daiichi Sankyo Company Ltd, Tokyo, Japan) was dissolved in DMSO and kept at −80°C in sealed vials. Immediately before use, R‐138727 was diluted in HEPES 10 mmol/L, NaCl 0.15 mol/L, pH 7.4 (HEPES‐saline). For all subsequent assays, whole blood was incubated for 30 minutes at 37°C with R‐138727 5 μmol/L, abciximab 3.0 μg/mL (ReoPro; Centocor), eptifibatide 0.9 μg/mL (Integrilin; Millennium Pharmaceuticals), R‐138727 5 μmol/L plus abciximab 3.0 μg/mL, R‐138727 5 μmol/L plus eptifibatide 0.9 μg/mL, or vehicle (DMSO diluted identically to R‐138727). R‐138727, abciximab, and eptifibatide concentrations used for these experiments were selected to correspond with the level of platelet inhibition achieved in patients treated with the current recommended dosing regimens: prasugrel at 60 mg loading dose and 10 mg/day maintenance dose;10 abciximab at 0.25 mg/kg intravenous bolus followed by continuous infusion of 0.125 μg/kg per minute;11 eptifibatide at 180 μg/kg intravenous bolus followed immediately by a continuous infusion of 2.0 μg/kg per minute and a second eptifibatide bolus of 180 μg/kg administered 10 minutes later.12 Concentrations of R‐138727, abciximab, and eptifibatide were calculated with respect to the plasma compartment of the blood to account for differences in hematocrit between donors. For selected experiments, platelet‐rich plasma (PRP) was prepared by centrifugation of treated whole blood for 10 minutes at 150g. For each assay subsequently described, 6 independent experiments were performed using blood collected from 6 different donors.
Platelet Aggregometry
Light transmission platelet aggregation was evaluated in a Chrono‐log aggregometer in PPACK‐anticoagulated PRP in response to ADP 20 μmol/L or collagen 20 μg/mL. The aggregation response was recorded for a total of 6 minutes using Aggro/Link software (Chrono‐log). Platelet counts were not adjusted before the use of PRP in aggregation.
Platelet Surface Expression of Activated GPIIb‐IIIa and P‐selectin
PPACK‐anticoagulated whole blood was incubated with FITC‐PAC1, PE anti‐CD62P, and PE‐Cy5 anti‐CD42a (as a platelet identifier) and either collagen 20 μg/mL plus ADP 20 μmol/L or no agonist (HEPES‐Tyrode's buffer) for 15 minutes at room temperature. Samples were then fixed by addition of 1% formaldehyde in HEPES‐saline. Flow cytometric analysis was performed in a calibrated Becton Dickinson FACSCalibur as previously described.13
Platelet Surface Binding of Annexin V
PPACK‐anticoagulated whole blood was diluted 1:10 in HEPES‐Tyrode's buffer and incubated with collagen 20 μg/mL plus ADP 20 μmol/L or no agonist (HEPES‐Tyrode's buffer) for 20 minutes at 37°C. Phosphatidylserine expression on the platelet surface was determined by annexin V binding as previously described.8 Briefly, after incubation with collagen plus ADP or buffer, samples were mixed with FITC‐conjugated annexin V and a PE‐Cy5 anti‐CD42a antibody (as a platelet identifier) in the presence of CaCl2 4 mmol/L, incubated for 15 minutes at room temperature, and fixed with 1% formaldehyde in HEPES‐saline. Flow cytometric analysis was performed in a calibrated Becton Dickinson FACSCalibur.
Tissue Factor Expression on Monocyte–Platelet Aggregates and Single Platelets
The presence of monocyte–platelet aggregates and the expression of tissue factor on monocyte–platelet aggregates and single platelets were detected, as previously described,14 in PPACK‐anticoagulated whole blood stimulated with collagen 20 μg/mL plus ADP 20 μmol/L or no agonist (HEPES‐saline). In brief, whole blood was incubated with agonists for 15 minutes at room temperature and then incubated with a mixture of PE anti‐CD42a 1 μg/mL for platelet identification, PE‐Cy5 anti‐CD14 for monocyte identification, and FITC‐conjugated anti–tissue factor mAb 10 μg/mL for 20 minutes at room temperature. After incubation, FACS Lysing solution (Becton Dickinson) was added to samples, and flow cytometric analysis was performed in a Becton Dickinson FACSCalibur.
Thrombin Generation
Hirudin‐anticoagulated whole blood was mixed for 15 minutes at 37°C with shaking with FXa 600 pmol/L and FVa 300 pmol/L combined with either collagen 20 μg/mL plus ADP 20 μmol/L or no agonist (HEPES‐Tyrode's buffer) in a 96‐well microtiter plate (total volume 100 μL, all concentrations are final concentrations in whole blood). The assay was stopped by the addition of 260 μL of EDTA 2 mmol/L in HEPES‐saline. Samples were centrifuged and thrombin generation was measured by the formation of F1.2 in the supernatant using the Enzygnost F1.2 ELISA.
Activated FXIII Generation
For FXIII assays, blood was collected from healthy volunteers into sodium citrate, 24 hours after ingestion of aspirin 325 mg. As described earlier, blood was treated with R‐138727, abciximab, and eptifibatide, and PRP was prepared via centrifugation. Sodium citrate–anticoagulated PRP was combined with FXIIIa substrate solution (fluorogenic FXIIIa substrate 56 μmol/L, CaCl2 20 mmol/L, FXa 60 pmol/L, FVa 30 pmol/L, and GPRP 10 mmol/L) and collagen 20 μg/mL plus ADP 20 μmol/L or no agonist (HEPES‐saline) in a 96‐well microtiter plate (all concentrations are final concentrations in PRP). Fluorescence was monitored (excitation at 313 nm and emission at 418 nm) in a microtiter plate reader for 45 minutes at 37°C with shaking. PPACK‐treated PRP was used as the plate blank.
Statistical Methods
Data analyses were performed by the authors using GraphPad Prism Version 5.0 and VassarStats: Website for Statistical Computation.15 Data are presented as mean±SEM from 6 independent experiments. The effects of R‐138727, the GPIIb‐IIIa antagonists, and the interactions between R‐138727 and the GPIIb‐IIIa antagonists were determined by 2‐factor repeated measures ANOVA (RM‐ANOVA) with P<0.05 considered statistically significant. Posttests to compare individual treatment conditions assumed samples were from normal distributions and were by Student's paired t test or by 1‐sample t test (for comparison with a normalized baseline result). To account for multiple comparisons, only posttest P values <0.0071 (Bonferroni correction) were considered significant.
Results
Inhibition of Platelet Aggregation by P2Y12 and GPIIb‐IIIa Antagonists
The P2Y12 antagonist R‐138727 has been previously shown to dose‐dependently inhibit ADP‐induced platelet aggregation.7 To investigate the combined effect of P2Y12 and GPIIb‐IIIa inhibition on a background of aspirin, platelet aggregation was studied in PRP from aspirin‐treated subjects, treated in vitro with R‐138727 alone or in combination with the GPIIb‐IIIa antagonists, abciximab or eptifibatide. Consistent with previous studies, when platelets were stimulated with ADP, aggregation was significantly inhibited in the presence of R‐138727 (Figure 1). Likewise, as expected, treatment with either GPIIb‐IIIa antagonist resulted in a marked decrease in ADP‐induced aggregation. However, the addition of either abciximab or eptifibatide to R‐138727 completely abrogated platelet aggregation (Figure 1). Two‐factor RM‐ANOVA of ADP‐induced platelet aggregation (Table) showed a significant effect of both R‐138727 (P<0.0001) and the GPIIb‐IIIa antagonists (P<0.0001) and a significant interaction between R‐138727 and the GPIIb‐IIIa antagonists (P=0.0009). When collagen was used as the agonist, the presence of R‐138727 resulted in a smaller, but still significant, decrease in maximal platelet aggregation (Figure 1). As with ADP‐induced aggregation, the GPIIb‐IIIa antagonists significantly inhibited collagen‐induced platelet aggregation compared with vehicle. Combined treatment with R‐138727 plus abciximab or eptifibatide resulted in a markedly reduced aggregation response compared with R‐138727 treatment alone (Figure 1). Furthermore, when R‐138727 was added to the GPIIb‐IIIa antagonists, platelet aggregation was decreased beyond the level of aggregation with GPIIb‐IIIa inhibition alone.
Figure 1.
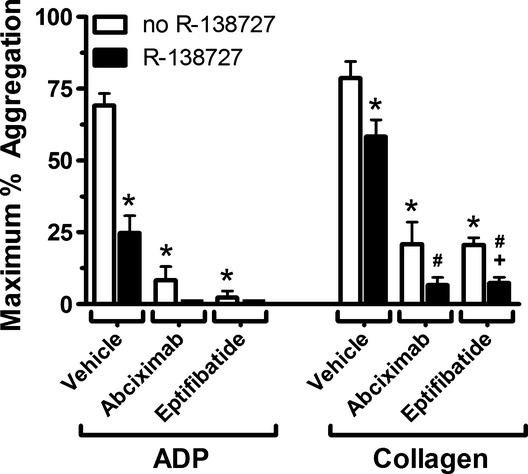
Effect of combined P2Y12 and GPIIb‐IIIa inhibition on ADP‐ or collagen‐induced platelet aggregation. Platelet‐rich plasma (PRP) was prepared from PPACK‐anticoagulated blood treated with vehicle or R‐138727 5 μmol/L in the absence or presence of abciximab 3 μg/mL or eptifibatide 0.9 μg/mL. PRP was stimulated with ADP 20 μmol/L or collagen 20 μg/mL and the extent of platelet aggregation was determined. Data are reported as mean± SEM from 6 experiments. *P<0.0071 vs no R‐138727, vehicle treatment; +P<0.0071 vs corresponding treatment in the absence of R‐138727; #P<0.0071 vs R‐138727, vehicle treatment. GP indicates glycoprotein; PPACK, d‐Phe‐Pro‐Arg‐chloromethylketone.
Table 1.
Statistical analysis of platelet activation, aggregation, and procoagulant responses to ADP 20 μmol/L and collagen 20 μg/mL in the presence of R‐138727 5 μmol/L, abciximab 3 μg/mL, or eptifibatide 0.9 μg/mL, and combinations thereof
| Factor | P Value | |
|---|---|---|
| Platelet aggregation—ADP (maximum % aggregation) | R‐138727 | <0.0001 |
| GPIIb‐IIIa antagonists | <0.0001 | |
| Interaction | 0.0009 | |
| Platelet aggregation—collagen (maximum % aggregation) | R‐138727 | 0.0021 |
| GPIIb‐IIIa antagonists | <0.0001 | |
| Interaction | 0.3960 | |
| Activated GPIIb‐IIIa—collagen+ADP (PAC1 MFI) | R‐138727 | <0.0001 |
| GPIIb‐IIIa antagonists | <0.0001 | |
| Interaction | <0.0001 | |
| Platelet surface P‐selectin—collagen+ADP (MFI) | R‐138727 | 0.0012 |
| GPIIb‐IIIa antagonists | 0.0131 | |
| Interaction | 0.0223 | |
| % MPAs—collagen+ADP | R‐138727 | 0.0008 |
| GPIIb‐IIIa antagonists | 0.0014 | |
| Interaction | 0.0004 | |
| Platelet fluorescence in MPAs—collagen+ADP (MFI) | R‐138727 | 0.0006 |
| GPIIb‐IIIa antagonists | 0.0002 | |
| Interaction | 0.0001 | |
| % Annexin V–positive platelets—collagen+ADP | R‐138727 | 0.0010 |
| GPIIb‐IIIa antagonists | <0.0001 | |
| Interaction | <0.0001 | |
| MPA tissue factor—collagen+ADP (MFI) | R‐138727 | 0.0007 |
| GPIIb‐IIIa antagonists | <0.0001 | |
| Interaction | 0.0001 | |
| Platelet tissue factor—collagen+ADP (MFI) | R‐138727 | <0.0001 |
| GPIIb‐IIIa antagonists | 0.2341 | |
| Interaction | 0.0012 | |
| Plasma F1.2—collagen+ADP (normalized) | R‐138727 | 0.0002 |
| GPIIb‐IIIa antagonists | 0.0102 | |
| Interaction | 0.0183 | |
| Relative FXIIIa activity—collagen+ADP (1/time to Vmax, normalized) | R‐138727 | 0.3542 |
| GPIIb‐IIIa antagonists | 0.0136 | |
| Interaction | 0.8089 |
Two‐factor ANOVA with repeated measures on both factors. GP indicates glycoprotein; MFI, mean fluorescence intensity; MPA, monocyte–platelet aggregate; Vmax, maximum velocity.
Platelet Surface Activated GPIIb‐IIIa and P‐selectin
Because platelet aggregation is dependent on the activation of GPIIb‐IIIa,16 the combined effect of P2Y12 and GPIIb‐IIIa inhibition on the expression and availability of activated GPIIb‐IIIa was evaluated in whole blood treated with R‐138727 alone or in combination with abciximab or eptifibatide. After stimulation with collagen plus ADP, platelet surface activated GPIIb‐IIIa expression, as reflected by mAb PAC1 binding, was, as expected, markedly increased compared with unstimulated samples (Figure 2A). R‐138727 treatment caused a substantial reduction in the level of surface activated GPIIb‐IIIa. The presence of either GPIIb‐IIIa antagonist significantly reduced PAC1 binding to activated GPIIb‐IIIa in collagen plus ADP–stimulated samples (Figure 2A). PAC1 binding was further inhibited when R‐138727 was added to abciximab‐ or eptifibatide‐treated samples, and combined GPIIb‐IIIa and P2Y12 antagonism resulted in significant inhibition beyond R‐138727 treatment alone (Figure 2A). In unstimulated (no agonist) samples with low baseline activated GPIIb‐IIIa, PAC1 binding was reduced by either GPIIb‐IIIa inhibitor and, more significantly, inhibited by the combination of R‐138727 plus abciximab or eptifibatide (Figure 2A).
Figure 2.
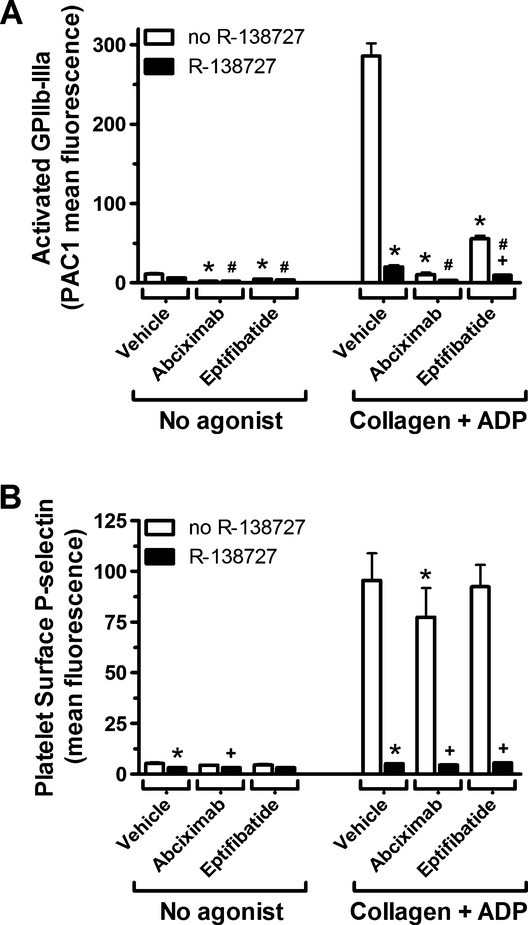
Inhibition of platelet surface expression of activated GPIIb‐IIIa and P‐selectin by R‐138727 plus abciximab or eptifibatide. PPACK‐anticoagulated whole blood treated with R‐138727, abciximab, or eptifibatide, and combinations thereof, was incubated with collagen 20 μg/mL plus ADP 20 μmol/L or no agonist and labeled with fluorescently‐conjugated antibodies for analysis by whole blood flow cytometry. A, Platelet surface activated GPIIb‐IIIa, as determined by platelet binding of the monoclonal antibody PAC1 (mean fluorescence intensity). B, Platelet surface expression of P‐selectin (mean fluorescence intensity). Data are shown as mean±SEM, n=6. *P<0.0071 vs no R‐138727, vehicle treatment; +P<0.0071 vs corresponding treatment in the absence of R‐138727; #P<0.0071 vs R‐138727, vehicle treatment. GP indicates glycoprotein; PPACK, d‐Phe‐Pro‐Arg‐chloromethylketone.
To further assess the level of platelet activation with combined P2Y12 and GPIIb‐IIIa inhibition, we evaluated the platelet surface expression of P‐selectin. Two‐factor RM‐ANOVA of collagen plus ADP–stimulated platelet surface P‐selectin (Table) showed a significant effect of R‐138727 (P=0.0012), the GPIIb‐IIIa antagonists (P=0.0131), and a significant interaction between R‐138727 and the GPIIb‐IIIa antagonists was present (P=0.0223). R‐138727 caused a large, significant decrease (≈95%, P=0.0011, paired t test) in platelet surface P‐selectin expression in collagen plus ADP–stimulated blood (Figure 2B); a smaller, significant decrease (≈19%, P=0.0067, paired t test) was observed with abciximab. No change in collagen plus ADP–stimulated P‐selectin expression was observed in the presence of eptifibatide. When abciximab or eptifibatide was used in combination with R‐138727, the decrease in P‐selectin expression was comparable to that observed with only R‐138727 treatment (Figure 2B).
Monocyte–Platelet Aggregates
As an additional marker of the level of platelet activation with combined P2Y12 and GPIIb‐IIIa inhibition, monocyte–platelet aggregates were measured with and without collagen plus ADP stimulation. In the absence of antiplatelet agents, as expected, collagen plus ADP increased the percentage of monocytes bound to platelets (monocyte–platelet aggregates) and the platelet fluorescence in monocyte–platelet aggregates (Figure 3A and 3B). By 2‐factor RM‐ANOVA, R‐138727, GPIIb‐IIIa antagonists, and the interaction between R‐138727 and GPIIb‐IIIa antagonists were highly significant for the collagen plus ADP–stimulated percentage of monocyte–platelet aggregates and the platelet fluorescence in monocyte–platelet aggregates (Table). In collagen plus ADP–stimulated samples, R‐138727 reduced the percentage of monocyte–platelet aggregates and the level of platelet fluorescence in the aggregates, indicating a reduced number of platelets in the aggregates (Figure 3). Although abciximab and eptifibatide each resulted in numerical increases in the percentage of monocyte–platelet aggregates and platelet fluorescence in monocyte–platelet aggregates, in posttests, only the abcximab‐induced increase in platelet fluorescence remained statistically significant. Addition of R‐138727 to abciximab abrogated this increase, reducing platelet fluorescence in monocyte–platelet aggregates to the level observed with R‐138727 treatment alone (Figure 3).
Figure 3.
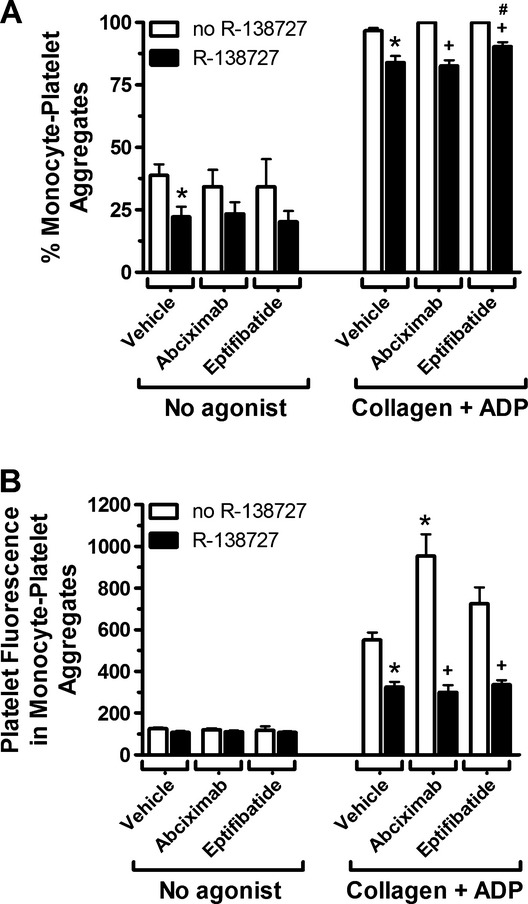
ADP plus collagen‐induced monocyte–platelet aggregates in the presence of P2Y12 and GPIIb‐IIIa antagonists. Whole blood anticoagulated with PPACK was stimulated with collagen 20 μg/mL plus ADP 20 μmol/L or no agonist, fluorescently‐labeled for monocytes and platelets, and analyzed flow cytometrically. A, Percentage of monocytes with platelet attached. B, Platelet fluorescence (mean fluorescence intensity) in monocyte–platelet aggregates. Results are mean±SEM (n=6). *P<0.0071 vs no R‐138727, vehicle treatment; +P<0.0071 vs corresponding treatment in the absence of R‐138727; #P<0.0071 vs R‐138727, vehicle treatment. GP indicates glycoprotein; PPACK, d‐Phe‐Pro‐Arg‐chloromethylketone.
Platelet Surface Expression of Phosphatidylserine
In addition to their role in aggregate formation, activated platelets promote coagulation through surface exposure of procoagulant phosphatidylserine.17 As expected, stimulation with collagen plus ADP increased the percentage of annexin V–positive platelets (Figure 4). Two‐factor RM‐ANOVA analysis (Table) showed a significant effect of both R‐138727 (P=0.0010) and the GPIIb‐IIIa antagonists (P<0.0001) and a significant interaction between R‐138727 and the GPIIb‐IIIa antagonists (P<0.0001). Individual treatment with R‐138727 or abciximab reduced annexin V binding in collagen plus ADP–stimulated samples (annexin V–positive platelets 22.9±2.7% with vehicle versus 9.5±1.6% for R‐138727, P=0.0004, and 14.7±1.9% for abciximab, P=0.0004), but no significant effect was observed with eptifibatide (P=0.0247 versus vehicle; nonsignificant with Bonferroni correction). No further inhibition was seen when abciximab or eptifibatide was added to R‐138727 treatment (Figure 4).
Figure 4.
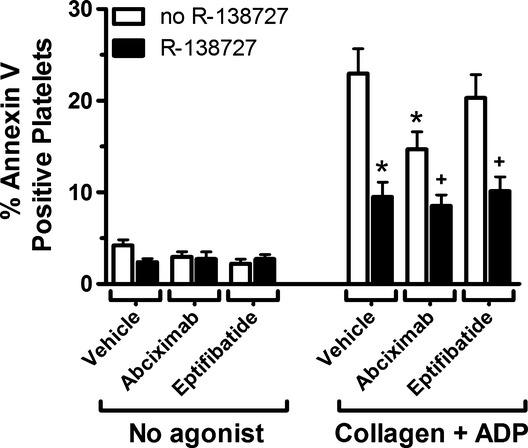
Effect of R‐138727 in combination with abciximab or eptifibatide on platelet surface phosphatidylserine exposure. Surface exposure of phosphatidylserine was determined flow cytometrically by annexin V binding of platelets from PPACK‐anticoagulated blood incubated with collagen 20 μg/mL plus ADP 20 μmol/L or no agonist. Data are shown as the percentage of platelets positive for annexin V staining, mean±SEM, n=6. *P<0.0071 vs no R‐138727, vehicle treatment; +P<0.0071 vs corresponding treatment in the absence of R‐138727; #P<0.0071 vs R‐138727, vehicle treatment. PPACK indicates d‐Phe‐Pro‐Arg‐chloromethylketone.
Tissue Factor Expression on Monocyte–Platelet Aggregates and Individual Platelets
Beyond platelet surface expression of phosphatidylserine, platelets and monocyte–platelet aggregates can stimulate coagulation through surface expression of tissue factor. In previous studies, treatment with R‐138727 decreased the level of tissue factor on monocyte–platelet aggregates.8 To extend these studies, we investigated the effect of R‐138727 combined with abciximab or eptifibatide on procoagulant surface expression of tissue factor. Stimulation with collagen plus ADP increased the level of tissue factor fluorescence on both monocyte–platelet aggregates and single platelets (Figure 5A and 5B). Two‐factor RM‐ANOVA analysis of tissue factor on monocyte–platelet aggregates (Table) showed significant effects of R‐138727 (P=0.0007), GPIIb‐IIIa antagonists (P<0.0001), and the interaction between R‐138727 and GPIIb‐IIIa antagonists (P=0.0001). Tissue factor on monocyte–platelet aggregates induced by collagen plus ADP was reduced by R‐138727 (Figure 5A). In parallel to the increase in monocyte–platelet aggregates observed with abciximab or eptifibatide (Figure 3), individual treatment with either GPIIb‐IIIa antagonist caused an increase in monocyte–platelet tissue factor (Figure 5A). The addition of R‐138727 to abciximab or eptifibatide reduced tissue factor levels on monocyte–platelet aggregates. When tissue factor on individual platelets stimulated with collagen plus ADP was examined, R‐138727 treatment caused significant inhibition, while the GPIIb‐IIIa antagonists had no significant effect (2‐factor RM‐ANOVA R‐138727, P<0.0001; GPIIb‐IIIa antagonists, P=0.2341; Table) on platelet tissue factor (Figure 5B).
Figure 5.
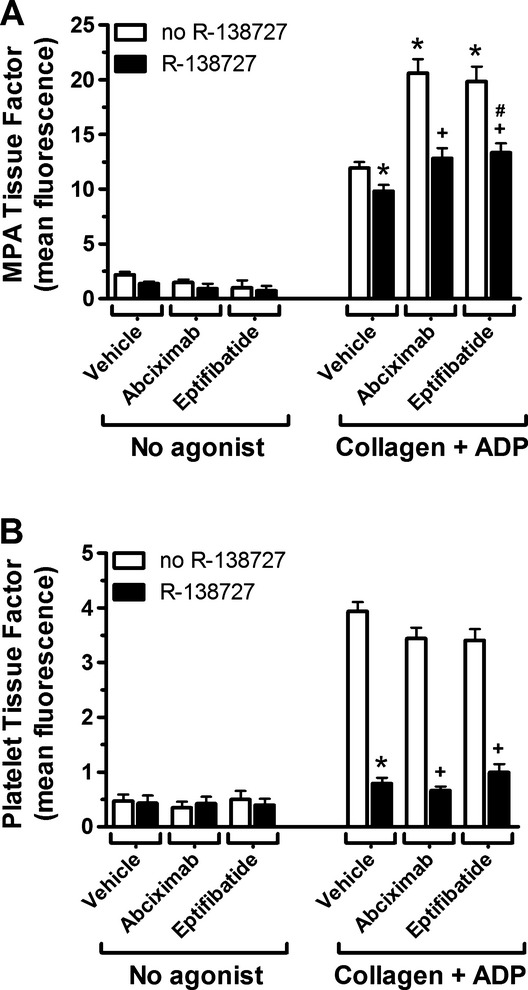
Tissue factor expression on monocyte–platelet aggregates (MPAs) and individual platelets in the presence of R‐138727 plus abciximab or eptifibatide. MPAs and individual platelets from PPACK‐anticoagulated blood stimulated with collagen 20 μg/mL plus ADP 20 μmol/L or no agonist were stained with an anti‐tissue factor antibody and analyzed flow cytometrically. A, Tissue factor on MPAs (fluorescence, arbitrary units). B, Tissue factor on individual platelets (fluorescence, arbitrary units). Results are reported as mean±SEM from 6 independent experiments. *P<0.0071 vs no R‐138727, vehicle treatment; +P<0.0071 vs corresponding treatment in the absence of R‐138727; #P<0.0071 vs R‐138727, vehicle treatment. PPACK indicates d‐Phe‐Pro‐Arg‐chloromethylketone.
Platelet‐Dependent Coagulation Factor Activation: F1.2 Generation and FXIIIa Activity
To determine the effects of combined platelet P2Y12 and GPIIb‐IIIa inhibition on coagulation, we investigated platelet‐dependent enhancement of thrombin generation and activation of FXIII in the presence of R‐138727 plus abciximab or eptifibatide. When thrombin generation was measured by the formation of F1.2 in whole blood, addition of collagen plus ADP, in the presence of priming concentrations of FVa and FXa, caused a marked increase in F1.2 compared with blood primed with FVa and FXa but without the addition of collagen plus ADP (Figure 6). Two‐factor RM‐ANOVA (Table) showed a significant effect of R‐138727 (P=0.0002) and the GPIIb‐IIIa antagonists (P=0.0102) and a significant interaction between R‐138727 and the GPIIb‐IIIa antagonists (P=0.0183). Collagen plus ADP–enhanced F1.2 levels were reduced by 38% by R‐138727 treatment compared with vehicle (P=0.0064, 1‐sample t test, Figure 6). Treatment with abciximab alone reduced the collagen plus ADP–dependent increase in F1.2 (P=0.0250, 1‐sample t test), while eptifibatide treatment enhanced collagen plus ADP–stimulated F1.2 (P=0.0482, 1‐sample t test), although neither effect reached statistical significance after Bonferroni correction. The combination of R‐138727 plus abciximab or eptifibatide reduced F1.2 compared with individual GPIIb‐IIIa antagonist treatment and resulted in a level of F1.2 that was comparable to R‐138727 treatment alone (Figure 6).
Figure 6.
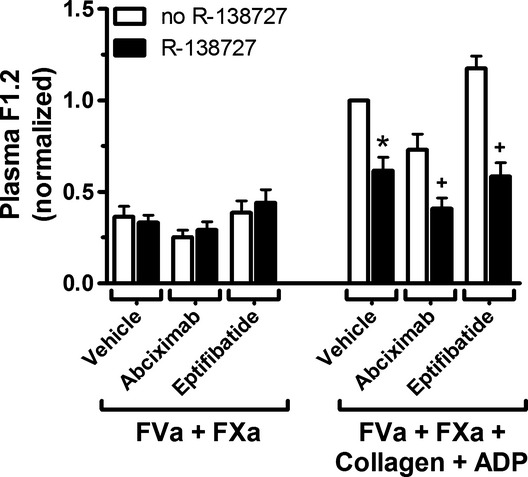
Inhibition of P2Y12 and GPIIb‐IIIa in platelet‐dependent thrombin generation. Hirudin‐anticoagulated blood was incubated with FXa 600 pmol/L and FVa 300 pmol/L with collagen 20 μg/mL plus ADP 20 μmol/L or with buffer. Thrombin generation was determined by the level of prothrombin fragment F1.2 as measured by ELISA. Results shown are normalized per subject to collagen plus ADP with no‐R138727, vehicle treatment; mean±SEM, n=6. *P<0.0071 vs no R‐138727, vehicle treatment; +P<0.0071 vs corresponding treatment in the absence of R‐138727; #P<0.0071 vs R‐138727, vehicle treatment. GP indicates glycoprotein; FVa and FXa, coagulation factors Va and Xa, respectively.
FXIIIa generation, primed by addition of FVa and FXa, was significantly increased by platelet activation with collagen plus ADP (Figure 7). Relative FXIIIa activity in collagen plus ADP–activated samples was not significantly affected by treatment with R‐138727 or eptifibatide alone (Figure 7). However, the relative FXIIIa activity was significantly, albeit modestly, decreased in the presence of abciximab (P=0.0015, 1‐sample t test, Figure 7).
Figure 7.
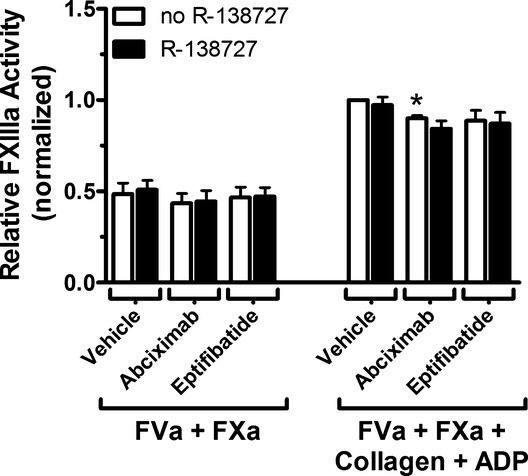
Platelet‐dependent FXIIIa generation in the presence of R‐138727 plus abciximab or eptifibatide. Sodium citrate–anticoagulated blood was incubated with solution of CaCl2, GPRP (to inhibit fibrin polymerization), FXa 60 pmol/L, and FVa 30 pmol/L combined with collagen 20 μg/mL plus ADP 20 μmol/L or no agonist. FXIIIa generation was monitored by the cleavage of a fluorogenic FXIIIa substrate, and the relative FXIIIa activity was reported as the reciprocal of the time to reach Vmax, normalized per subject by the value obtained for a vehicle‐treated sample stimulated with collagen plus ADP. Results are mean±SEM, from 6 experiments. *P<0.0071 vs no R‐138727, vehicle treatment; +P<0.0071 vs corresponding treatment in the absence of R‐138727; #P<0.0071 vs R‐138727, vehicle treatment. GPRP indicates Gly‐Pro‐Arg‐Pro; FVa and FXa, coagulation factors Va and Xa, respectively; Vmax, maximum velocity.
Discussion
The main findings of this study are as follows. (1) The combination of P2Y12 and GPIIb‐IIIa antagonists, at whole blood concentrations that demonstrate pharmacological efficacy, resulted in greater inhibition of ADP– and collagen–induced platelet aggregation and collagen plus ADP–induced platelet surface activated GPIIb‐IIIa than with either antagonist alone (Figures 1 and 2A). (2) The level of tissue factor on monocyte–platelet aggregates was increased by GPIIb‐IIIa inhibitors, and the increase was significantly reduced by the active metabolite of prasugrel, R‐138727 (Figure 5A). (3) R‐138727 and abciximab each significantly inhibited collagen plus ADP–stimulated platelet surface expression of phosphatidylserine (Figure 4) and reduced thrombin generation (Figure 6). In contrast, eptifibatide alone increased, albeit modestly, platelet activation–dependent thrombin generation (Figure 6). The combination of R‐138727 with eptifibatide reduced the level of thrombin generation to that seen with R‐138727 alone. (4) Abciximab, but not eptifibatide, reduced FXIIIa activation downstream from platelet activation with collagen plus ADP (Figure 7).
Clinical Evidence Suggesting a Benefit From Combined GPIIb‐IIIa and P2Y12 Inhibition
The presently described additive effects of GPIIb‐IIIa antagonists and prasugrel's active metabolite on platelet activation, aggregation, and procoagulant activities have not previously been reported. While current indications for GPIIb‐IIIa antagonist use are limited,3,18 broader application of GPIIb‐IIIa antagonists in combination with P2Y12 antagonists may have clinical benefit. For example, when prasugrel was administered with or without the GPIIb‐IIIa antagonist tirofiban in ST‐segment elevation acute myocardial infarction patients on aspirin, platelet inhibition was suboptimal (as measured by ADP‐induced aggregation) for at least 2 hours with prasugrel alone, but the addition of tirofiban led to a significantly higher degree of platelet inhibition.6 Furthermore, a recent meta‐analysis of randomized trials of patients undergoing elective PCI with stents and periprocedural thienopyridines, assigned to randomly receive a GPIIb‐IIIa antagonist or control, show a clinical benefit of GPIIb‐IIIa inhibitors (both abciximab and small‐molecule inhibitors).2 Specifically, GPIIb‐IIIa antagonists added on a background of aspirin and a thienopyridine were shown to reduce nonfatal myocardial infarction, without an increase in major bleeding, but with an increase in minor bleeding. These studies combined with the present study suggest that GPIIb‐IIIa antagonists are beneficial in inhibiting platelet function when used in combination with P2Y12 antagonists.
Augmented Inhibition of Platelet Activation, Aggregation, and Procoagulant Activities by Combined Use of R‐138727 and GPIIb‐IIIa Antagonists
Because light transmission platelet aggregation has been the de facto gold standard for platelet function testing throughout the development of the GPIIb‐IIIa antagonists and newer P2Y12 antagonists,19 the additive inhibitory effect of R‐138727 plus abciximab or eptifibatide on collagen– and ADP–stimulated platelet aggregation is not surprising. The additional inhibition of platelet aggregation and platelet surface activated GPIIb‐IIIa expression by combined P2Y12 and GPIIb‐IIIa antagonists may in part explain the clinical benefit resulting from their combined use in the meta‐analysis described earlier.2 Downstream effects of combined inhibition with abciximab and R‐138727 observed in the present study [reduced annexin V–positive platelets (Figure 4) and reduced conversion of prothrombin to thrombin with concomitant release of F1.2 (Figure 6)] are also likely to be mechanistically linked to the clinical benefit observed in the meta‐analysis.2 The results of the present study are similar to that reported for the combination of cangrelor, a reversible P2Y12 antagonist and abciximab or tirofiban, which resulted in additive inhibition of platelet aggregate formation, dense granule secretion, soluble CD40 ligand release, annexin V binding, and procoagulant microparticle formation, and to a study of in vivo clopidogrel plus in vitro GPIIb‐IIIa inhibition that demonstrated additive inhibition of ADP‐ or collagen‐induced platelet aggregation and fibrinogen binding to GPIIb‐IIIa.20–21
Enhanced Procoagulant Status Following Treatment With GPIIb‐IIIa Antagonists Alone, and Ablation by R‐138727
The results of previous studies on the effect of GPIIb‐IIIa inhibitors on monocyte–platelet aggregate formation and tissue factor on monocyte–platelet aggregates are inconsistent, with GPIIb‐IIIa inhibitors reported to both augment and inhibit these processes.22–25 In agreement with the present results, separate studies have shown that abciximab enhanced ADP‐induced platelet fluorescence in monocyte–platelet aggregates,22 and eptifibatide enhanced tissue factor expression on monocytes.23 The enhanced procoagulant status observed after GPIIb‐IIIa antagonist treatment may be relevant to the increased thrombotic complications observed with oral GPIIb‐IIIa antagonists.26 In the current study, whole blood stimulated with collagen plus ADP in the presence of either abciximab or eptifibatide appears to be in a procoagulant state, as evidenced by increased exposure of tissue factor antigen on monocyte–platelet aggregates (Figure 5A), together with an increase in platelet mass associated with each monocyte (Figure 3B). Indeed, platelet activation–dependent prothrombinase activity is modestly enhanced in the presence of eptifibatide (Figure 6). However, abciximab under the same conditions reduced prothrombinase activity (Figure 6), thus demonstrating a difference in the ability of eptifibatide (a 1000 Da, small molecule, reversible GPIIb‐IIIa antagonist) and abciximab (a much larger [50 000 Da], tight‐binding GPIIb‐IIIa antagonist) to modulate platelet‐dependent procoagulant activity.
Prothrombin has been shown to bind to GPIIb‐IIIa and this binding accelerates thrombin generation.27 With its larger size, abciximab (and other antibody inhibitors) may not only block prothrombin binding to GPIIb‐IIIa but may also sterically hinder other binding reactions on the platelet surface that are important for thrombin generation, such as coagulation factor binding to surface‐exposed phosphatidylserine. This concept is in agreement with a previous study, showing that abciximab more strongly inhibited annexin V and factor V/Va binding to the platelet surface than eptifibatide.28 The molecular size difference and specific molecular interactions between each antagonist and GPIIb‐IIIa may explain the variable response in prothrombin activation. Because tissue factor activation is modulated by protein disulfide‐isomerase,29 it is also tempting to speculate that eptifibatide and abciximab differentially affect this activation.
In the present study, the addition of R‐138727 to GPIIb‐IIIa antagonists resulted in a significant decrease compared with GPIIb‐IIIa inhibitors alone in the percentage of monocyte–platelet aggregates, tissue factor expression on monocyte–platelet aggregates, and thrombin generation. This is in agreement with a previous study on clopidogrel and abciximab which reported that the addition of clopidogrel to abciximab treatment ablated the abciximab‐induced increase in monocyte–platelet aggregates.22 Taken together, the presently‐described ablation by prasugrel's active metabolite of the GPIIb‐IIIa antagonist‐induced increase of tissue factor on monocyte–platelet aggregates and the eptifibatide‐induced increase in thrombin generation, suggests that inhibition of the platelet P2Y12 ADP receptor may reduce the prothrombotic effects of GPIIb‐IIIa antagonists.
Study Limitations and Strengths
This study used blood from healthy subjects rather than from acute coronary syndrome patients, the population for whom these drugs have an FDA‐approved indication, to minimize the variables of comedications and the degree of disease. However, all healthy donors were treated with aspirin to correspond to the clinical conditions where prasugrel and GPIIb‐IIIa antagonists might be coadministered. The power to detect treatment differences is limited by the small sample size (n=6); however, significant effects of GPIIb‐IIIa and/or P2Y12 inhibition were detected for each end point, even after application of the conservative Bonferroni correction.
Conclusions
The presently described complementary effects of abciximab and prasugrel's active metabolite on collagen plus ADP–induced platelet activation, aggregation, and procoagulant activity suggest that the combined use of abciximab and prasugrel may, to a greater degree than with either agent alone, reduce and destabilize thrombus formation in vivo. The GPIIb‐IIIa antagonist‐induced increase of tissue factor on monocyte–platelet aggregates and the eptifibatide‐induced increase in thrombin generation were attenuated by prasugrel's active metabolite, suggesting a novel mechanism for reducing the prothrombotic effects of these GPIIb‐IIIa antagonists.
Sources of Funding
This study was supported in part by a grant from Eli Lilly and Daiichi Sankyo. Dr. Berny‐Lang was supported by an NIH T32 Training Grant (5T32HL007574‐31). This work was conducted with support from Harvard Catalyst|The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health award 8UL1TR000170‐05 and financial contributions from Harvard University and its affiliated academic health care centers). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, or the National Institutes of Health.
Disclosures
Drs. Berny‐Lang and Barnard report no conflicts. Dr. Jakubowski is an employee and shareholder of Eli Lilly and Company. Dr. Sugidachi is an employee of Daiichi Sankyo Co., Ltd. Dr. Michelson received research grants from GLSynthesis and Lilly/Daiichi Sankyo; and is a member of data monitoring committee for clinical trials of prasugrel sponsored by Lilly/Daiichi Sankyo. Dr. Frelinger III received research grants from GLSynthesis and Lilly/Daiichi Sankyo and is a consultant for Eli Lilly.
Acknowledgments
The authors acknowledge the statistical support of Carter Petty, Harvard Catalyst Biostatistics Program, and technical contributions of Youfu Li, Anu Nigam, and Michael Lampa.
References
- 1.Michelson AD. Advances in antiplatelet therapy. Hematol Am Soc Hematol Educ Program. 2011; 2011:62-69 [DOI] [PubMed] [Google Scholar]
- 2.Winchester DE, Wen X, Brearley WD, Park KE, Anderson RD, Bavry AA. Efficacy and safety of glycoprotein IIb/IIIa inhibitors during elective coronary revascularization: a meta‐analysis of randomized trials performed in the era of stents and thienopyridines. J Am Coll Cardiol. 2011; 57:1190-1199 [DOI] [PubMed] [Google Scholar]
- 3.Kristensen SD, Wurtz M, Grove EL, De CR, Huber K, Moliterno DJ, Neumann FJ. Contemporary use of glycoprotein IIb/IIIa inhibitors. Thromb Haemost. 2012; 107:215-224 [DOI] [PubMed] [Google Scholar]
- 4.Kushner FG, Hand M, Smith SC, Jr, King SB, III, Anderson JL, Antman EM, Bailey SR, Bates ER, Blankenship JC, Casey DE, Jr, Green LA, Jacobs AK, Hochman JS, Krumholz HM, Morrison DA, Ornato JP, Pearle DL, Peterson ED, Sloan MA, Whitlow PL, Williams DO. 2009 Focused updates: ACC/AHA guidelines for the management of patients with ST‐elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Catheter Cardiovasc Interv. 2009; 74:E25-E68 [DOI] [PubMed] [Google Scholar]
- 5.Azmoon S, Angiolillo DJ. Switching antiplatelet regimens: alternatives to clopidogrel in patients with acute coronary syndrome undergoing PCI: a review of the literature and practical considerations for the interventional cardiologist. Catheter Cardiovasc Interv. 2013; 81:232-242 [DOI] [PubMed] [Google Scholar]
- 6.Valgimigli M, Tebaldi M, Campo G, Gambetti S, Bristot L, Monti M, Parrinello G, Ferrari R. Prasugrel versus tirofiban bolus with or without short post‐bolus infusion with or without concomitant prasugrel administration in patients with myocardial infarction undergoing coronary stenting: the FABOLUS PRO (Facilitation through Aggrastat By drOpping or shortening Infusion Line in patients with ST‐segment elevation myocardial infarction compared with or on top of PRasugrel given at loading dOse) trial. JACC Cardiovasc Interv. 2012; 5:268-277 [DOI] [PubMed] [Google Scholar]
- 7.Frelinger AL, III, Jakubowski JA, Li Y, Barnard MR, Fox ML, Linden MD, Sugidachi A, Winters KJ, Furman MI, Michelson AD. The active metabolite of prasugrel inhibits ADP‐stimulated thrombo‐inflammatory markers of platelet activation: influence of other blood cells, calcium, and aspirin. Thromb Haemost. 2007; 98:192-200 [PubMed] [Google Scholar]
- 8.Frelinger AL, III, Jakubowski JA, Li Y, Barnard MR, Linden MD, Tarnow I, Fox ML, Sugidachi A, Winters KJ, Furman MI, Michelson AD. The active metabolite of prasugrel inhibits adenosine diphosphate‐ and collagen‐stimulated platelet procoagulant activities. J Thromb Haemost. 2008; 6:359-365 [DOI] [PubMed] [Google Scholar]
- 9.Judge HM, Buckland RJ, Sugidachi A, Jakubowski JA, Storey RF. The active metabolite of prasugrel effectively blocks the platelet P2Y12 receptor and inhibits procoagulant and pro‐inflammatory platelet responses. Platelets. 2008; 19:125-133 [DOI] [PubMed] [Google Scholar]
- 10.EFFIENT (Prasugrel) Tablets. 2011Indianapolis, IN: Eli Lilly and Company [Google Scholar]
- 11.REOPRO (Abciximab) for Intravenous Administration. 2005Indianapolis, IN: Centocor B.V., Eli Lilly and Company [Google Scholar]
- 12.INTEGRELIN (Eptifibatide) Injection for Intravenous Administration. 2011Whitehouse Station, NJ: Merck & Company, Inc [Google Scholar]
- 13.Berny‐Lang MA, Barnard MR, Frelinger AL, III, Michelson AD. In: Michelson AD. (ed.). Flow cytometry. Platelets. 20133rd edSan Diego, CA: Elsevier/Academic Press; 581-602 [Google Scholar]
- 14.Barnard MR, Linden MD, Frelinger AL, III, Li Y, Fox ML, Furman MI, Michelson AD. Effects of platelet binding on whole blood flow cytometry assays of monocyte and neutrophil procoagulant activity. J Thromb Haemost. 2005; 3:2563-2570 [DOI] [PubMed] [Google Scholar]
- 15.Lowry R. Two‐factor ANOVA with repeated measures on both factors VassarStats: Website for Statistical Computation. Available at: http://vassarstats.net. Accessed March 5, 2013.
- 16.Bledzka K, Pesho MM, Ma Y‐Q, Plow EF. In: Michelson AD. (ed.). Integrin alphaIIb‐beta3. Platelets. 20133rd edSan Diego, CA: Elsevier/Academic Press; 233-248 [Google Scholar]
- 17.Bouchard BA, Silveira JR, Tracy PB. In: Michelson AD. (ed.). Interactions between platelets and the coagulation system. Platelets. 20133rd edSan Diego, CA: Elsevier/Academic Press; 425-451 [Google Scholar]
- 18.Armstrong PC, Peter K. GPIIb/IIIa inhibitors: from bench to bedside and back to bench again. Thromb Haemost. 2012; 107:808-814 [DOI] [PubMed] [Google Scholar]
- 19.Michelson AD. Methods for the measurement of platelet function. Am J Cardiol. 2009; 103:20A-26A [DOI] [PubMed] [Google Scholar]
- 20.Klinkhardt U, Kirchmaier CM, Westrup D, Graff J, Mahnel R, Breddin HK, Harder S. Ex vivo–in vitro interaction between aspirin, clopidogrel, and the glycoprotein IIb/IIIa inhibitors abciximab and SR121566A. Clin Pharmacol Ther. 2000; 67:305-313 [DOI] [PubMed] [Google Scholar]
- 21.Judge HM, Buckland RJ, Holgate CE, Storey RF. Glycoprotein IIb/IIIa and P2Y12 receptor antagonists yield additive inhibition of platelet aggregation, granule secretion, soluble CD40L release and procoagulant responses. Platelets. 2005; 16:398-407 [DOI] [PubMed] [Google Scholar]
- 22.Klinkhardt U, Graff J, Harder S. Clopidogrel, but not abciximab, reduces platelet leukocyte conjugates and P‐selectin expression in a human ex vivo in vitro model. Clin Pharmacol Ther. 2002; 71:176-185 [DOI] [PubMed] [Google Scholar]
- 23.Scholz T, Zhao L, Temmler U, Bath P, Heptinstall S, Losche W. The GPIIb/IIIa antagonist eptifibatide markedly potentiates platelet‐leukocyte interaction and tissue factor expression following platelet activation in whole blood in vitro. Platelets. 2002; 13:401-406 [DOI] [PubMed] [Google Scholar]
- 24.Steiner S, Seidinger D, Huber K, Kaun C, Minar E, Kopp CW. Effect of glycoprotein IIb/IIIa antagonist abciximab on monocyte‐platelet aggregates and tissue factor expression. Arterioscler Thromb Vasc Biol. 2003; 23:1697-1702 [DOI] [PubMed] [Google Scholar]
- 25.Zhao L, Bath PM, May J, Losche W, Heptinstall S. P‐selectin, tissue factor and CD40 ligand expression on platelet‐leucocyte conjugates in the presence of a GPIIb/IIIa antagonist. Platelets. 2003; 14:473-480 [DOI] [PubMed] [Google Scholar]
- 26.Leebeek FW, Boersma E, Cannon CP, van de Werf FJ, Simoons ML. Oral glycoprotein IIb/IIIa receptor inhibitors in patients with cardiovascular disease: why were the results so unfavourable? Eur Heart J. 2002; 23:444-457 [DOI] [PubMed] [Google Scholar]
- 27.Byzova TV, Plow EF. Networking in the hemostatic system. Integrin alphaIIbbeta3 binds prothrombin and influences its activation. J Biol Chem. 1997; 272:27183-27188 [DOI] [PubMed] [Google Scholar]
- 28.Furman MI, Krueger LA, Frelinger AL, III, Barnard MR, Mascelli MA, Nakada MT, Michelson AD. GPIIb‐IIIa antagonist‐induced reduction in platelet surface factor V/Va binding and phosphatidylserine expression in whole blood. Thromb Haemost. 2000; 84:492-498 [PubMed] [Google Scholar]
- 29.Ruf W. Role of thiol pathways in TF procoagulant regulation. Thromb Res. 2012; 129suppl 2:S11-S12 [DOI] [PMC free article] [PubMed] [Google Scholar]