

Abstract
A general, highly selective asymmetric redox-relay oxidative Heck reaction using achiral or racemic acyclic alkenols and boronic acid derivatives is reported. This reaction delivers remotely functionalized arylated carbonyl products from acyclic alkenol substrates, with excellent enantioselectivity under mild conditions, bearing a range of useful functionality. A preliminary mechanistic investigation suggests that the regioselectivity of the initial migratory insertion is highly dependent on the electronic nature of the boronic acid and more subtle electronic effects of the alkenyl alcohol.
An existing challenge in synthetic chemistry is setting chiral centers via carbon–carbon bond formation remote to functional groups.1 Besides directly functionalizing specific C–H bonds in an enantioselective manner, addition of carbon nucleophiles to alkenes using a metal catalyst is an especially attractive approach as the alkene is an easily accessible, robust functional group. This strategy requires addition of the carbon nucleophile–metal species to the alkene, which is often electronically biased towards site-selective addition (e.g., a conjugated alkene). The resultant metal-alkyl complex needs to be efficiently transformed in order to avoid deleterious reaction pathways available to metal-alkyls. One successful tactic is the metal-catalyzed conjugate addition of carbon-nucleophiles to α,β-unsaturated carbonyls wherein the metal-enolate formed in the process is hydrolyzed to reform the active catalyst for a subsequent catalytic cycle (Figure 1a).2 These methods allow for the formation of a wide range of β-substituted carbonyl products in high enantiomeric excess.
Figure 1.

a) Enantioselective β-functionalization reactions. b) Enantioselective redox-relay Heck reactions. c) Mechanistic analysis.
We have recently reported an alternative conceptual approach to setting remote chiral centers using an alkenyl substrate.3 Specifically, we have developed an enantioselective Heck-type reaction4–6 of racemic acyclic alkenol substrates using aryl diazonium salts as the coupling partners, which delivers β-aryl carbonyl products similarly to conjugate addition methods (Figure 1b).3 The approach also allows direct access to γ- or δ-substituted aryl carbonyl products in high enantioselectivity by using a redox-relay strategy7 wherein the unsaturation in the alkene is migrated towards the alcohol.8 This transformation occurs presumably through an iterative β-hydride elimination/migratory insertion process to ultimately be trapped by formal oxidation of the alcohol resulting in a carbonyl product (Figure 1c).9
While this method leads to high enantioselectivity for a range of alkenol substrates (mainly allylic and homoallylic alcohols), a significant question arises as to why preferential insertion is observed at the site distal from the alkyl alcohol.10 Additionally, the use of aryl diazonium salts limits both the scope and potentially the practical application of this method. Therefore, we became interested in developing an oxidative variant of the enantioselective redoxrelay Heck reaction using ubiquitous aryl boronic acid derivatives11 as the aryl source to improve the synthetic utility and also to begin to probe the origin of site selective migratory insertion. Herein, we report an enantioselective redox-relay oxidative Heck reaction with acyclic alkenols and aryl boronic acids, delivering remotely functionalized carbonyl products in high enantioselectivity and generally high site selectivity.
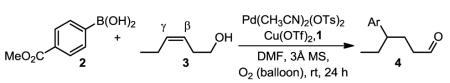
We began our investigation by exploring our previously developed catalytic system for oxidative Heck reactions of electronically nonbiased olefins, which use .12 These conditions employ a cationic Pd salt ligated with an N-heterocyclic carbene, and catalytic Cu(OTf)2 under an O2 atmosphere. To initiate the optimization, the N-heterocyclic carbene was replaced with the chiral pyridine oxazoline ligand 1,3,13 resulting in poor conversion of a cis-homoallylic alcohol 3, albeit with good mass balance for the desired product (entry 1, Table 1). Poor conversion in Pd-catalyzed aerobic oxidations is often attributed to catalyst deactivation through aggregation of Pd metal during the process. This is a result of retarded oxidation of Pd(0) to Pd(II) with O2 (or Cu (II)), which can be circumvented by the addition of molecular sieves (MS).14 Indeed, the addition of 3Å MS significantly improved the reaction outcome with enhanced yields of the desired product (entries 2–4). Increasing the boronic acid and catalyst loadings gave complete conversion in 82% yield (entry 5), 19:1 regioselectivity (γ:β), and excellent enantioselectivity (er: 99:1). Of note, removing Cu(OTf)2 significantly reduces the yield and, in the absence of Pd, no significant reaction is observed.
Table 1.
Reaction optimization.
| Entry | Pd/Cu/ligand (mol%) |
2 (equiv.) |
3Å MS (mg/mmol) |
conv. (%)a |
yield (%)a |
|---|---|---|---|---|---|
| 1 | 5/5/11 | 2 | -- | 17 | 15 |
| 2 | 5/5/11 | 2 | 40 | 58 | 50 |
| 3 | 5/5/11 | 2 | 150 | 80 | 70 |
| 4 | 5/5/11 | 2 | 400 | 78 | 67 |
| 5 | 6/6/13 | 3 | 150 | <99 | 82b (99:1 er)c |
Determined by GC analysis using an internal standard.
isolated yield.
er value was determined with SFC after reducing the resulting aldehyde to the primary alcohol.
With this catalytic system in hand, we initially investigated a wide array of arylboronic acids with homoallylic alcohols substrates (Table 2), which deliver γ-aryl carbonyl chiral building blocks in uniformly high enantioselectivity (er up to 99:1) with moderate to good yields. In general, the observed site selectivity (γ vs β) for alkene functionalization is good to excellent. High site- and enantioselectivity is observed for ortho-substituted aryl boronic acids (4a, 4b). In the case of meta or para-substituted aryl boronic acids, electron-deficient examples afford high site selectivity (4c–h). In contrast, electron-rich arylboronic acids give considerably lower levels of site selectivity, although in high enantioselectivity (4j–l). Interestingly, the minor isomeric product is also formed in high enantioselectivity (4l), which has several important mechanistic implications, as discussed below. A racemic secondary alkenyl alcohol (4m) performs similarly as does an E-alkene, which yields the other enantiomer as the major product (4n). Scaling of this method from 0.3 to 10 mmol resulted in very similar results (4). Finally, several heteroaromatic boronic acid derivatives were evaluated (4o–q). In all cases, excellent site and enantioselectivity is observed although the conversion of the alkene is poor possibly due to observed higher levels of homocoupling and phenol formation for these boronic acids.
Table 2.
Scope of the enantioselective redox-relay Heck reaction of homoallylic alcohols and aryl boronic acids.

|
Yield are reported as a combination of isomers and reaction performed on 0.5 mmol scale. Enatioselectivity determined SFC equipped with a chiral column.
Ratio of γ to β–product as detemined by either 1H NMR or gas chromatography.
Enatioselectivity of the β-product is 99:1 er.
Reaction performed on 10 mmol scale.
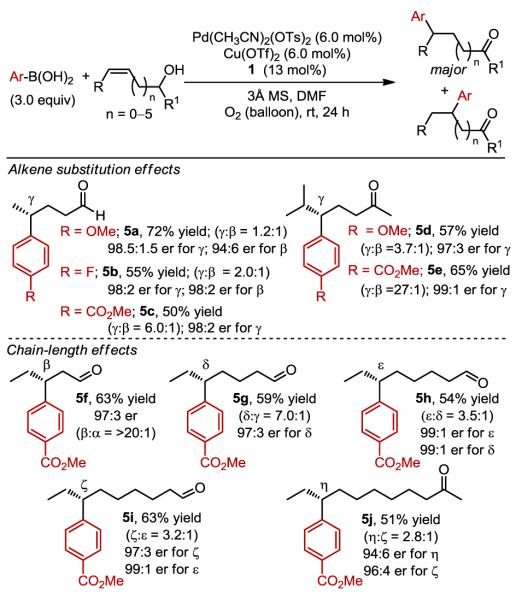
A series of both primary and racemic secondary substituted alkenyl alcohols were selected as substrates to probe whether a steric effect can influence the site and enantioselectivities (Table 3, 5a–e). Excellent enantioselectivity is observed in all cases, although an apparent steric effect influences site selectivity, with smaller groups leading to more of the β-insertion product (compare 5a (-Me), 4l (-Et), 5d (-iPr)). Of note, this minor product is formed in similar enantioselectivity to that of the major product. Next, the effect of chain-length (i.e., the distance from the alcohol to alkene) was probed by systematically evaluating ethyl-substituted alkenyl alcohols (Table 3, 5f–j). Again, the enantioselectivity is consistently high for all substrates evaluated, suggesting that the alcohol is not involved directly in face selection. This is also supported by the successful use of racemic alcohol substrates (5d, 5e, 5j). A clear trend emerges in terms of site selectivity for alkene insertion: as the alcohol is further removed from the alkene the selectivity is diminished. As an important synthetic note, many of these enantiomerically enriched products would be very cumbersome to synthesize using conventional approaches as the chiral centers are quite remote from the carbonyl.
Table 3.
Evaluation of various alkenol substrates.
|
Yields are reported as a combination of isomers. Enantioselectivity determined by SFC equipped with a chiral column. Ratio of γ to β-product as determined by either 1H NMR or gas chromatography.
In many asymmetric catalytic reactions, enantioselectivity is highly sensitive to the nature of the substrates used in the reaction.15 In this case, enantioselectivity is essentially independent of the nature of both reaction partners. In contrast, the site selectivity of the alkene insertion is controlled both by the nature of the arylboronic acid and the substitution and chain length of the alkenyl alcohol. To quantify the electronic effect of aryl boronic acids on the site selection, a linear free energy relationship was identified using Hammett σ-values of both meta and para-substituted boronic acids versus the log of the ratio of site selectivity, which is proportional to relative rates of isomer formation (Figure 2).16 The resultant Hammett plot has a ρ-value of 1.42 suggesting that the orientation of insertion is sensitive to the electronic nature of the Pd-Ar species formed (vide infra). In conjunction with this correlation is the trend of site selectivity as a function of chain-length (Table 3). To probe the origin of this interesting observation, the relative 13C chemical shifts for each alkene carbon were compared.17 For all cases, the most downfield shifted carbon is distal from the alcohol. Interestingly, correlating the Δδ 13C chemical shifts versus the log of the ratio of site selectivity reveals a clear trend. Although various scenarios cannot be ruled out, this observation is also suggestive of electronic effects governing site selectivity.
Figure 2.

Left: Plot of Log of site-selectivity versus Hammett σ-values derived from the scope presented in Table 2. Right: Plot of Log of site-selectivity versus Δδ 13C chemical shift.
Stereochemical outcome of cis-homoallyic alcohols
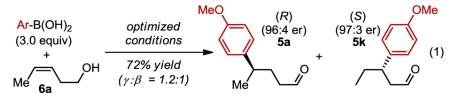 |
Stereochemical outcome of cis-allyic alcohols
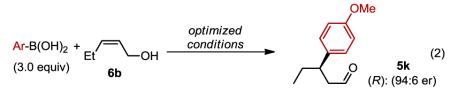 |
Considering the above discussion, an important question concerning the relationship of site selection to face selection arises as high enantiomeric excess is also observed for minor isomers analyzed (eq. 1). Therefore, the absolute configuration was determined for addition to cis-homoallylic alcohol 6a revealing that the enantiomer formed from γ-insertion product 5a is (R) whereas the β-insertion product 5k is (S) (see supporting information for details). Addition to an allylic alcohol 6b also yields product 5k as the only product in high enantioselectivity (eq. 2). In this case though, the opposite enantiomer is observed to that of 5k formed from a homoallylic substrate.3
Taken together, these data suggest that as the opposite faces of the alkene are presented to the catalyst a highly enantioselective insertion takes place but at different alkene carbons (Scheme 1). For the major pathway, the alkene presumably undergoes migratory insertion in the orientation as depicted in the transformation of A→B to account for the observed enantioselectivity. Of particular note, the site selectivity is decreased as the Pd-aryl species becomes less electrophilic (i.e., when the aryl group is more electron-rich). This is coupled to diminished site selectivity as a function of a less electronically-biased alkene, which suggests that an electronic bias is required for enhanced site-selectivity and that this is a highly sensitive event considering the magnitude of the ρ-value. An interesting feature of the minor insertion pathway is that the redox-relay product is formed even though this would formally require translation of the metal through the initially fashioned C–C bond. As this product is also formed in high enantioselectivity as the opposite enantiomer, the initial migratory insertion likely proceeds with the opposite orientation of the alkene as depicted in D→E. To form the aldehyde product, β-hydride elimination should yield the trisubstituted alkene in F followed by insertion of the resultant Pd–H to deliver G. Subsequent repetitive events ultimately will yield the aldehyde. The high level of enantioselection suggests that the Pd–H does not dissociate from the alkene, which may be attributed to the electrophilic nature of the catalyst. At this stage, simple stereochemical models cannot account for the excellent enantioselectivity observed as well as why the site selection operates in concert with face selection. These issues will require significant mechanistic investigations, which are currently underway.
Scheme 1.
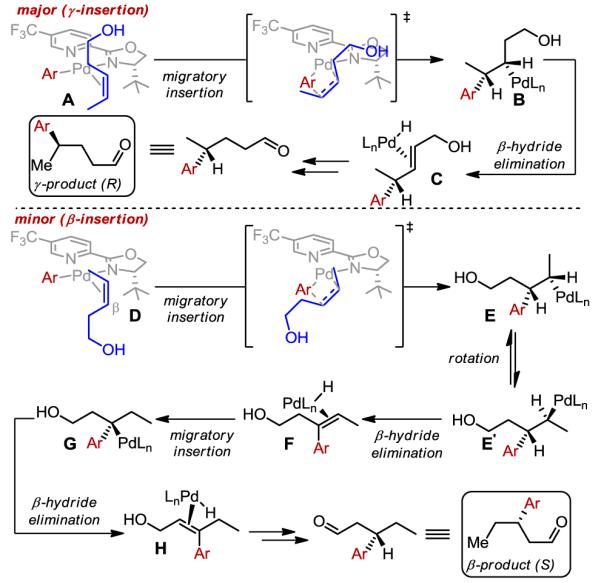
Mechanistic analysis of stereochemical course.
In conclusion, we report a highly enantioselective oxidative Heck reaction of alkenyl alcohols that operates through a redox-relay process. The products formed using this method would be difficult to access rapidly using traditional approaches. Preliminary efforts to understand the origin of site selectivity in this process are reported, wherein Hammett and other correlative techniques suggest that electronic effects play a major role in controlling site selection. Understanding the rules governing migratory insertion of relatively unbiased alkenes has not been extensively defined. Research is underway to probe this important organometallic mechanistic question, apply this method to synthetic endeavors, and expand the concept of redox-relay Heck reactions to new substrate and reaction types.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS RO1 GM063540).
Footnotes
Supporting Information. Experimental procedures and characterization data for new substances. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1) (a).Zultanski SL, Fu GC. J. Am. Chem. Soc. 2011;133:15362. doi: 10.1021/ja2079515. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smith SW, Fu GC. J. Am. Chem. Soc. 2009;131:14231. doi: 10.1021/ja9061823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2) (a).For selected reviews, see: Sun Y-W, Zhu P-L, Xu Q, Shi M. RSC Adv. 2013;3:3153. Hawner C, Alexakis A. Chem. Commun. 2010;46:7295. doi: 10.1039/c0cc02309d. Harutyunyan SR, den Hartog T, Geurts K, Minnaard AJ, Feringa BL. Chem. Rev. 2008;108:2824. doi: 10.1021/cr068424k. Hayashi T, Yamasaki K. Chem. Rev. 2003;103:2829. doi: 10.1021/cr020022z.
- (3).Werner EW, Mei T-S, Burckle AJ, Sigman MS. Science. 2012;338:1455. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4) (a).For the selected reviews, see: Mc Cartney D, Guiry P. J. Chem. Soc. Rev. 2011;40:5122. doi: 10.1039/c1cs15101k. Shibasaki M, Vogl EM, Ohshima T. Adv. Synth. Catal. 2004;346:1533. Dounay AB, Overman LE. Chem. Rev. 2003;103:2945. doi: 10.1021/cr020039h.
- (5).For an early example of intermolecular asymmetric Heck reaction with acyclic olefins, see: Yonehara K, Mori K, Hashizume T, Chung K-G, Ohe K, Uemura S. J. Organomet. Chem. 2000;603:40.
- (6) (a).For recent examples of intermolecular asymmetric Heck reactions of acyclic electronically biased olefins, see: Yoo KS, O’Neil J, Sakaguchi S, Giles R, Lee JH, Jung KW. J. Org. Chem. 2010;75:95. doi: 10.1021/jo901977n. Yoo KS, Park CP, Yoon CH, Sakaguchi S, O’Neil J, Jung KW. Org. Lett. 2007;9:3933. doi: 10.1021/ol701584f.
- (7) (a).For selected examples of using a redox relay strategy, see: Renata H, Zhou Q, Baran PS. Science. 2013;339:59. doi: 10.1126/science.1230631. Burns NZ, Baran PS, Hoffmann RW. Angew. Chem. Int. Ed. 2009;48:2854. doi: 10.1002/anie.200806086. For examples of Pd relays, see: Aspin S, Goutierre A-S, Larini P, Jazzar R, Baudoin O. Angew. Chem. Int. Ed. 2012;51:10808. doi: 10.1002/anie.201206237. Stokes BJ, Opra SM, Sigman MS. J. Am. Chem. Soc. 2012;134:11408. doi: 10.1021/ja305403s.
- (8) (a).For examples of isomerization/migration in Heck-type reactions, see: Kochi T, Hamasaki T, Aoyama Y, Kawasaki J, Kakiuchi F. J. Am. Chem. Soc. 2012;134:16544. doi: 10.1021/ja308377u. Crawley ML, Phipps KM, Goljer I, Mehlmann JF, Lundquist JT, Ullrich JW, Yang C, Mahaney PE. Org. Lett. 2009;11:1183. doi: 10.1021/ol900036y. Berthiol F, Doucet H, Santelli M. Tetrahedron. 2006;62:4372. Wang Y, Dong X, Larock RC. J. Org. Chem. 2003;68:3090. doi: 10.1021/jo026716p. Bouquillon S, Ganchegui B, Estrine B, Hénin F, Muzart J. J. Organomet. Chem. 2001;634:153. Larock RC, Leung W-Y, Stolz-Dunn S. Tetrahedron Lett. 1989;30:6629.
- (9).Sigman MS, Werner EW. Acc. Chem. Res. 2012;45:874. doi: 10.1021/ar200236v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10) (a).For examples of highly regioselective Heck reactions, see: Qin L, Ren X, Lu Y, Li Y, Zhou J. Angew. Chem. Int. Ed. 2012;51:5915. doi: 10.1002/anie.201201806. Werner EW, Sigman MS. J. Am. Chem. Soc. 2011;133:9692. doi: 10.1021/ja203164p. Zhu C, Falck JR. Angew. Chem. Int. Ed. 2011;50:6626. doi: 10.1002/anie.201101857. Delcamp JH, Brucks AP, White MC. J. Am. Chem. Soc. 2008;130:11270. doi: 10.1021/ja804120r. Mo J, Xu L, Ruan J, Liu S, Xiao J. Chem. Commun. 2006:3591. doi: 10.1039/b608033b.
- (11) (a).For selected examples of oxidative Heck reactions, see: Zheng C, Wang D, Stahl SS. J. Am. Chem. Soc. 2012;134:16496. doi: 10.1021/ja307371w. Zhang X, Fan S, He C-Y, Wan X, Min Q-Q, Yang J, Jiang Z-X. J. Am. Chem. Soc. 2010;132:4506. doi: 10.1021/ja908434e. Yoo KS, Yoon CH, Jung KW. J. Am. Chem. Soc. 2006;128:16384. doi: 10.1021/ja063710z. Andappan MMS, Nilsson P, von Schenck H, Larhed M. J. Org. Chem. 2004;69:5212. doi: 10.1021/jo049434t. Jung YC, Mishra RK, Yoon CH, Jung KW. Org. Lett. 2003;5:2231. doi: 10.1021/ol034458s. Parrish JP, Jung YC, Shin SI, Jung KW. J. Org. Chem. 2002;67:7127. doi: 10.1021/jo020159p. Du X, Suguro M, Hirabayashi K, Mori A, Nishikata T, Hagiwara N, Kawata T, Okeda T, Wang HF, Fugami K, Kosugi M. Org. Lett. 2001;3:3313. doi: 10.1021/ol016529y.
- (12).Werner EW, Sigman MS. J. Am. Chem Soc. 2010;132:13981. doi: 10.1021/ja1060998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13) (a).For recent examples of using pyridine/quinoline oxazoline ligands in Pd-catalyzed reactions, see: Kikushima K, Holder JC, Gatti M, Stoltz BM. J. Am. Chem. Soc. 2011;133:6902. doi: 10.1021/ja200664x. McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org. Lett. 2011;13:2830. doi: 10.1021/ol200784y. Jensen KH, Pathak TP, Zhang Y, Sigman MS. J. Am. Chem. Soc. 2009;131:17074. doi: 10.1021/ja909030c. Michel BW, Camelio AM, Cornell CN, Sigman MS. J. Am. Chem. Soc. 2009;131:6076. doi: 10.1021/ja901212h.
- (14) (a).For the studies on roles of molecular sieves in Pd-catalyzed aerobic oxidations, see: Steinhoff BA, King AE, Stahl SS. J. Org. Chem. 2006;71:1861. doi: 10.1021/jo052192s. Nishimura T, Maeda Y, Kakiuchi N, Uemura S. J. Chem. Soc., Perkin Trans. 2000;1:4301.
- (15).For an example, see: Harper KC, Vilardi SC, Sigman MS. J. Am. Chem. Soc. 2013;135:2482. doi: 10.1021/ja4001807.
- (16).Hansch C, Leo A, Taft RW. Chem. Rev. 1991;91:165. [Google Scholar]
- (17) (a).Verma R, Hansch C. Chem. Rev. 2011;111:2865. doi: 10.1021/cr100125d. [DOI] [PubMed] [Google Scholar]; (b) Neuvonen H, Neuvonen K, Koch A, Kleinpeter E, Pasanen P. J. Org. Chem. 2002;67:6995. doi: 10.1021/jo020121c. [DOI] [PubMed] [Google Scholar]; (c) Jones RG, Partington P. J. Chem. Soc., Faraday Transactions. 1972;2:2087. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.