

Abstract
p38 is a mitogen-activated protein kinase and mediates cell growth, cell differentiation, and synaptic plasticity. The aim of this study is to determine the extent to which p38 plays a role in maintaining mitochondrial respiration in male and female mice under a normal condition. To achieve this aim, we have generated transgenic mice that lack p38 in cerebellar Purkinje neurons by crossing Pcp2 (Purkinje cell protein 2)-Cre mice with p38loxP/loxP mice. Mitochondria from cerebellum were then isolated from the transgenic and wild-type mice to measure mitochondrial respiration using XF24 respirometer. The mRNA and protein expression of cytochrome c oxidase (COX) in cerebellum were also measured using RT-PCR and immunoblot methods. Separately, HT22 cells were used to determine the involvement of 17β-estradiol (E2) and COX in mitochondrial respiration. The genetic knockout of p38 in Purkinje neurons suppressed the mitochondrial respiration only in male mice and increased COX expression only in female mice. The inhibition of COX by sodium azide (SA) sharply suppressed mitochondrial respiration of HT22 cells in a manner that was protected by E2. These data suggest that p38 is required for the mitochondrial respiration of male mice. When p38 is below a normal level, females may maintain mitochondrial respiration through COX up-regulation.
1. Introduction
Mitochondrial oxidative phosphorylation is essential for aerobic organisms, as it provides the bulk of usable energy in the form of ATP. Mitochondrial ATP production is driven by transferring electrons across mitochondrial electron transfer complexes (ETC I-IV). In humans or experimental animals, mitochondrial defects are often observed in a variety of CNS disorders and brain aging [26], partly due to mitochondrial and nuclear gene mutations [5, 6]. A sex difference in mitochondrial function has been determined, with female rat brain maintaining mitochondrial function to a greater degree than males [8].
p38 is a mitogen-activated protein kinase and is involved in inflammation, cell growth, and cell death [20, 21]. There are four isoforms of p38: α, β, δ, and γ. p38α and p38β are expressed in neurons and glial cells, whereas p38δ and p38γ are exclusively expressed in immune cell types [28, 29]. Of the p38 isoforms, the activities of both p38α and p38β are remarkably high in normal adult mouse brain including cerebral cortex, hippocampus, cerebellum, and few nuclei of the brainstem. This indicates that α and β isoforms of p38 are primarily responsible for most of the constitutive p38 activity in brain [18]. The excessive p38 activation occurs during the stress of alcohol withdrawal and stroke, whereas the inhibition of p38 promotes neuronal survival [10, 16]. While the aberrant activation of p38 is associated with pathological conditions, a certain level of p38 appears essential for cellular and neuronal survival. For instance, p38 mediates synaptic plasticity, memory formation, gene regulation, and brain-derived neurotrophic factor production [25]. p38 also mediates the stimulating effect of Purkinje cell protein 2 (Pcp2) on the cellular differentiation [8]. Although p38 is mainly located in cytosol, studies have demonstrated the effects of p38 on mitochondria. p38 activation is associated with free radical production [22]. p38 inhibits the adverse effect of a nuclear receptor (PPARγ) coactivator on mitochondrial respiration in muscle [7]. These studies suggest that there is a crosstalk between cytosolic p38 and mitochondria.
In the current study, we intend to determine whether p38 plays a positive or negative role in maintaining mitochondrial respiration under a normal condition and whether there is a sex difference in this effect of p38. To reach this aim, we have generated transgenic mice that lack p38 in cerebellar Purkinje cells and assessed mitochondrial respiration, cytochrome c oxidase [(COX) ETC IV], and the effect of estrogen on these parameters.
2. Materials and methods
2.1 Chemicals
Analytic grade reagents were purchased from IDT Company (San Jose, CA), the Jackson Laboratory (Bar Harbor, Maine), Sigma Aldrich (St. Louis, MO), Cellsignaling Technology (Danvers, MA), Seahorse Bioscience (North Billerica, MA), Invitrogen (Grand Island, NY), and Abcam (Cambridge, MA).
2.2 Purkinje cell-specific deletion of p38
Among p38 isoforms, we selected p38α because it is the most abundant isozyme in the brain and the best characterized isoform [9, 24]. To avoid lethality, we employed a conditional transgenic mouse system to down-regulate Purkinje p38 genes using the Cre/loxP system and Pcp2 promoter (Purkinje neuron specific marker) [8]. Transgenic mice (Pcp2-Cre mice) that express Cre recombinase under the control of the Pcp2 (Jackson Laboratory) were cross-mated with floxed-p38α mice to generate the Pcp2-Cre+/-/p38loxP/loxP mice. The mice with floxed-p38 were kindly provided by Boehringer Ingelheim Inc. The p38 floxed allele was generated by homologous recombination of embryonic stem cells in which two sites of ATG containing Purkinje p38 sequence were flanked by loxP [3, 23] and excised in the presence of Pcp2-Cre. When pups reached 21 days old, the tips of the tail were collected for identification of genotype. All mice were sacrificed at the age of two months.
2.3 Genotyping procedure for Pcp2 and p38
DNA was isolated by incubating tail samples overnight at 55°C in proteinase K buffer. Primer sequences were as follows: for Pcp2-Cre transgene forward, 5′- GCG GTC TGG CAG TAA AAA CTA TC-3′; for Pcp2-Cre transgene reverse, 5′- GTG AAA CAG CAT TGC TGT CAC TT-3′; for Pcp2-Cre internal positive control forward, 5′- CTA GGC CAC AGA ATT GAA AGA TCT-3′; for Pcp2-Cre internal positive control reverse, 5′-GTA GGT GGA AAT TCT AGC ATC ATC C-3′; loxP-flanked p38α allele: 5′-TCCTACGAGCGTCGGCAAGGTG-3′ and 5′-ACTCCCCGAGAGTTCCTGCCTC-3′. Sequential denaturing (96°C, 30 sec), annealing (52°C, 1 min) and extension (72°C, 1 min) were repeated 35 times for genotyping the Pcp2-Cre transgene. The program of 30 cycles of denaturing (94°C for 30 sec), annealing (58°C, 30 sec), and extension (72°C, 45 sec) was used to genotype the p38α alleles.
2.4 Immunohistochemical detection of p38 in cerebellum
Mice were anesthetized with isoflorane and perfused with 0.9% saline. The formalin-fixed and paraffin-embedded left hemispheres were cut into 8 μm-thick slices on a microtome. The slices were deparaffinized in xylene, rehydrated through a series of graded ethanol solutions, and washed with PBS. The slices were subsequently moisturized at 95°C and incubated with primary antibody, polyclonal rabbit anti-p38α (1:50) overnight at 4°C. The slices were then incubated with broad spectrum poly HRP conjugate for 40 min at room temperature. The antigen-antibody bindings were visualized with a diaminobenzidine color reaction for 10 min. The slides were further rinsed, dehydrated through a series of graded ethanol and xylene, and mounted with Permount. All photographs were taken with a compatible Zeiss digital camera.
2.5 Mitochondrial respiration
Mitochondria were isolated from cerebellum by conventional differential centrifugation [15], diluted with assay solution (KCL, KH2PO4, MgCL2, HEPES, EGTA, and FA-free BSA), and transferred into XF24 cell culture plate. The samples were centrifuged (4 °C, at 3000 ×g), added with succinate (substrate) and rotenone (ETC I inhibitor), incubated (37 °C), and loaded into the XF24 respirometer. ADP and oligomycin were then injected into the XF injection ports. State II and III mitochondrial respirations are defined as oxygen consumption rate (OCR) before and after ADP is added to stimulate respiration, respectively. State IV respiration is defined as OCR in the absence of ADP or any metabolic inhibitors [2].
2.6 Real-time PCR to measure COX subunit IV (COXIV) mRNA
COX is composed of 13 subunits. Subunit IV was measured because of its abundant expression and sensitivity [14, 27]. Total RNA was isolated from cerebellum using Trizol reagent following the manufacturer's instructions. RNA was converted to cDNA by adding random primers and Superscript III reverse transcriptase. Real-time PCR was conducted to analyze gene expression using an ABI PRISM 7000 sequence detection system with TaqMan primers. Primer sequences were as following: for COXIV forward, 5′-GTTCAGTTGTACCGCATCCA-3′; for COXIV reverse, 5′-TTGTCATAGTCCCACTTGGC-3′.
2.7 Immunoblotting
Cerebellar mitochondrial lysate was used to detect COX subunit IV protein. Protein samples were loaded into 12% polyacrylamide gel and then transferred onto polyvinylidene fluoride membranes. Blots were probed overnight with mouse monoclonal primary antibody against COX subunit IV (1:1000) and incubated with horseradish peroxidase-conjugated anti-mouse secondary antibody for one hour. Bands were detected using UVP bioimaging system and quantified by the image densitometer. Voltage dependent anion channel1 (VDAC1) was used as a positive loading control for the mitochondrial protein level.
2.8 Mitochondrial respiration in HT22 cells
HT22 cells were seeded into XF24 plates at the density of 15000/well, and divided into the control group, the sodium azide (SA) (a COX inhibitor) group, and the SA + E2 group. HT22 cells were treated with 0.5 mM SA and/or E2 ranging from 0.01 to 1.0 μM for 5 hours. OCR was then measured as mentioned above.
3. Statistical analysis
All numerical data are expressed as mean ± SEM. Results of XF24 assay and immunoblots were analyzed by t-test or two-way ANOVA. Post-hoc Tukey's test was then conducted to detect an individual group difference. p value was set less than 0.05 to indicate statistical significance.
4. Results
4.1 Purkinje cell-specific deletion of p38
The results from genotyping reveal the DNA images of Pcp2-Cre at 100 base pair, homozygous p38loxP/loxP at 414 base pair, and an internal positive control (DNA quality control) at 324bp (Figure 1A). Immunohistochemistry results reveal that wild-type mice (C57BL/6 mice) and transgenic mice (Pcp2-Cre-/-/p38loxP/loxP) without Pcp2-Cre (Figure 1A-b) show p38 positive stains (dark brown deposits) in the Purkinje neurons along the Purkinje layer. By comparison, the p38 markers do not appear in transgenic mice (Figure 1A-a) that express both Pcp2-Cre and homozygous p38loxP/loxP, proving that p38 has been successfully knocked out in Purkinje neurons.
Figure 1.
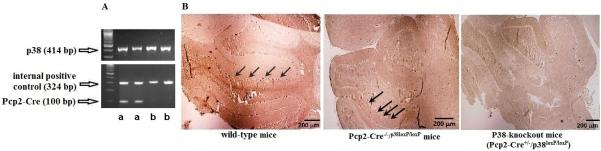
Purkinje cell-specific deletion of p38. Pcp2-Cre mice were cross-mated with floxed-p38α mice. Animal tail samples and PCR method were used to identify genotypes. Figure 1A illustrates the DNA images of Pcp2-Cre, homozygous p38loxP/loxP, and an internal positive control (DNA quality control) at 100, 414, and 324 base pair (bp), respectively. In Figure 1B, the cerebellar sections of these mice were immunostained using p38α antibody. Wild-type mice (C57BL/6 mice) and transgenic mice (Pcp2-Cre-/-/p38loxP/loxP) without Pcp2-Cre (Figure 1A-b) show p38 positive stains (dark brown deposits) in the Purkinje neurons along the Purkinje layer. The p38 markers do not appear in transgenic mice that express both Pcp2-Cre and p38loxP/loxP(Pcp2-Cre+/-/p38loxP/loxP) (Figure 1A-a).
4.2 Mitochondrial respiration
p38-knockout did not significantly alter mitochondrial respiration in female mice (Figure 2A) but significantly suppressed mitochondrial respiration in male mice (p < 0.0001) (Figure 2B). p38-knockout did not alter different states (II-IV) of mitochondrial respiration. For wild-type mice, the mitochondrial respiration (state II) was higher in female (937.00±62.17 pMoles/min) than male mice (625.20±10.90 pMoles/min) (p < 0.0001) (Figure not shown). These results indicate that p38-knockout suppresses the mitochondrial respiration only in male mice.
Figure 2.
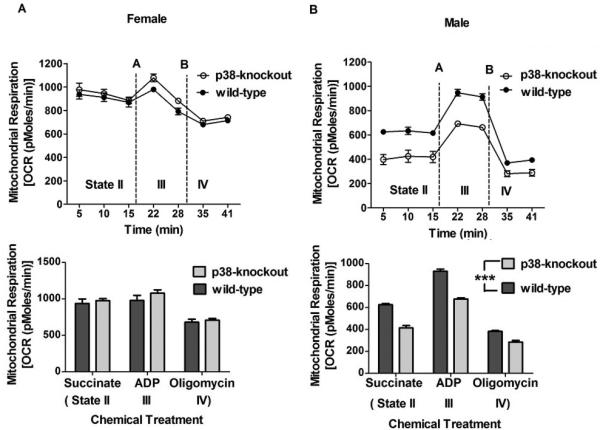
Mitochondrial respiration in wild-type and p38-knockout mice. Mitochondria isolated from cerebellum were used to assess mitochondrial respiration (OCR) using XF respirometer. OCR did not differ between wild-type and p38-knockout female mice but was lower in p38-knockout male mice than wild-type male mice (***p <0.0001). Succinate, ADP, and oligomycin were added to mitochondrial samples to determine the effect of p38-knockout on a different state (II-IV) of mitochondrial respiration. p38-knockout did not affect the mitochondrial respiration states. All data are represented as mean ± SEM. N= 5 or 6 mice/group.
4.3 COX subunit IV mRNA and protein expression
The effect of p38-knockout on COX expression was tested in male and female mice. Both mRNA and protein levels of COX subunit IV were increased in p38-knockout female mice compared to wild-type female mice (Figure 3A and 3B). By comparison, neither mRNA nor protein content of COX subunit IV was altered in p38-knockout male mice. These results indicate that p38-knockout increases COX only in female mice.
Figure 3.
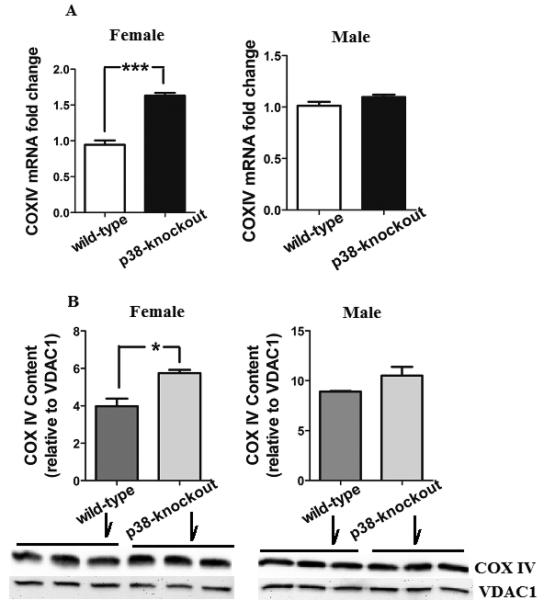
mRNA and protein expressions of COX subunit IV. Cerebellar mitochondria were used to assess the levels of COX subunit IV mRNA and protein using RT-PCR and immunoblot, respectively. Compared to wild-type mice, a significant increase in the levels of COX-IV mRNA (***p <0.001) and protein (*p <0.05) were found in p38-knockout female mice but not in male mice. Voltage dependent anion channel1 (VDAC1) was used as a positive loading control for the mitochondrial protein level. All data are represented as mean ± SEM. N= 5 or 6 mice/group.
4.4 The effect of E2 and COX on mitochondrial respiration
Mitochondrial respiration was suppressed (p < 0.01) when cells were treated with a COX inhibitor (SA) alone (Figure 4). This effect of SA was blunted in the presence of E2, suggesting that E2 promotes mitochondrial respiration through COX activation. Among test doses (0.01-1.0 μM), the lowest dose of E2 that was able to exert the protection was 0.1 μM (p < 0.0001). E2 treatment alone at 0.1 and 1.0 μM didn't increase mitochondrial respiration (data not shown).
Figure 4.
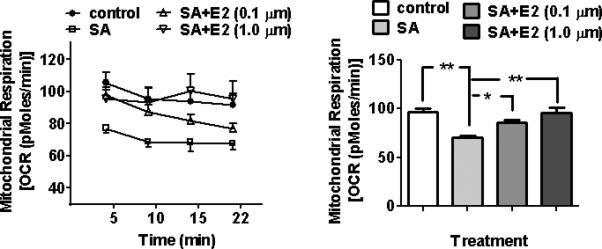
Effects of E2 and COX on mitochondrial respiration. HT22 cells were treated with a COX inhibitor (0.5 mM sodium azide (SA)) with or without E2 (0.1 μM or 1.0 μM). SA sharply decreased mitochondrial respiration (**p <0.01) and this effect of SA was blunted by E2 (*p <0.01, **p < 0.01). N=5 wells/group.
5. Discussion
The key finding of the current study is that p38 influences mitochondria differently between male and female mice. p38-knockout suppresses mitochondrial respiration only in male mice and increases COX expression only in female mice. These data suggest that a certain level of p38 is required for the mitochondrial respiration of male mice. When p38 is below a necessary level for mitochondrial respiration, females may have capacity to maintain mitochondrial respiration through up-regulating COX.
Sex difference in mitochondria has been reported in previous studies in which females have age-associated oxidative damage to a smaller degree than males due to more efficient mitochondrial respiration. The sex difference is also reportedly attributed to a higher level of glutathione peroxidase (a H2O2 quencher) and uncoupling protein 5 (free radical inhibitor) in females than males. The disruption of ETC III impairs mitochondrial respiration of male mice and shortened their life-span compared to female mice [12]. Compared to males, mitochondria from female rats generate less hydrogen peroxide, more reduced glutathione (antioxidant), and more 16S rRNA of which expression is inversely correlated with aging [1]. Ovariectomy abolishes whereas E2 replacement reinstates the sex differences [1]. These studies suggest that female mitochondria might be equipped with more effective protective mechanisms than male mitochondria. Our current observations of greater mitochondrial respiration in wild-type female mice than male mice essentially agree with these studies.
p38 is a signaling protein kinase and transduces neuronal signals. Accordingly, p38 is abundantly expressed in neurons and synapsis, mediating synaptic plasticity and gene regulation [25]. While excessive p38 activation is associated with pathological conditions, a certain level of p38 might be required for maintaining mitochondrial respiration. Our transgenic mice hardly show p38 in Purkinje neurons, certainly indicating that their p38 is well below a normal level. It is of interest that p38 deletion suppresses mitochondrial respiration only in male mice and increases COX only in female mice. A potential explanation is that p38 deletion may trigger a compensatory mechanism in female mitochondria to up-regulate COX and subsequently enhance mitochondrial respiration. E2 may play a role in this process by increasing COX as E2 increases COX activity [14]. With this scenario, male mitochondria in the absence of p38 may be less able to respire in part due to insufficient E2 for COX up-regulation. Alternatively, females might be generally more resistant to p38 alteration than males as hypoxia increased p38 activation in male but not in female cardiac fibroblasts [30].
The up-regulating effect of E2 on COX has been demonstrated in our previous study in which E2-implantation increased COX activity in ovariectomized rats [14, 15]. We conducted an in vitro experiment to test whether and how E2 plays a role in sex-dependent mitochondrial respiration. When cells were exposed to a COX inhibitor, they lost mitochondrial respiration that was restored in the presence of E2 (0.1 and 1.0 μM). These data suggest that COX activation mediates mitochondrial respiration and E2 acts as a COX activator. However, E2 lost such protective effect at a low physiologically relevant concentration (0.01μM). These results are somewhat puzzling because E2 at a physiological concentration protects COX from stress associated with alcohol withdrawal in rats [14, 15]. This discrepancy may result from a difference between in vivo and in vitro conditions. For instance, when E2 concentration is below a certain level, this cell line may become less sensitive to E2's protection for COX. Alternatively, the protective dose of E2 may differ depending upon the nature of stress. Others also report that E2 reverses age-related loss of COX cofactors [13]. E2 protects mitochondria from trauma-hemorrhage through COX up-regulation [11]. Although there may be a differential dose effect of E2, these studies strengthen the idea that E2's protection for COX helps female mice to maintain mitochondrial respiration. How cytosolic p38 affects mitochondria is still an open question. One possibility is that p38 activation induces its substrate (p53) to translocate into mitochondria [17]. p53 in turn eliminates unhealthy mitochondrial proteins [17, 19], thereby enhancing mitochondrial functions. If E2 increases p53 mRNA level [6], E2 in female mice might enable such p53's mito-protection.
In conclusion, our findings provide empirical evidence that p38-knockout in the major neurons of cerebellum sex-specifically regulates mitochondrial respiration of this brain area. The current study may provide a new mechanistic insight involving p38 into sex-dependent mitochondrial respiration.
HIGHLIGHTS.
p38-knockout creates a sex difference in mitochondrial respiration.
p38-knockout suppresses mitochondrial respiration in male mice and increases COX expression in female mice.
Estrogen protects against mitochondrial respiratory impairment.
Acknowledgement
We wish to thank Wenjun Li for his technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borras C, Gambini J, Vina J. Mitochondrial oxidant generation is involved in determining why females live longer than males. Front Biosci. 2007;12:1008–1013. doi: 10.2741/2120. [DOI] [PubMed] [Google Scholar]
- 2.Boveris A, Costa L, Cadenas E. The mitochondrial production of oxygen radicals and cellular aging. . Understanding the Process of Aging: The Roles of Mitochondria, Free Radicals, and Antioxidants. Marcel Dekker, Inc.; New York: 1999. [Google Scholar]
- 3.Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi D, Hwang S, Lee E, Yoon S, Yoon BK, Bae D. Expression of mitochondria-dependent apoptosis genes (p53, Bax, and Bcl-2) in rat granulosa cells during follicular development. J Soc Gynecol Investig. 2004;11:311–317. doi: 10.1016/j.jsgi.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 5.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 6.Edmond JC. Mitochondrial disorders. Int Ophthalmol Clin. 2009;49:27–33. doi: 10.1097/IIO.0b013e3181a8de58. [DOI] [PubMed] [Google Scholar]
- 7.Fan M, Rhee J, St-Pierre J, Handschin C, Puigserver P, Lin J, Jaeger S, Erdjument-Bromage H, Tempst P, Spiegelman BM. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev. 2004;18:278–289. doi: 10.1101/gad.1152204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guan J, Luo Y, Denker BM. Purkinje cell protein-2 (Pcp2) stimulates differentiation in PC12 cells by Gbetagamma-mediated activation of Ras and p38 MAPK. Biochem J. 2005;392:389–397. doi: 10.1042/BJ20042102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo G, Bhat NR. p38alpha MAP kinase mediates hypoxia-induced motor neuron cell death: a potential target of minocycline's neuroprotective action. Neurochem Res. 2007;32:2160–2166. doi: 10.1007/s11064-007-9408-8. [DOI] [PubMed] [Google Scholar]
- 10.Horstmann S, Kahle PJ, Borasio GD. Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J Neurosci Res. 1998;52:483–490. doi: 10.1002/(SICI)1097-4547(19980515)52:4<483::AID-JNR12>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh YC, Yu HP, Suzuki T, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Upregulation of mitochondrial respiratory complex IV by estrogen receptor-beta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma-hemorrhage. J Mol Cell Cardiol. 2006;41:511–521. doi: 10.1016/j.yjmcc.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Hughes BG, Hekimi S. A mild impairment of mitochondrial electron transport has sex-specific effects on lifespan and aging in mice. PLoS One. 2011;6:e26116. doi: 10.1371/journal.pone.0026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones TT, Brewer GJ. Critical age-related loss of cofactors of neuron cytochrome C oxidase reversed by estrogen. Exp Neurol. 2009;215:212–219. doi: 10.1016/j.expneurol.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung ME, Agarwal R, Simpkins JW. Ethanol withdrawal posttranslationally decreases the activity of cytochrome c oxidase in an estrogen reversible manner. Neurosci Lett. 2007;416:160–164. doi: 10.1016/j.neulet.2007.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung ME, Ju X, Metzger DB, Simpkins JW. Ethanol withdrawal hastens the aging of cytochrome c oxidase. Neurobiol Aging. 2012;33:618, e621–632. doi: 10.1016/j.neurobiolaging.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung ME, Ju X, Simpkins JW, Metzger DB, Yan LJ, Wen Y. Ethanol withdrawal acts as an age-specific stressor to activate cerebellar p38 kinase. Neurobiol Aging. 2011;32:2266–2278. doi: 10.1016/j.neurobiolaging.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitamura N, Nakamura Y, Miyamoto Y, Miyamoto T, Kabu K, Yoshida M, Futamura M, Ichinose S, Arakawa H. Mieap, a p53-inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PLoS One. 2011;6:e16060. doi: 10.1371/journal.pone.0016060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SH, Park J, Che Y, Han PL, Lee JK. Constitutive activity and differential localization of p38alpha and p38beta MAPKs in adult mouse brain. J Neurosci Res. 2000;60:623–631. doi: 10.1002/(SICI)1097-4547(20000601)60:5<623::AID-JNR7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 19.Liu H, Pedram A, Kim JK. Oestrogen prevents cardiomyocyte apoptosis by suppressing p38alpha-mediated activation of p53 and by down-regulating p53 inhibition on p38beta. Cardiovasc Res. 2011;89:119–128. doi: 10.1093/cvr/cvq265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mielke K, Herdegen T. JNK and p38 stresskinases--degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol. 2000;61:45–60. doi: 10.1016/s0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- 21.Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 22.Park GB, Kim YS, Lee HK, Song H, Kim S, Cho DH, Hur DY. Reactive oxygen species and p38 MAPK regulate Bax translocation and calcium redistribution in salubrinal-induced apoptosis of EBV-transformed B cells. Cancer Lett. 2011;313:235–248. doi: 10.1016/j.canlet.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 23.Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, Chi JT, Yan Z. p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS One. 2009;4:e7934. doi: 10.1371/journal.pone.0007934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sudo T, Yagasaki Y, Hama H, Watanabe N, Osada H. Exip, a new alternative splicing variant of p38 alpha, can induce an earlier onset of apoptosis in HeLa cells. Biochem Biophys Res Commun. 2002;291:838–843. doi: 10.1006/bbrc.2002.6529. [DOI] [PubMed] [Google Scholar]
- 25.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 26.Trifunovic A, Larsson NG. Mitochondrial dysfunction as a cause of ageing. J Intern Med. 2008;263:167–178. doi: 10.1111/j.1365-2796.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- 27.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Shen B, Lin A. Novel strategies for inhibition of the p38 MAPK pathway. Trends Pharmacol Sci. 2007;28:286–295. doi: 10.1016/j.tips.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Zhao X, Eghbali-Webb M. Gender-related differences in basal and hypoxia-induced activation of signal transduction pathways controlling cell cycle progression and apoptosis, in cardiac fibroblasts. Endocrine. 2002;18:137–145. doi: 10.1385/ENDO:18:2:137. [DOI] [PubMed] [Google Scholar]