

Abstract
Spiroplasma is a genus of wall-less, low-GC, Gram-positive bacteria with helical morphology. As commensals or pathogens of plants, insects, ticks, or crustaceans, they are closely related with mycoplasmas and form a monophyletic group (Spiroplasma–Entomoplasmataceae–Mycoides) with Mycoplasma mycoides and its relatives. In this study, we report the complete genome sequences of Spiroplasma chrysopicola and S. syrphidicola from the Chrysopicola clade. These species form the sister group to the Citri clade, which includes several well-known pathogenic spiroplasmas. Surprisingly, these two newly available genomes from the Chrysopicola clade contain no plectroviral genes, which were found to be highly repetitive in the previously sequenced genomes from the Citri clade. Based on the genome alignment and patterns of GC-skew, these two Chrysopicola genomes appear to be relatively stable, rather than being highly rearranged as those from the Citri clade. Phylogenetic analyses suggest that the susceptibility to plectroviral invasion probably originated in the common ancestor of the Citri clade or one of its subclades. This susceptibility may be attributed to the absence of antiviral systems found in the Chrysopicola clade. Using the virus-free genomes of the Chrysopicola clade as references, we inferred the putative viral integration sites in the Citri genomes. Comparisons of syntenic regions suggest that the extensive viral invasion in the Citri clade promoted genome rearrangements and expansions. More importantly, the viral invasion may have facilitated horizontal gene transfers that contributed to adaptation in the Citri clade.
Keywords: Citri–Chrysopicola–Mirum clade, Mollicutes, plectrovirus, Spiroplasma chrysopicola, Spiroplasma syrphidicola, viral insertion
Introduction
Spiroplasma (Spiroplasmataceae, Entomoplasmatales) is a genus of low-GC, Gram-positive, wall-less bacteria that are distinguished from other genera in the class Mollicutes by their helical morphology. The majority of spiroplasmas are found to be commensals of insects or plants, whereas a small number of species are pathogens of plants, insects, and crustaceans (Gasparich et al. 2004; Gasparich 2010). Phylogenetic analyses based on 16 S rDNA suggested that Entomoplasmatales (Spiroplasma, Entomoplasma, and Mesoplasma) and Mycoplasmatales (Mycoplasma and Ureaplasma) form a major clade of Mollicutes (Mycoplasmatales–Entomoplasmatales), whereas the other major clade includes Acholeplasma and “Candidatus Phytoplasma” (Gasparich et al. 2004). The traditionally delimited Spiroplasma and Mycoplasma were found to be nonmonophyletic (Gasparich et al. 2004; Volokhov et al. 2012). Mycoplasma mycoides and several other mycoplasmas form a monophyletic group within a clade that contains entomoplasmas and mesoplasmas (Mycoides–Entomoplasmataceae), which, in turn, is nested within a clade that otherwise comprised spiroplasmas. This latter group (Spiroplasma–Entomoplasmataceae–Mycoides) has three additional clades, including the basal Ixodetis, the Apis sister to the Mycoides–Entomoplasmataceae clade, and the Citri–Chrysopicola–Mirum (Gasparich et al. 2004; Lo et al. 2013).
The Citri–Chrysopicola–Mirum clade includes the only three phytopathogenic spiroplasmas discovered to date: Spiroplasma citri (Saglio et al. 1973), S. phoeniceum (Saillard et al. 1987), and S. kunkelii (Whitcomb et al. 1986), the causative agents of citrus stubborn disease, periwinkle yellowing, and corn stunt disease, respectively. Similar to the phytopathogenic phytoplasmas, all these phytopathogenic spiroplasmas are transmitted by leafhopper vectors (Saillard et al. 1987). This clade also contains species pathogenic to arthropods, including honeybees (S. melliferum; Clark et al. 1985), Drosophila (S. poulsonii; Williamson et al. 1999), crabs (S. eriocheiris; Wang et al. 2004), and shrimps (S. penaei; Nunan et al. 2005). Because of the great economical importance and early discovery of S. citri and S. melliferum, they have been extensively used for studies on Spiroplasma biology and genomics. Previous genome sequencing efforts revealed that the genomes of these two species contain highly repetitive plectrovirus-related fragments (Carle et al. 2010; Alexeev et al. 2012; Lo et al. 2013). Because these viral fragments can be relatively large in size (∼7 kb) and comprise more than 20% of these genomes, the three available Spiroplasma genomes to date are in draft assemblies rather than complete sequences. In addition, similar plectroviral genes were found in the survey sequence tags (Bai and Hogenhout 2002) and in an 85-kb segment (Zhao et al. 2003) of the S. kunkelii genome, suggesting that the presence of plectroviral sequences may be a common characteristic of Spiroplasma. This is in sharp contrast to the sequenced genomes of Mycoplasma and Mesoplasma, where such repetitive regions of viral origin are not found.
Compared with the plant-pathogenic phytoplasmas and vertebrate-pathogenic mycoplasmas, spiroplasmas occupying diverse ecological niches are relatively understudied. Additionally, previous genome sequencing projects for spiroplasmas have only focused on the three species mentioned earlier, and a complete Spiroplasma genome sequence is still lacking. To have a better understanding of the evolution of the plectroviral sequences and their impacts on the Spiroplasma genomes, we chose two other species from the Citri–Chrysopicola–Mirum clade for whole-genome sequencing. These two species, S. chrysopicola (Whitcomb et al. 1997) isolated from a deerfly (Chrysops sp.) and S. syrphidicola (Whitcomb et al. 1996) from a syrphid fly (Eristalis arbustorum), have not been reported to be pathogenic to insects or plants. They constitute a monophyletic group distinct from the phytopathogenic or entomopathogenic species in the Citri–Chrysopicola–Mirum clade (Gasparich et al. 2004; Lo et al. 2013). Therefore, their genomes are good references for comparisons with the previously sequenced Spiroplasma genomes.
Materials and Methods
DNA Extraction and Whole-Genome Shotgun Sequencing
The bacterial strains, S. chrysopicola DF-1T (ATCC 43209) and S. syrphidicola EA-1T (ATCC 33826), were obtained from the American Type Culture Collection. For each species, 20 ml R2 medium (Moulder et al. 2002) containing 200 µl culture was incubated at 30 °C for 72 h. DNA extractions were carried out using the Wizard Genomic DNA Purification Kit (Promega). One paired-end library (insert size = ∼155 bp) and one mate-pair library (insert size = ∼4.5 kb) were prepared for each species. Approximately 0.7-Gb 101-bp reads were sequenced for each library on the HiSeq 2000 platform (Illumina, USA) by a commercial sequencing service provider (Yourgene Bioscience, Taiwan).
Genome Assembly and Annotation
For each Spiroplasma species, the paired-end and mate-pair reads were used for de novo assembly by the program ALLPATHS-LG release 42781 (Gnerre et al. 2011). The assembled scaffolds were used as the starting point for our iterative assembly improvement process (Lo et al. 2013). For each iteration, we mapped all raw reads from the two libraries to the existing scaffolds using BWA v0.6.2 (Li and Durbin 2009) and visualized the results with IGV v2.1.24 (Robinson et al. 2011). Neighboring scaffolds with mate-pair support for continuity were combined, and reads overhanging at margins of contigs or scaffolds were used to extend the assembly and to fill gaps. The MPILEUP program in the SAMTOOLS v0.1.18 package (Li et al. 2009) was used to identify polymorphic sites. Polymerase chain reaction and primer walking were used to confirm these polymorphic sites and the sequences of repetitive regions. Mapping of the raw reads onto the final assembly using BWA resulted in 300- and 171-fold coverage of mate-pair reads (mapping quality of at least 37) and 586- and 645-fold coverage of pair-end reads (mapping quality of 60) for S. chrysopicola and S. syrphidicola, respectively.
The complete genome sequences were processed using RNAmmer (Lagesen et al. 2007), tRNAscan-SE (Lowe and Eddy 1997), and PRODIGAL (Hyatt et al. 2010) for gene prediction. The gene name and description for the protein-coding genes were assigned based on the orthologous genes identified by OrthoMCL (Li et al. [2003]; e value ≤ 1 × 10−15) in S. melliferum IPMB4A, S. melliferum KC3, and S. citri GII3-3X and BlastP (Altschul et al. 1997; Camacho et al. 2009) searches against the National Center for Biotechnology Information (NCBI) nonredundant (nr) protein database (Benson et al. 2012). For functional categorization, all protein sequences were annotated by utilizing the KAAS tool (Moriya et al. 2007) provided by the KEGG database (Kanehisa and Goto 2000; Kanehisa et al. 2010) using the bidirectional best hit method and a set of selected reference genomes as described in Lo et al. (2013). The KEGG orthology assignment was further mapped to the COG functional category assignment (Tatusov et al. 1997, 2003). Genes that did not have any COG functional category assignment were assigned to a custom category (category X). CRISPRfinder (Grissa et al. 2007) was used to search for CRISPRs (clustered regularly interspaced short palindromic repeats) in the genomes. Open reading frames overlapping with CRISPR repeats were removed from the gene list, as the typical repeat region do not encode proteins (Haft et al. 2005). Positions of genes and viral insertions (see later), GC-skew, and GC content were drawn using Circos (Krzywinski et al. 2009).
Molecular Phylogenetic Inference
Previous phylogenetic analyses of the Citri–Chrysopicola–Mirum clade were either less comprehensive in terms of taxon sampling or based solely on 16S rDNA, which did not provide sufficient variable characters to resolve every node. Therefore, we constructed a phylogeny based on a combined data set of 16S rDNA and DNA-directed RNA polymerase subunit beta (rpoB), a single-copy gene with phylogenetic utility for Mycoplasmataceae (Volokhov et al. 2012) and spiroplasmas (Bi et al. 2008). Sequences of the two genes were retrieved for selected species of the Mycoplasmatales–Entomoplasmatales clade with an emphasis on the Citri–Chrysopicola–Mirum clade (supplementary table S1, Supplementary Material online), aligned using MUSCLE v3.8 (Edgar 2004) with the default settings, and concatenated into a single data set. A maximum likelihood phylogeny was inferred using PhyML v3.0 (Guindon and Gascuel 2003) with the GTR + I + G model and six substitution rate categories. Bootstrap supports were estimated from 1,000 samples of alignment generated by the SEQBOOT program of PHYLIP v3.69 (Felsenstein 1989).
Genomic Analyses
We aligned the genomes of S. chrysopicola and S. syrphidicola using Mauve 2.3.1 (Darling et al. 2010) and calculated their nucleotide sequence identity from a concatenated MUSCLE alignment of single-copy genes using the DNADIST program of PHYLIP. To have a better understanding of the distribution and impacts of the plectroviral insertions in Spiroplasma genomes, we examined the syntenic regions between the Citri and Chrysopicola clades based on the OrthoMCL homologous gene clustering results and identified syntenic regions in the Citri clade with plectroviral genes in the middle or at the margin. We define synteny as the occurrence of at least four orthologous genes in the same order. Because most of the plectroviral genes in the S. melliferum IPMB4A assembly were removed from the contigs due to unresolvable polymorphisms (Lo et al. 2013), this analysis was focused on S. melliferum KC3 and S. citri. The occurrence of plectroviral genes between two syntenic regions was regarded as a single viral insertion and mapped on the genome maps of S. chrysopicola and S. syrphidicola in the middle between the two regions. If a syntenic region could only be found on one side of the plectroviral genes, the viral insertion was mapped next to the ortholog nearest to the plectroviral genes.
We compared the gene content among the five sequenced Spiroplasma genomes by using M. mycoides (NC_005364) and M. penetrans (NC_004432) as the outgroups. Protein-coding genes were clustered using OrthoMCL as described earlier. Amino acid sequences of single-copy genes were extracted, aligned with MUSCLE, concatenated, and used for PhyML phylogenetic analyses with the LG model (Le and Gascuel 2008). Because the genomes of the two S. melliferum strains are approximately 99.9% identical at the nucleotide level based on shared single-copy genes (Lo et al. 2013), we combined these two partial genomes into a pan-genome to better represent the complete genome of S. melliferum. This approach is not expected to bias the results of gene content comparisons, because we focus on homologous gene clusters rather than individual genes. For the whole-genome phylogenetic analyses, genes from the strain IPMB4A were used.
Results and Discussion
Phylogenetic Relationships within the Citri–Chrysopicola–Mirum Clade
A maximum likelihood phylogeny was inferred from a combined data set of 16S rDNA and rpoB (fig. 1). All nodes received a bootstrap support of at least 75%. The phylogeny indicates that the Mycoplasmatales–Entomoplasmatales clade is divided into two groups, the Spiroplasma–Entomoplasmataceae–Mycoides clade, and a clade formed by the rest of the mycoplasmas. The Spiroplasma–Entomoplasmataceae–Mycoides clade is further divided into the Ixodetis, Mycoides–Entomoplasmataceae, Apis, and Citri–Chrysopicola–Mirum clades. In general, this phylogeny of spiroplasmas and mycoplasmas is consistent with previous analyses based only on 16S rDNA (Gasparich et al. 2004; Lo et al. 2013) but provides stronger support for each of the major nodes. Additionally, with a more comprehensive sampling, our phylogeny provides new insights into the relationships within the Citri–Chrysopicola–Mirum clade. Previous analyses based on 16S rDNA either failed to resolve the relationships among S. kunkelii, S. phoeniceum and the clade formed by S. citri and S. melliferum (Wang et al. 2004) or suggested that S. kunkelii was more closely related to S. citri and S. melliferum than S. phoeniceum (Bi et al. 2008; Lo et al. 2013). However, our result strongly indicates that S. phoeniceum and S. kunkelii are more closely related to each other than either is to the clade of S. citri and S. melliferum. For the purposes of this study, we further divided the Citri–Chrysopicola–Mirum clade into three subclades named after the specific epithets of the three component species chosen by Gasparich et al. (2004): the Mirum clade (S. mirum and S. eriocheiris), the Chrysopicola clade (S. chrysopicola and S. syrphidicola), and the Citri clade, of which all characterized species except for S. insolitum are known to be pathogenic.
Fig. 1.—

Molecular phylogeny of the Mycoplasmatales–Entomoplasmatales clade. The maximum-likelihood phylogeny was based on 16S rDNA and rpoB sequences, with emphasis on the Citri–Chrysopicola–Mirum clade. All nodes, except where indicated, received 100% bootstrap support. Taxa with genome sequences in GenBank are highlighted in bold, with genome sizes indicated in parentheses. The genome sizes of the species that do not have complete genome sequences available are estimates based on pulsed-field gel electrophoresis. The occurrence of plectroviral fragments in the chromosome is indicated: “N”: no plectroviral fragments in complete genome sequences; “Y”: plectroviral fragments comprising a significant proportion of genome sequences; “?”: presence of plectroviral fragments in the chromosome indicated by Southern hybridization (Spiroplasma phoeniceum) or susceptibility to plectroviral infection (S. poulsonii).
Genome Features of S. chrysopicola and S. syrphidicola
The genomes of S. chrysopicola and S. syrphidicola were sequenced to completion (fig. 2; table 1). They are approximately 1.1 Mb in size, which are much smaller than the estimated genome sizes of the Citri clade spiroplasmas (1.82 Mb in S. citri [Carle et al. 2010], 1.38 Mb in S. melliferum IPMB4A [Lo et al. 2013], and 1.55 Mb in S. kunkelii [Dally et al. 2006]; fig. 1). The GC contents of these two species (28.8–29.2%) are higher than those of the Citri clade (25.9–27.5%) and the Mycoides–Entomoplasmataceae clade (24.0% in M. mycoides and 27.0% in Mesoplasma florum). This agrees with the finding based on biochemically determined GC contents that S. chrysopicola and S. syrphidicola appeared to be at the high end of the Spiroplasma range (Whitcomb et al. 1996, 1997; Gasparich et al. 2004). The genomes of both species demonstrate GC-skew inversions near dnaA, the first gene downstream of oriC in most bacteria (Mott and Berger 2007), and at about the opposite position of dnaA in the genome (fig. 2A). In between these two inversions, the GC-skew remains positive for the first half of the genome and negative for the second except for an 11-kb segment near the replication origin. These two halves roughly correspond to asymmetry in transcription polarity (rings 2 and 3 in fig. 2A). Together, these indicate that the two genomes have been relatively stable, probably with few recent rearrangements. On the contrary, several complete genomes of mycoplasmas or phytoplasmas exhibit anomalous GC-skew patterns (Minion et al. 2004; Westberg et al. 2004; Bai et al. 2006), which are related to rearrangements caused by a high proportion of repetitive sequences (Westberg et al. 2004; Bai et al. 2006). Another characteristic of the S. chrysopicola and S. syrphidicola genomes is related to the gene organization around the oriC. The gene order rpnA-rpmH-oriC-dnaA-dnaN is conserved in Bacillus, Acholeplasma, M. penetrans, M. mycoides, and Mes. florum. In the Chrysopicola and Citri clades, however, guaB is located between rpmH and oriC, which seems to be a synapomorphy of these spiroplasmas in the Citri–Chrysopicola–Mirum clade.
Fig. 2.—

Genome maps and alignment of Spiroplasma chrysopicola and S. syrphidicola. (A) Genome maps of S. chrysopicola and S. syrphidicola. Rings from the outermost to the center: 1) scale marks, 2) protein-coding genes on the forward strand, 3) protein-coding genes on the reverse strand, 4) tRNA (purple) and rRNA (red) genes, 5) putative sites of viral insertions in S. melliferum KC3 (red) relative to the respective genomes, 6) putative sites of viral insertions in S. citri (green) relative to the respective genomes, 7) protein-coding genes unique to the respective genomes, 8) GC skew, and 9) GC content. Protein-coding genes are color coded according to their COG categories. The positions of the genomic regions shown in figure 3A and B are indicated by arrows. (B) Whole-genome alignment between S. chrysopicola and S. syrphidicola.
Table 1.
Genome Assembly Statistics
| Spiroplasma chrysopicola DF-1 | S. syrphidicola EA-1 | S. melliferum IPMB4A | S. melliferum KC3 | S. citri GII3-3X | |
|---|---|---|---|---|---|
| GenBank accession | CP005077 | CP005078 | AMGI01000001– AMGI01000024 | AGBZ01000001– AGBZ01000004 | AM285301– AM285339 |
| Number of chromosomal contig(s) | 1 | 1 | 24 | 4 | 39 |
| Combined size of chromosomal contig(s) (bp) | 1,123,322 | 1,107,344 | 1,098,846 | 1,260,174 | 1,525,756 |
| Estimated chromosomal size (bp) | — | — | 1,380,000 | 1,430,000 | 1,820,000 |
| Estimated coverage (%) | — | — | 79.6 | 88.1 | 83.8 |
| G + C content (%) | 28.8 | 29.2 | 27.5 | 27.0 | 25.9 |
| Coding density (%) | 89.0 | 90.4 | 85.1 | 83.0 | 80.2 |
| Protein-coding genesa | 1,009 | 1,006 | 932 | 1,222 | 1,905 |
| Length distribution (Q1/Q2/Q3)(a.a.) | 171/278/425 | 176/279/425 | 176/280/440 | 119/233/376 | 83/149/286 |
| Plectrovirus proteinsb | 0 | 0 | 11 | 132 | 375 |
| Hypothetical proteins | 394 | 410 | 337 | 485 | 519 |
| Annotated pseudogenesa | 6 | 3 | 12 | 12 | 401 |
| rRNA operon | 1 | 1 | 1 | 1 | 1 |
| tRNA | 33 | 32 | 32 | 31 | 32 |
| Number of plasmids | 0 | 0 | 0 | 4 | 7 |
aFor S. chrysopicola, S. syrphidicola, and S. melliferum IPMB4A, putative pseudogenes were annotated with the “/pseudo” tag in gene feature as suggested by the NCBI GenBank guidelines and were not counted in the total number of protein-coding genes. For S. melliferum KC3 and S. citri, putative pseudogenes were annotated by adding the term “truncated” in the CDS product description field and were included in the total number of protein-coding genes.
bMost of the plectrovirus-related regions were excluded from the final S. melliferum IPMB4A assembly due to unresolvable polymorphism, resulting in a lower number of plectroviral genes (Lo et al. 2013).
The two genomes of the Chrysopicola clade are largely syntenic, except for a 41-kb block of mainly hypothetical genes around position 1,000 kb in the S. chrysopicola genome (fig. 2B). This block corresponds to an inverted block centered at position 500 kb of the S. syrphidicola genome. Interestingly, the positive-to-negative GC-skew inversion is not right opposite the negative-to-positive inversion in the S. chrysopicola genome, resulting in a smaller half with positive GC-skew (fig. 2A). In addition, the positive-to-negative inversion is 43 kb upstream of the theoretical replication terminus (right opposite the origin). Taken together, these suggest that the 41-kb block was recently inverted and translocated to its present location in the lineage leading to S. chrysopicola, resulting in asymmetrical halves with positive and negative GC-skew.
Both S. chrysopicola and S. syrphidicola have a single rRNA operon (16S-23S-5S), which is consistent with the finding that only one rRNA operon was found in S. citri, S. melliferum, and S. kunkelii (Dally et al. 2006; Carle et al. 2010; Alexeev et al. 2012; Lo et al. 2013). This is different from the Mycoides–Entomoplasmataceae clade, where both M. mycoides (Westberg et al. 2004) and Mes. florum contain two rRNA operons with the same 16S-23S-5S organization. Genes related to the helical morphology of Spiroplasma (Kürner et al. 2005), fib (fibril protein) and mreB (cell shape determining protein), are found in the two species. It should be noted that, as in the partial genomes of S. citri and S. melliferum, there are five copies of mreB (mreB1–mreB5) in both species, whereas there are only three copies in Bacillus subtilis (Jones et al. 2001). In addition, the five mreB paralogs are arranged in a conserved order in both S. chrysopicola and S. syrphidicola, with mreB1 located 18 kb and 13 kb upstream of the rRNA operon, respectively, and mreB2–mreB3 2 kb upstream of mreB4–mreB5, which is 10 kb from guaB (the last gene before the replication origin). Interestingly, the cluster of genes from mreB2 to mreB5 comprises the first 7 kb of the 11 kb segment with positive GC-skew near the replication origin (fig. 2A). The mreB paralogs, along with other genes in this segment, are transcribed from a strand different from that of the flanking genes. In S. citri, mreB1 is located in a small contig bounded by plectroviral sequences, whereas in both S. melliferum strains, mreB1 is located 20 kb upstream of the rRNA operon in the same contig. The other four mreB paralogs in both S. melliferum and S. citri are arranged in the same order as in the Chrysopicola clade and the distance between mreB5 and guaB is 10 kb in S. melliferum and 13 kb in S. citri due to the presence of a plectroviral fragment in between. The conservation in the copy number, gene order, and genomic positions of the mreB orthologs between the Citri and Chrysopicola clades suggest that their role as cytoskeletal filaments (Kürner et al. 2005) is essential for these spiroplasmas. In contrast to the Citri–Chrysopicola–Mirum clade, no mreB homolog has been reported from the Mycoides–Entomoplasmataceae clade, which is closely related with spiroplasmas, but characterized by pleomorphic or coccoid morphology (Tully et al. 1994; Manso-Silvá et al. 2009).
Although extensive gene decay was reported for the S. citri genome (21% of the annotated coding sequences are truncated) (Carle et al. 2010), such phenomenon is not found in S. chrysopicola, S. syrphidicola, or S. melliferum. Similar gene decay is also found in an 85-kb segment of the S. kunkelii genome (Zhao et al. 2003), which, similar to the S. citri genome, has a truncated copy of pyrP (encoding uracil permease). However, this gene is intact in S. melliferum, the sister species of S. citri, and in the Chrysopicola clade. The observation of gene decay in leafhopper-transmitted phytopathogenic S. citri and S. kunkelii, but not in other closely related spiroplasmas, suggests that these vector-borne pathogenic species may have smaller effective population size and suffer elevated levels of genetic drift (Kuo et al. 2009).
Origin and Evolution of Viral Invasion in Spiroplasma
The most striking genome feature of S. chrysopicola and S. syrphidicola is that they do not contain any trace of plectroviral genes (table 1), which were found to be abundant in the genomes of S. citri (Carle et al. 2010), S. kunkelii (Bai and Hogenhout 2002; Zhao et al. 2003), and S. melliferum (Alexeev et al. 2012; Lo et al. 2013). In our OrthoMCL results (supplementary table S2, Supplementary Material online), no protein-coding gene from S. chrysopicola or S. syrphidicola was grouped with any viral gene of S. citri or S. melliferum. We used the amino acid and nucleotide sequences of large plectroviral fragments from the Citri clade as queries and performed TBlastN and BlastN searches against the genomes of S. chrysopicola and S. syrphidicola. However, no significant hit was found.
The plectroviral fragments in the S. citri genome have been linked to genome rearrangements (Ye et al. 1996; Lo et al. 2013). Even though the recA gene, which is involved in homologous recombination, is known to have been pseudogenized in S. citri and S. melliferum (Marais et al. 1996; Carle et al. 2010; Lo et al. 2013), genome alignment between these two closely related species (99.0% genome-wide nucleotide sequence identity, calculated based on a concatenated alignment of 529 single-copy genes that contains 547,687 aligned sites) revealed extensive genome rearrangements (Lo et al. 2013). These seemingly contrary observations may be explained by a hypothesis that the extensive rearrangements have occurred soon after the divergence of S. citri and S. melliferum, whereas the losses of recA in these species are relatively recent events (Marais et al. 1996; Lo et al. 2013). Moreover, a comparison between S. citri strains that have been propagated in laboratory for 10 years has found several large-scale changes in their chromosomal organization (Ye et al. 1996), suggesting high levels of genome instability even in the absence of a functional copy of recA.
In contrast to the S. citri–S. melliferum comparison, only one small translocation was observed between S. chrysopicola and S. syrphidicola (fig. 2B). This result is surprising because the latter pair is much more divergent (92.2% genome-wide nucleotide sequence identity based on the alignment described earlier). The GC-skew pattern (fig. 2A) further indicates that the genomes of the Chrysopicola clade have been relatively stable. Together, these suggest that the absence of plectroviral fragments in these two spiroplasmas is unlikely to be the result of recent loss of viral fragments but is due to the lack of viral invasion throughout the lineage history of the Chrysopicola clade.
In addition to the three Spiroplasma species that have partial genome sequences available (i.e., S. melliferum, S. citri, and S. kunkelii), the presence of viral fragments was identified in S. phoeniceum based on Southern hybridization (Renaudin et al. 1988). Thus, the susceptibility to viral invasion appeared to be a derived state that originated in the common ancestor of this subgroup in the Citri clade (fig. 1). However, additional evidences suggest that this susceptibility is likely to originate in the common ancestor of the entire Citri clade. First, viruses with similar genome characteristics (single-stranded circular DNA) and morphology to those of S. citri viruses have been isolated from Drosophila-associated spiroplasmas such as S. poulsonii (Oishi et al. 1984; Cohen et al. 1987). Second, based on the genome sizes estimated by pulsed-field gel electrophresis (fig. 1 and table 1; Carle et al. 1995; Williamson et al. 1997, 1999; Dally et al. 2006; Bi et al. 2008; Lo et al. 2013), all characterized lineages in the Citri clade appear to have experienced genome expansions. Compared with the Chrysopicola clade, the genomes of the Citri clade are larger by 0.26 Mb (S. melliferum) to 0.92 Mb (S. poulsonii). Because plectroviral genes comprise approximately 20% of protein-coding genes in the assembled parts of the S. citri genome (∼84% of the whole genome; table 1) and most of the unassembled parts probably correspond to plectroviral fragments (Carle et al. 2010), plectroviral fragments could account for up to 35% of the S. citri genome. This clearly indicates that the viral fragments are a major contributor to the larger genome sizes observed in the Citri clade. This observation is intriguing among Mollicutes because genome reduction linked with host association (Ochman and Davalos 2006; Kuo et al. 2009; McCutcheon and Moran 2012) was found in mycoplasmas and phytoplasmas (Chen et al. 2012).
To the best of our knowledge, plectroviral invasion has not been reported for Mollicutes outside the Citri clade. Plectroviruses with homologous genes of S. citri plectroviruses (Sha et al. 2000) and a similar replication mechanism (Dickinson and Townsend 1984) were isolated from Acholeplasma (Gourlay and Wyld 1973; Steinick et al. 1980), another insect/plant-associated Mollicutes genus distantly related to spiroplasmas (fig. 1). Although Southern blot hybridization suggested that plectrovirus genomes seemed to be present in the cellular DNA of several A. laidlawii strains (Just et al. 1989), no viral invasion was reported for the complete genome of A. laidlawii PG-8A (Lazarev et al. 2011). However, other types of repetitive sequences have been found to promote chromosomal rearrangements in various Mollicutes lineages. For example, insertion sequences comprise 13% of the M. mycoides genome and are linked to high genomic plasticity (Westberg et al. 2004). Additionally, the potential mobile units in phytoplasmas are associated with the genome instability observed in these lineages (Bai et al. 2006; Wei et al. 2008; Toruño et al. 2010).
It should be noted that the absence of viral invasion in the Chrysopicola and Mycoides–Entomoplasmataceae clades is unlikely to be attributed to differences in ecological niches. Plectroviruses have been isolated and shown to infect spiroplasmas from the dipteran genus Drosophila, implying that these viruses might also be found in the hosts of S. chrysopicola and S. syrphidicola. Additionally, Mes. florum (originally described as Acholeplasma florum) was isolated from floral surfaces, where acholeplasmas could be found (Tully et al. 1994), and is likely to be exposed to plectroviruses, but it does not contain any plectroviral fragments. Therefore, the susceptibility to viral invasion seems to have a physiological or genetic basis that requires further investigation.
Dynamics of Insertions and Losses of Viral Fragments
By comparing the syntenic regions among the available Spiroplasma genomes, we are able to visualize the distribution of plectroviral fragments in S. citri or S. melliferum relative to the complete genomes of the Chrysopicola clade (fig. 2A). Except for an approximately 100-kb region extending from the replication origin and an approximately 150-kb region encompassing most of the ribosomal protein genes, the plectroviral fragments are extensively distributed throughout these genomes. Additionally, the numbers of insertions drawn in figure 2A are probably underestimates, because we only mapped viral fragments that are present in the assembled parts of the S. citri and S. melliferum genomes and that are bordered by syntenic regions found in the Chrysopicola clade.
Because several plectroviral insertion sites are shared among strains within S. citri (Bébéar et al. 1996) and within S. melliferum (Alexeev et al. 2012; Lo et al. 2013), these plectroviral fragments are assumed to be inheritable. However, between-species comparisons indicate that few insertion sites are shared between S. citri and S. melliferum (fig. 2A). Moreover, these two species do not have viral fragments in the positions corresponding to those in the 85-kb segment of the S. kunkelii genome (Zhao et al. 2003). The observations of these species-specific viral insertions are in agreement with the findings that plectroviruses are commonly isolated from either S. citri (Cole et al. 1973) or S. melliferum (Liss and Cole 1981) and that plectroviral insertion into the host chromosome has been observed in laboratory-cultured strains (Dickinson and Townsend 1985; Sha et al. 1995). These suggest that integration of plectroviral genomes into the bacterial chromosome is an ongoing process in natural populations of these spiroplasmas. New insertions, rather than differential losses, may thus account for the high proportion of species-specific viral insertion sites in S. citri (24 out of the 30 mapped) and S. melliferum (10 out of the 15 mapped). Considering the close evolutionary relationship (99.0% genome-wide nucleotide sequence identity) between these two species, viral insertions appear to have occurred at a high rate in both lineages.
However, the rapid accumulation of viral insertions seems to be counterbalanced by losses of viral fragments. If viral invasion follows an insertion-only model and the last common ancestor of the Citri clade was susceptible to viral invasion and had a genome size like that of the present-day Chrysopicola clade (1.1 Mb), 10 unique insertions in S. melliferum since its divergence from S. citri would translate into 374 insertions in the lineage leading to S. melliferum since the last common ancestor of the Citri clade (based on the branch lengths in fig. 1 and assuming a constant rate of new insertions). If the size of every insertion is 6.8 kb (Sha et al. 2000), the present-day S. melliferum would have a genome size of 3.6 Mb, of which 70% comprised plectroviral fragments. As the genomes of S. citri and S. melliferum are not overwhelmed by viral fragments and have not attained such large sizes, it implies that losses of viral fragments are also taking place at a comparable rate to counteract viral invasion. The removal of these repetitive sequences may be partially driven by the intrinsic deletional bias in bacteria that maintains their genome compactness (Kuo and Ochman 2009), as indicated by the presence of truncated viral genes (Carle et al. 2010) and partial viral fragments (Bébéar et al. 1996).
The dynamics of insertions and losses seems to have resulted in greater variability of genome sizes in the susceptible species. Three closely related S. citri strains maintained in different hosts could have a difference in genome size up to 270 kb (Ye et al. 1996). Furthermore, this difference was the result of laboratory propagation within a period of merely 10 years. In agreement with the difference in distribution of viral fragments between S. citri and S. melliferum, this also suggests that the gains and losses of viral fragments are highly dynamic.
Impacts of Viral Fragments: A Closer Look
Several previous studies suggested that the plectroviral fragments in S. citri and S. melliferum may be associated with chromosomal rearrangements and horizontal gene transfers (Ye et al. 1992, 1996; Alexeev et al. 2012; Lo et al. 2013). To test this hypothesis, we compared the syntenic regions between genomes of the Citri and Chrysopicola clades. These comparisons allowed us to establish the directionality of rearrangement events. Moreover, the gene-level resolution provided by the genome sequences allowed us to identify boundaries of viral integration and candidates of horizontally transferred genes. Many genes unique to either or both S. citri and S. melliferum were indeed found in the vicinity of plectroviral fragments. For example, examination of one insertion site in S. melliferum KC3 revealed nine such genes (fig. 3A). These genes include a set of type I restriction enzyme and methylase (hsdR and hsdM), as well as a phosphotransferase system (PTS) component, a lysophospholipase (yqkD), and a glycerophosphoryl diester phosphodiesterase family protein (glpQ). In another example, 11 genes unique to S. citri are found in the vicinity of S. citri-specific plectroviral insertion sites (fig. 3B). It is notable that genes unique to S. citri are also found at the boundaries of contigs, which are supposed to be bordered by repetitive plectroviral sequences (Carle et al. 2010).
Fig. 3.—

Examples of syntenic regions with plectroviral genes in the Citri clade. Annotated plectroviral genes are highlighted in red with labels indicating the ORF number assignments. Hypothetical genes with unknown functions are in gray. Genes within plectroviral sequences, where not indicated, are found in both Spiroplasma citri and S. melliferum (†). Putative viral insertion positions are indicated by red arrowheads. The corresponding regions in S. syrphidicola contain the same genes in the same order as in S. chrysopicola and are not shown for simplicity. In (A), the corresponding region in S. citri has other plectroviral fragments in the vicinity (fig. 2) and is scattered into different contigs.
Association of viral fragments with genes unique to the Citri clade does not unambiguously indicate that the genes were brought in along with the viral genome. However, the range of genome sizes (6.8 kb [Sha et al. 2000] to 8.5 kb [Dickinson and Townsend 1984]) and nonconservation in gene contents of plectroviruses (Sha et al. 2000) do suggest they may carry bacterial DNA through imprecise excision of prophage genome from the host chromosome. Furthermore, plectroviruses may be able to invade different strains or even different species of the Citri clade, as indicated by the experimental infection of various S. citri strains by viruses isolated from S. melliferum (Sha et al. 1995) and the infection of Drosophila-associated spiroplasmas by viruses isolated from other Drosophila spiroplasma strains (Oishi et al. 1984; Cohen et al. 1987). Because virulence-related genes could be transferred among bacteria by viruses (Beres et al. 2002), the ability to infect different spiroplasmas indicates that plectroviruses may help to spread virulence factors among different Citri species that share similar niches, such as the three leafhopper-transmitted phytopathogenic spiroplasmas (Liu et al. 1983; Whitcomb et al. 1986; Saillard et al. 1987).
The example in figure 3B also demonstrates possible rearrangement events unique to S. citri. In this scenario, where the gene order in S. melliferum and the Chrysopicola clade represents the ancestral state, a viral fragment was inserted into between celC and upp (fig. 2A). Another viral fragment was inserted into between fur and proS with an inverted orientation. Subsequent recombination(s) between these two inserted fragments replaced the genes downstream of celC for fur and its downstream genes. Other rearrangement events may have occurred involving genes downstream of tyrS, such that only the segment from fur to tyrS was rearranged from its original position (downstream of proS) to the position downstream of celC. According to the physical mapping (Ye et al. 1992; Carle et al. 2010), the effect of these rearrangements would correspond to a translocation across a genomic distance of approximately 240 kb from one side of oriC to the other.
Comparative Analyses of Gene Content
To better understand the potential impacts of viral invasion on the gene content of susceptible genomes, we compared the content of homologous gene clusters in the four Spiroplasma species with sequenced genomes (fig. 4 and supplementary table S2, Supplementary Material online). With M. mycoides and M. penetrans as the outgroups, the presence/absence patterns of homologous gene clusters were mapped onto the whole-genome phylogeny (fig. 5 and supplementary table S3, Supplementary Material online). The genes unique to the four species from the Citri and Chrysopicola clades include mreB and fib, which are related to the spiroplasma morphology (Kürner et al. 2005), and spi (spiralin gene), which encodes an abundant lipoprotein in the plasma membrane (Killiny et al. 2005) that appears to be unique to the Citri–Chrysopicola–Mirum clade (Meng et al. 2010). We found that treB, encoding a PTS component for transporting trehalose, the main hemolymph sugar in insects (Becker et al. 1996), is uniquely absent in the Chrysopicola clade. Because spiroplasmas in insect hemolymph must rely on trehalose (André et al. 2003), the lack of treB indicates that both S. chrysopicola and S. syrphidicola may be unable to proliferate within hemolymph. It has been shown that multiplication within hemolymph and subsequent invasion to other tissues are highly related to the pathogenicity of S. citri to its insect hosts (Liu et al. 1983). Together, these may explain why both S. citri and S. melliferum are pathogenic to their insect hosts (Liu et al. 1983; Clark et al. 1985), whereas there is no report of entomopathogenicity for S. chrysopicola and S. syrphidicola.
Fig. 4.—
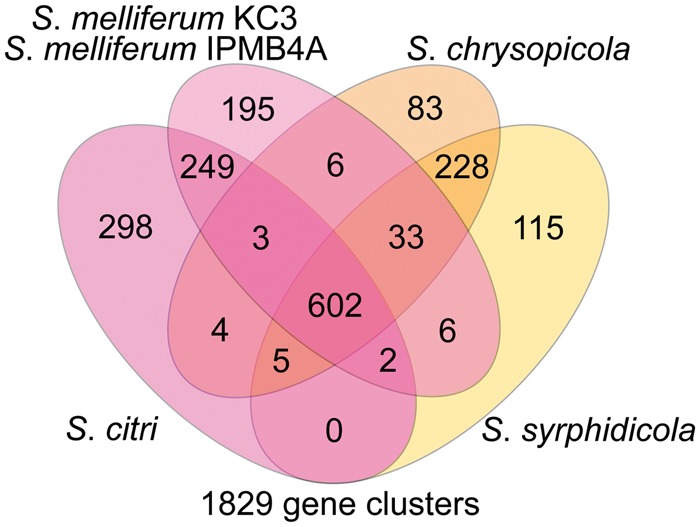
Numbers of shared and unique gene clusters among Spiroplasma chrysopicola, S. syrphidicola, S. citri, and S. melliferum. Genomes of the two S. melliferum strains (IPMB4A and KC3) were analyzed as a single pan-genome of S. melliferum. For detailed lists of these orthologous gene clusters, see supplementary table S2, Supplementary Material online.
Fig. 5.—

Phylogenetic distribution pattern of homologous gene clusters. The organismal phylogeny is inferred from the concatenated protein alignment of 267 single-copy genes shared by all species (with 106,639 aligned amino acid sites). All internal nodes received 100% bootstrap support based on 1,000 resampling and maximum likelihood inference. The numbers in parentheses after species names indicate the number of gene clusters found in each species. The numbers above a branch and preceded by a “+” sign indicate the number of homologous gene clusters that are uniquely present in all daughter lineages; the numbers below a branch and preceded by a “−” sign indicate the number of homologous gene clusters that are uniquely absent. For example, 135 gene clusters are shared by Spiroplasma chrysopicola and S. syrphidicola and do not contain any homolog from any of the other four species compared; similarly, nine gene clusters are missing in these two Spiroplasma species but are present in all other four species. For detailed lists of these orthologous gene clusters, see supplementary table S3, Supplementary Material online.
Additionally, the genomes of the Citri and Chrysopicola clades differ in their content of antiviral systems. A Type I restriction/modification (R/M) system is intact in S. chrysopicola (SCHRY_v1c02520-2540) and apparently truncated in S. citri (SPICI01B_067-69), whereas another Type I R/M system is present in S. melliferum KC3 (SPM_0320-325; fig. 3A). However, only S. chrysopicola and S. syrphidicola contain genes for a Type II R/M system (SCHRY_v1c02610, SCHRY_v1c06730-6740; SSYRP_v1c02700, SSYRP_v1c06950-6960). A CRISPR system is only found at approximately 1,010 kb of the S. syrphidicola genome (CRISPR-associated [cas] genes: SSYRP_v1c09370-9400; CRISPR repeats: positions 1,005,965–1,007,784). This is the first CRISPR system reported from the Spiroplasma–Entomoplasmataceae–Mycoides clade of Mollicutes. Although other CRISPR systems are found in Mycoplasma and Ureaplasma (Makarova et al. 2011), this S. syrphidicola CRISPR system is more similar to that of Ruminococcus albus (Clostridiales), which was the top hit for the putative cas genes (cas1, cas2, and cas9) in Basic Local Alignment Search Tool searches against the NCBI nr database. One interesting observation is that the position of the CRISPR system coincides with the 41-kb block in S. chrysopicola (fig. 2B), which appears to have been rearranged to its present position only recently based on the GC-skew pattern (fig. 2A). It indicates that the recent rearrangement event may have caused the loss of the CRISPR system in S. chrysopicola. Therefore, it is possible that this system might have been present in the common ancestor of the Chrysopicola clade or even of the Chrysopicola and Citri clades. The presence/absence patterns of the Type II R/M system and the CRISPR system, both of which provide potential defense against virus attack (Pingoud et al. 2005; Bhaya et al. 2011), indicate that their absence may be related to susceptibility of the Citri clade to viral invasion. Further evidences are needed to ascertain whether the loss of either of these systems occurred at the root of the Citri clade and resulted in susceptibility to viral invasion.
Another interesting finding is that, compared with the Chrysopicola clade, lineages within the Citri clade have more unique gene clusters. Whereas the Chysopicola clade has 135 unique gene clusters, the clade of S. melliferum and S. citri has 162. This is mainly due to the presence of viral genes, which account for 32 of the 162 unique gene clusters. In addition, S. melliferum (210) and S. citri (437) each has more unique clusters than either S. chrysopicola (82) or S. syrphidicola (107). A portion of the clusters unique to S. melliferum (34) or S. citri (96) corresponds to plectroviral genes, which is consistent with the finding that they both have many unique viral insertions (fig. 2A). However, the majority of the unique gene clusters (176/210 in S. melliferum and 341/437 in S. citri) are not of viral origin. This comes as a surprise when we consider that S. melliferum and S. citri are much less divergent from each other than S. chrysopicola and S. syrphidicola (figs. 1 and 5). This may be accounted for by the potential role of viral invasion in promoting gene gains through horizontal transfer. The high insertion rates of viral fragments, coupled with differentiation in ecological niches between S. citri and S. melliferum, may explain why they accumulated high numbers of lineage-specific genes shortly after their divergence.
Building a Model for Viral Invasion
Based on the phylogenetic and comparative genome analyses in the previous sections, we proposed a model for the genome evolution of spiroplasma species susceptible to viral invasion as a summary of this study (fig. 6). We hypothesize that the common ancestor of the Chrysopicola and Citri clades was resistant to virus attack and had few or no viral fragments in its genome, probably similar to the genomes of present-day S. chrysopicola and S. syrphidicola. After the divergence of the Chrysopicola clade, the common ancestor of the susceptible species lost its antiviral system (R/M, CRISPR, or other systems) and started to accumulate viral fragments in the genome. The insertion of these viral fragments has several consequences, including increased genome sizes, homologous recombination between similar copies of viral fragments, and acquisitions of nonviral foreign DNA via phage-mediated horizontal gene transfers. As new viral fragments are inserted into the genome, old fragments are lost due to a mutational bias toward deletions, which counterbalances the high insertion rate. The loss of viral fragments implies that the older, viral fragment-mediated rearrangements may no longer be recognizable from the current distribution of viral fragments. As a result of the accumulated effects of viral invasion, these spiroplasmas are characterized by larger and more variable genome sizes, nonconserved genome organization, and higher numbers of lineage-specific genes. In contrast, the genomes from the Chrysopicola clade remain relatively small and have few incidences of rearrangements. This model may serve as a working hypothesis for genome evolution of susceptible spiroplasmas. More studies, including genome sequencing for other species from the Citri (S. phoeniceum, S. poulsonii, S. penaei, and S. insolitum) and Mirum (S. eriocheiris and S. mirum) clades and experimental infection of S. chrysopicola and S. syrphidicola with plectroviruses to examine their antiviral capacity, will be needed to further test this hypothesis.
Fig. 6.—

A model for the genome evolution in spiroplasmas susceptible to viral invasion.
Supplementary Material
Supplementary tables S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank the DNA Analysis Core Laboratory (Institute of Plant and Microbial Biology, Academia Sinica) for providing Sanger sequencing service. This work was supported by the research grants from the Institute of Plant and Microbial Biology at Academia Sinica and the National Science Council of Taiwan (NSC 101-2621-B-001-004-MY3) to C.H.K.
Literature Cited
- Alexeev D, et al. Application of Spiroplasma melliferum proteogenomic profiling for the discovery of virulence factors and pathogenicity mechanisms in host-associated spiroplasmas. J Proteome Res. 2012;11:224–236. doi: 10.1021/pr2008626. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André A, Maccheroni W, Doignon F, Garnier M, Renaudin J. Glucose and trehalose PTS permeases of Spiroplasma citri probably share a single IIA domain, enabling the spiroplasma to adapt quickly to carbohydrate changes in its environment. Microbiology. 2003;149:2687–2696. doi: 10.1099/mic.0.26336-0. [DOI] [PubMed] [Google Scholar]
- Bai X, et al. Living with genome instability: the adaptation of phytoplasmas to diverse environments of their insect and plant hosts. J Bacteriol. 2006;188:3682–3696. doi: 10.1128/JB.188.10.3682-3696.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Hogenhout SA. A genome sequence survey of the mollicute corn stunt spiroplasma Spiroplasma kunkelii. FEMS Microbiol Lett. 2002;210:7–17. doi: 10.1111/j.1574-6968.2002.tb11153.x. [DOI] [PubMed] [Google Scholar]
- Bébéar C-M, Aullo P, Bové J-M, Renaudin J. Spiroplasma citri virus SpV1: characterization of viral sequences present in the spiroplasmal host chromosome. Curr Microbiol. 1996;32:134–140. [Google Scholar]
- Becker A, Schlöder P, Steele JE, Wegener G. The regulation of trehalose metabolism in insects. Experientia. 1996;52:433–439. doi: 10.1007/BF01919312. [DOI] [PubMed] [Google Scholar]
- Benson DA, et al. GenBank. Nucleic Acids Res. 2012;40:D48–D53. doi: 10.1093/nar/gkr1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beres SB, et al. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci U S A. 2002;99:10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in Bacteria and Archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- Bi K, Huang H, Gu W, Wang J, Wang W. Phylogenetic analysis of Spiroplasmas from three freshwater crustaceans (Eriocheir sinensis, Procambarus clarkia and Penaeus vannamei) in China. J Invertebr Pathol. 2008;99:57–65. doi: 10.1016/j.jip.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle P, et al. Partial chromosome sequence of Spiroplasma citri reveals extensive viral invasion and important gene decay. Appl Environ Microbiol. 2010;76:3420–3426. doi: 10.1128/AEM.02954-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle P, Laigret F, Tully JG, Bové JM. Heterogeneity of genome sizes within the genus Spiroplasma. Int J Syst Bacteriol. 1995;45:178–181. doi: 10.1099/00207713-45-1-178. [DOI] [PubMed] [Google Scholar]
- Chen L-L, Chung W-C, Lin C-P, Kuo C-H. Comparative analysis of gene content evolution in phytoplasmas and mycoplasmas. PLoS One. 2012;7:e34407. doi: 10.1371/journal.pone.0034407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TB, et al. Spiroplasma melliferum, a new species from the honeybee (Apis mellifera) Int J Syst Bacteriol. 1985;35:296–308. [Google Scholar]
- Cohen AJ, Williamson DL, Oishi K. SpV3 viruses of Drosophila spiroplasmas. Israel J Med Sci. 1987;23:429–433. [PubMed] [Google Scholar]
- Cole RM, Tully JG, Popkin TJ, Bové JM. Morphology, ultrastructure, and bacteriophage infection of the helical mycoplasma-like organism (Spiroplasma citri gen. nov., sp. nov.) cultured from “stubborn” disease of citrus. J Bacteriol. 1973;115:367–386. doi: 10.1128/jb.115.1.367-386.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dally EL, et al. Physical and genetic map of the Spiroplasma kunkelii CR2-3x chromosome. Can J Microbiol. 2006;52:857–867. doi: 10.1139/w06-044. [DOI] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson MJ, Townsend R. Characterization of the genome of a rod-shaped virus infecting Spiroplasma citri. J Gen Virol. 1984;65:1607–1610. [Google Scholar]
- Dickinson MJ, Townsend R. Lysogenisation of Spiroplasma citri by a type 3 spiroplasmavirus. Virology. 1985;146:102–110. doi: 10.1016/0042-6822(85)90056-x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP—Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Gasparich GE. Spiroplasmas and phytoplasmas: microbes associated with plant hosts. Biologicals. 2010;38:193–203. doi: 10.1016/j.biologicals.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Gasparich GE, et al. The genus Spiroplasma and its non-helical descendants: phylogenetic classification, correlation with phenotype and roots of the Mycoplasma mycoides clade. Int J Syst Evol Microbiol. 2004;54:893–918. doi: 10.1099/ijs.0.02688-0. [DOI] [PubMed] [Google Scholar]
- Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlay RN, Wyld SG. Isolation of Mycoplasmatales virus-laidlawii 3, a new virus infecting Acholeplasma laidlawii. J Gen Virol. 1973;19:279–283. doi: 10.1099/0022-1317-19-2-279. [DOI] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comp Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LJF, Carballido-López R, Errington J. Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell. 2001;104:913–922. doi: 10.1016/s0092-8674(01)00287-2. [DOI] [PubMed] [Google Scholar]
- Just W, Silva Cardoso M, Lorenz A, Klotz G. Release of mycoplasmavirus L1 upon transfection of Acholeplasma laidlawii with homologous and heterologous viral DNA. Arch Virol. 1989;107:1–13. doi: 10.1007/BF01313873. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killiny N, Castroviejo M, Saillard C. Spiroplasma citri spiralin acts in vitro as a lectin binding to glycoproteins from its insect vector Circulifer haematoceps. Phytopathology. 2005;95:541–548. doi: 10.1094/PHYTO-95-0541. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C-H, Moran NA, Ochman H. The consequences of genetic drift for bacterial genome complexity. Genome Res. 2009;19:1450–1454. doi: 10.1101/gr.091785.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C-H, Ochman H. Deletional bias across the three domains of life. Genome Biol Evol. 2009;1:145–152. doi: 10.1093/gbe/evp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kürner J, Frangakis AS, Baumeister W. Cryo-electron tomography reveals the cytoskeletal structure of Spiroplasma melliferum. Science. 2005;307:436–438. doi: 10.1126/science.1104031. [DOI] [PubMed] [Google Scholar]
- Lagesen K, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarev VN, et al. Complete genome and proteome of Acholeplasma laidlawii. J Bacteriol. 2011;193:4943–4953. doi: 10.1128/JB.05059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss A, Cole R. Spiroplasmavirus group 1: isolation, growth, and properties. Curr Microbiol. 1981;5:357–362. [Google Scholar]
- Liu HY, Gumpf DJ, Oldfield GN, Calavan EC. The relationship of Spiroplasma citri and Circulifer tenellus. Phytopathology. 1983;73:585–590. [Google Scholar]
- Lo W-S, Chen L-L, Chung W-C, Gasparich G, Kuo C-H. Comparative genome analysis of Spiroplasma melliferum IPMB4A, a honeybee-associated bacterium. BMC Genomics. 2013;14:22. doi: 10.1186/1471-2164-14-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manso-Silvá L, et al. Mycoplasma leachii sp. nov. as a new species designation for Mycoplasma sp. bovine group 7 of leach, and reclassification of Mycoplasma mycoides subsp. mycoides LC as a serovar of Mycoplasma mycoides subsp. capri. Int J Syst Evol Microbiol. 2009;59:1353–1358. doi: 10.1099/ijs.0.005546-0. [DOI] [PubMed] [Google Scholar]
- Marais A, Bové JM, Renaudin J. Characterization of the recA gene regions of Spiroplasma citri and Spiroplasma melliferum. J Bacteriol. 1996;178:7003–7009. doi: 10.1128/jb.178.23.7003-7009.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- Meng Q, et al. Identification and characterization of spiralin-like protein SLP25 from Spiroplasma eriocheiris. Vet Microbiol. 2010;144:473–477. doi: 10.1016/j.vetmic.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Minion FC, et al. The genome sequence of Mycoplasma hyopneumoniae strain 232, the agent of swine mycoplasmosis. J Bacteriol. 2004;186:7123–7133. doi: 10.1128/JB.186.21.7123-7133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott ML, Berger JM. DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol. 2007;5:343–354. doi: 10.1038/nrmicro1640. [DOI] [PubMed] [Google Scholar]
- Moulder RW, French FE, Chang CJ. Simplified media for spiroplasmas associated with tabanid flies. Can J Microbiol. 2002;48:1–6. doi: 10.1139/w01-128. [DOI] [PubMed] [Google Scholar]
- Nunan LM, Lightner DV, Oduori MA, Gasparich GE. Spiroplasma penaei sp. nov., associated with mortalities in Penaeus vannamei, Pacific white shrimp. Int J Syst Evol Microbiol. 2005;55:2317–2322. doi: 10.1099/ijs.0.63555-0. [DOI] [PubMed] [Google Scholar]
- Ochman H, Davalos LM. The nature and dynamics of bacterial genomes. Science. 2006;311:1730–1733. doi: 10.1126/science.1119966. [DOI] [PubMed] [Google Scholar]
- Oishi K, Poulson DF, Williamson DL. Virus-mediated change in clumping properties of Drosophila SR spiroplasmas. Curr Microbiol. 1984;10:153–158. [Google Scholar]
- Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell Mol Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PubMed] [Google Scholar]
- Renaudin J, Bodin-Ramiro C, Bové JM. Spiroplasma citri virus SpV1-78, a non-lytic rod-shaped virus with single-stranded, circular DNA: presence of viral sequences in the spiroplasma genome. In: Timmer LW, Garnsey SM, Navarro L, editors. Proceedings of the 10th Conference of the International Organization of Citrus Virologists. Riverside (CA): University of California Press; 1988. pp. 285–290. [Google Scholar]
- Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglio P, et al. Spiroplasma citri gen. and sp. n.: a mycoplasma-like organism associated with “stubborn” disease of citrus. Int J Syst Bacteriol. 1973;23:191–204. [Google Scholar]
- Saillard C, et al. Spiroplasma phoeniceum sp. nov., a new plant-pathogenic species from Syria. Int J Syst Bacteriol. 1987;37:106–115. [Google Scholar]
- Sha Y, Melcher U, Davis RE, Fletcher J. Resistance of Spiroplasma citri lines to the virus SVTS2 is associated with integration of viral DNA sequences into host chromosomal and extrachromosomal DNA. Appl Environ Microbiol. 1995;61:3950–3959. doi: 10.1128/aem.61.11.3950-3959.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha Y, Melcher U, Davis R, Fletcher J. Common elements of spiroplasma plectroviruses revealed by nucleotide sequence of SVTS2. Virus Genes. 2000;20:47–56. doi: 10.1023/a:1008108106916. [DOI] [PubMed] [Google Scholar]
- Steinick LE, Wieslander A, Johansson KE, Liss A. Membrane composition and virus susceptibility of Acholeplasma laidlawii. J Bacteriol. 1980;143:1200–1207. doi: 10.1128/jb.143.3.1200-1207.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Toruño TY, Seruga Musić M, Simi S, Nicolaisen M, Hogenhout SA. Phytoplasma PMU1 exists as linear chromosomal and circular extrachromosomal elements and has enhanced expression in insect vectors compared with plant hosts. Mol Microbiol. 2010;77:1406–1415. doi: 10.1111/j.1365-2958.2010.07296.x. [DOI] [PubMed] [Google Scholar]
- Tully JG, et al. Taxonomic descriptions of eight new non-sterol-requiring mollicutes assigned to the genus Mesoplasma. Int J Syst Bacteriol. 1994;44:685–693. doi: 10.1099/00207713-44-4-685. [DOI] [PubMed] [Google Scholar]
- Volokhov DV, Simonyan V, Davidson MK, Chizhikov VE. RNA polymerase beta subunit (rpoB) gene and the 16S-23S rRNA intergenic transcribed spacer region (ITS) as complementary molecular markers in addition to the 16S rRNA gene for phylogenetic analysis and identification of the species of the family Mycoplasmataceae. Mol Phylogen Evol. 2012;62:515–528. doi: 10.1016/j.ympev.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Wang W, et al. A spiroplasma associated with tremor disease in the Chinese mitten crab (Eriocheir sinensis) Microbiology. 2004;150:3035–3040. doi: 10.1099/mic.0.26664-0. [DOI] [PubMed] [Google Scholar]
- Wei W, Davis RE, Jomantiene R, Zhao Y. Ancient, recurrent phage attacks and recombination shaped dynamic sequence-variable mosaics at the root of phytoplasma genome evolution. Proc Natl Acad Sci U S A. 2008;105:11827–11832. doi: 10.1073/pnas.0805237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westberg J, et al. The genome sequence of Mycoplasma mycoides subsp. mycoides SC type strain PG1T, the causative agent of contagious bovine pleuropneumonia (CBPP) Genome Res. 2004;14:221–227. doi: 10.1101/gr.1673304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb RF, et al. Spiroplasma kunkelii sp. nov.: characterization of the etiological agent of corn stunt disease. Int J Syst Bacteriol. 1986;36:170–178. [Google Scholar]
- Whitcomb RF, et al. Spiroplasma syrphidicola sp. nov., from a syrphid fly (Diptera: Syrphidae) Int J Syst Bacteriol. 1996;46:797–801. doi: 10.1099/00207713-46-3-797. [DOI] [PubMed] [Google Scholar]
- Whitcomb RF, et al. Spiroplasma chrysopicola sp. nov., Spiroplasma gladiatoris sp. nov., Spiroplasma helicoides sp. nov., and Spiroplasma tabanidicola sp. nov., from Tabanid (Diptera: Tabanidae) flies. Int J Syst Bacteriol. 1997;47:713–719. [Google Scholar]
- Williamson DL, et al. Spiroplasma platyhelix sp. nov., a new mollicute with unusual morphology and genome size from the dragonfly Pachydiplax longipennis. Int J Syst Bacteriol. 1997;47:763–766. doi: 10.1099/00207713-47-3-763. [DOI] [PubMed] [Google Scholar]
- Williamson DL, et al. Spiroplasma poulsonii sp. nov., a new species associated with male-lethality in Drosophila willistoni, a neotropical species of fruit fly. Int J Syst Bacteriol. 1999;49:611–618. doi: 10.1099/00207713-49-2-611. [DOI] [PubMed] [Google Scholar]
- Ye F, Melcher U, Rascoe J, Fletcher J. Extensive chromosome aberrations in Spiroplasma citri strain BR3. Biochem Genet. 1996;34:269–286. doi: 10.1007/BF02399947. [DOI] [PubMed] [Google Scholar]
- Ye F, et al. A physical and genetic map of the Spiroplasma citri genome. Nucleic Acids Res. 1992;20:1559–1565. doi: 10.1093/nar/20.7.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, et al. Gene content and organization of an 85-kb DNA segment from the genome of the phytopathogenic mollicute Spiroplasma kunkelii. Mol Genet Genomics. 2003;269:592–602. doi: 10.1007/s00438-003-0878-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.