

Abstract
How introns are lost from eukaryotic genomes during evolution remains an enigmatic question in biology. By comparative genome analysis of five Caenorhabditis and eight Drosophila species, we found that the likelihood of intron loss is highly influenced by the degree of sequence homology at exon–intron junctions: a significant elevated degree of microhomology was observed for sequences immediately flanking those introns that were eliminated from the genome of one or more subspecies. This determinant was significant even at individual nucleotides. We propose that microhomology-mediated DNA repair underlies this phenomenon, which we termed microhomology-mediated intron loss. This hypothesis is further supported by the observations that in both species 1) smaller introns are preferentially lost over longer ones and 2) genes that are highly transcribed in germ cells, and are thus more prone to DNA double strand breaks, display elevated frequencies of intron loss. Our data also testify against a prominent role for reverse transcriptase-mediated intron loss in metazoans.
Keywords: intron evolution, DNA repair, intron loss
Introduction
Introns are noncoding DNA sequences of ambiguous function that in eukaryotes interrupt exons and are removed from pre-mRNA by the splice machinery prior to translation. A question that has puzzled biologists already for over 30 years is how introns are introduced, maintained and lost from the genomes of eukaryotes. The “intron early theory” proposes that most introns were already present before eukaryotes and prokaryotes diverged, in the genome of their common ancestor. Subsequently, prokaryotes lost their introns and eukaryotes retained (at least some of) their introns. In an alternative model, known as the “intron late theory,” introns were proposed to have emerged solely within the eukaryote lineage and accumulated in genomes over evolutionary time, especially in species that do not experience selection pressure for small genome size. The most early ancestral eukaryotic progenitor is assumed to contain already many introns, prior to initial divergence, based on the existence of introns in homologous genes across early diverged species (Fedorov et al. 2002; Rogozin et al. 2003; Stajich et al. 2007).
Although genomes of some vertebrate species contain more than 100,000 introns, others have extremely few: the genome of the parasite Giardia lamblia, as an example, contains only two introns (Li et al. 2009), which may be explained by extensive intron loss in time. The increased availability of sequenced genomes has revealed, however, that rates of intron gain and loss can differ greatly between groups of species (Rogozin et al. 2003; Coghlan and Wolfe 2004; Nielsen et al. 2004; Roy and Hartl 2006; Coulombe-Huntington and Majewski 2007a; Li et al. 2009; Farlow et al. 2010; Zhang et al. 2010; Colbourne et al. 2011; Fawcett et al. 2012).
In numerous species, a clear tendency can be observed toward introns being lost (Rogozin et al. 2003; Nielsen et al. 2004; Coulombe-Huntington and Majewski 2007a; Zhang et al. 2010; Fawcett et al. 2012) and various intron-loss mechanisms have been proposed. Reverse transcription of mRNA and subsequent recombinational integration of the produced cDNA into the genome, also known as reverse transcriptase-mediated intron loss (RTMIL), has been suggested to explain cases where introns are lost while the surrounding exonic sequence remained perfectly intact (Roy 2006). A prediction from a model where reverse transcriptase starts at the 3′-ends of mRNA is a bias of intron loss towards the 3′-side (as cDNA synthesis would not always reach the 5′-end of the mRNA, is expected). A trend toward more frequent loss of 3′-positioned introns was observed in Drosophila (Yenerall et al. 2011) and Arabidopsis (Fawcett et al. 2012). More recently, modified versions of RTMIL were proposed, for example, where the 3′-end of an mRNA folds back on itself to serve as a primer for reverse transcription (Feiber et al. 2002; Niu et al. 2005). These models predict that adjacent introns will be more frequently lost than dispersed ones. For example in fungi numerous cases of intron loss could now be explained by this model (Croll and McDonald 2012). No evidence was found in favor of this hypothesis in the nematode Caenorhabditis elegans (Roy and Gilbert 2005).
We wondered whether another previously hypothesized mechanism of intron loss, that is, error-prone DNA repair, could be responsible for the precise loss of introns from genomes. This thought was triggered when we anecdotally observed substantial sequence homology at the exon–intron junction of an intron in the pcn-1 locus that was lost in C. elegans, but was still present in several other nematode species. In such cases, loss of the intronic sequence could be the result of DNA double-strand break (DSB) repair, guided by sequence homology near the break sites, as we previously have witnessed homology-driven DSB repair leading to intron-size deletions in C. elegans cells (Pontier and Tijsterman 2009). The likelihood of a small deletion leading to the exact removal of an intron is very low, but may be enhanced in cases where flanking sequences are homologous. We thus hypothesized that homologous sequences at the intron–exon junctions may direct repair of sporadic intronic DSBs leading to precise excision of the intron, a notion supported by glimpses of sequence homology surrounding introns that are uniquely present in the nematode C. briggsae (Kent and Zahler 2000), as if these sequences facilitated intron removal from the C. elegans genome.
Here, we have constructed data sets of conserved introns using either five Caenorhabditis or eight Drosophila species to uncover the mechanisms that are responsible for intron loss during evolution. Our large data set allowed us to look indepth into the current models of intron loss during evolution, even up to chromosome resolution, which was not possible until recently.
Materials and Methods
Protein Alignments
Using the Ensembl Perl application program interface, alignments of protein sequences of C. elegans, C. briggsae, C. remanei, C. brenneri, and C. japonica were retrieved (version 59, Kersey et al. 2012). Intron positions were re-inserted into the protein sequences and subsequent analysis was performed using custom Perl scripts. For Drosophila, the same analysis was performed for D. simulans, D. sechellia, D. melanogaster, D. yakuba, D. erecta, D. ananassae, D. pseudoobscura, and D. willistoni (version 59, Kersey et al. 2012).
Inferring Intron Loss
We restricted our analysis to regions of genes that were highly conserved: Introns were included only if 15 amino acids on both sides of the intron were at least 50% identical across all species. Next, we identified all cases where an intron was lost at least once in four species; the evolutionary most distinct species C. japonica was used as an outgroup. The principle of Dollo parsimony was applied to the set of introns to distinguish parallel intron losses from intron gains. C. japonica and D. willistoni were used as outgroups in the Caenorhabditis and Drosophila analysis, respectively.
Results
Intron Loss and Gain in Caenorhabditis and Drosophila
We retrieved alignments of all protein sequences from C. elegans, C. briggsae, C. remanei, C. brenneri, and C. japonica and re-inserted intron positions based on genome annotations. We restricted our analysis to regions of genes that were highly conserved: introns were only included if 15 amino acids on both sides of the intron were at least 50% identical across all species. Next, we identified all cases where an intron was lost at least once in four species; the evolutionary most distinct species C. japonica was used as an outgroup. Within 11,343 highly conserved loci, we found 27,488 conserved introns. By further analyzing the conserved intron set, we found 2,753 cases of intron loss and 778 cases of potential intron gain; 19,444 introns had remained perfectly stable. 2,351 intron losses and 596 gains were found within a single species and 402 losses and 182 gains were located at ancestral nodes (fig. 1A). Dollo parsimony was used to discriminate intron loss from intron gain. Independent parallel loss of the same intron was favored as an explanation over parallel gain of an intron in different species. If both loss and gain could explain an intron event, it was discarded from our analysis. The same analysis was performed for eight Drosophila species (fig. 1B).
Fig. 1.—
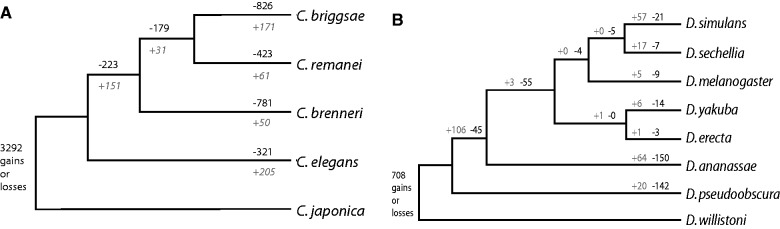
Intron dynamics in Caenorhabditis and Drosophila subspecies. (A) Phylogenetic tree of Caenorhabditis species with number of introns lost (black) and gained (gray) [(B) as in (A)], but now for the Drosophila species. Genetic distances are not drawn to scale.
No RTMIL in C. elegans and D. melanogaster
Although RTMIL has been proposed to explain cases of precise intron loss in Drosophila (Coulombe-Huntington and Majewski 2007b; Yenerall et al. 2011) and other species (Roy 2006), no evidence was found previously for this mechanism in C. elegans (Roy and Gilbert 2005). To further test this conclusion, we investigated our larger data set, which also include additional nematode and fly species for two RTMIL predictions: preferential loss of 3′ over 5′ introns and preferential loss of adjacent introns over ones located more dispersed. Although we observed a slight nonrandom distribution of intron loss, where the 3′-end of a locus is more susceptible than the 5′-end (supplementary fig. S1A and B, Supplementary Material online), we noticed that this bias is fully explained by a single peak of retained introns at the utmost 5′ side. We argue that this phenomenon can be best explained by the notion that sequence elements regulating gene expression are frequently located in the first intron in C. elegans (Bradnam and Korf 2008) and Drosophila (Haddrill et al. 2005) genes (supplementary fig. S1C and D, Supplementary Material online). Deletion of these introns may thus be under negative selection pressure (Ho et al. 2001; Bradnam and Korf 2008). We also failed to find support for the other projection of RTMIL, which is that pairs of adjacent introns are more frequently lost than dispersed pairs. Using the method published in Roy and Gilbert (2005), including Bonferroni correction for multiple testing, we found no difference in the number of expected and observed lost pairs of adjacent introns in C. elegans and C. brenneri. A small, but statistical difference was found in C. briggsae and C. remanei (P < 0.01, supplementary fig. S1E, Supplementary Material online). The same analysis for Drosophila led to a surprising conclusion: we found a statistical difference only for D. pseudoobscura (P < 0.05). In the other six Drosophila species, the number of cases of adjacent intron pair loss was not different from random chance (supplementary fig. S1F, Supplementary Material online). Because D. pseudoobscura has been used to argue a role for RTMIL in flies (Coulombe-Huntington and Majewski 2007b), we wished to nuance that conclusion. Our data indicate that there is no support for a profound role of RTMIL in intron evolution in nematodes and flies, despite the notion of few atypical cases in flies where RTMIL seems the most logical explanation (Yenerall et al. 2011).
Microhomology Is a Determinant for Intron Loss
We next addressed the hypothesis of microhomology-mediated DNA repair underlying the disappearance of introns. We predicted that introns that were lost during evolution were more frequently surrounded by microhomologous sequences at their exon–intron borders, than those that were retained. In other words: Is microhomology a determinant of intron loss? We restricted our analysis to the consensus splice donor (GT) and acceptor (AG) sequences and the immediately flanking two nucleotides of exonic sequences. Other intronic nucleotides as well as the wobble base (defined here as the nucleotide occupying the third position in a codon) of coding triplets were excluded. The rationale for eliminating the wobble position is as follows: As soon as an intron is lost, wobble bases surrounding the intron–exon junction lose their potential function in splicing. As a consequence, selection pressure on such noncoding nucleotides, if present, is likely lost together with the intron. The nature of the base at the time of analysis is therefore not informative as to the nature of the base at the time of intron loss. Thus, while the wobble bases may have contributed to the degree of microhomology at the time of intron loss, we eliminated them from our analysis. We subsequently determined the degree of homology by comparing the consensus splice donor nucleotides GT to the two outermost 5′-nucleotides of the 3′ exon, and the consensus acceptor nucleotides AG to the two outermost 3′-nucleotides of the 5′ exon. Identical nucleotides scored 1, nonidentical scored 0. Noncoding wobble bases were omitted; hence, the score window is maximized to 3. Figure 2B strikingly demonstrates that introns have indeed been more susceptible to being lost from genomes when they were flanked with homologous exon/intron junctions. Although the group of retained introns in Caenorhabditis had a homology score of 1.37, lost introns scored 1.59 (with a scale from 0 to 3, ranging from no to perfect homology). Moreover, introns that were lost multiple times independently, scored even higher: 1.78 and 1.90 for 2 and 3 times being lost, respectively (P < 0.001 for each lost group compared with the retained group, χ2 test, df = 3). Phase one introns were excluded in this graph because they have a maximum score of 2 upon wobble base removal (supplementary fig. S2, Supplementary Material online). Figure 2D shows that sequence homology at each individual position of the junction contributed to the higher rates of intron loss in Caenorhabditis.
Fig. 2.—

MMIL. (A) Schematic representation of the intron–exon junction alignment. For all intronic positions, the degree of homology was determined by comparing the consensus splice donor nucleotides GT to the 2 outermost 5′-nucleotides of the 3′ exon and the consensus acceptor nucleotides AG to the 2 outermost 3′-nucleotides of the 5′ exon. Identical nucleotides scored 1, nonidentical scored 0. Noncoding wobble bases were omitted; hence, the score window is maximized to 3. (B) The degree of intron–exon junction homology for intronic positions that suffered from 0, 1, 2, or 3 cases of intron loss. χ2 test (df = 3) was used to compare zero-lost group (n = 73,853) with the groups containing one loss (n = 1,832): P < 0.001, two losses (n = 528): P < 0.001, and three losses (n = 120): P < 0.001. (C) The degree of intron/exon junction homology for Drosophila intronic positions that suffered from zero (n = 99,864) or one or more (n = 1,385) losses (χ2 test, df = 3, P < 0.001). Homology scores for individual nucleotide positions as depicted in figure 3A for (D) Caenorhabditis and (E) Drosophila. *P < 0.05, **P < 0.01 and ***P < 0.001. (F) A microhomology-mediated end-joining mechanism for intron loss.
To investigate the generality of this phenomenon, we performed a similar analysis on eight sequenced Drosophila species, resulting in a similar outcome: introns were more frequently lost when they had matching intron–exon junctions (fig. 2C and E; supplementary fig. S3, Supplementary Material online). In Drosophila, the group of retained introns has a homology ranking of 1.37, lost introns score 1.69 (P < 0.001, χ2 test, df = 3).
Increased Likelihood of Loss for Small Introns
Sequence homology adjacent to DSBs is used in at least two error-prone DNA repair pathways, that is, single-strand annealing and microhomology-mediated end-joining, the latter of which requires just a few identical bases on either side of the break (Decottignies 2007; Pontier and Tijsterman 2009). Such pathways preferably use homologous sequence in close proximity to the DSB (McVey and Lee 2008), and if DSB repair underlies the precise loss of introns, we expect shorter introns to be more prone to being lost. Because we earlier reasoned that the first introns in nematodes and flies possibly contain regulatory sequences and thus generally have greater length, we excluded all 5′ introns from our results. Our prediction was indeed met: We found smaller introns disappear at higher rates, both in Caenorhabditis (fig. 3A) and in Drosophila (fig. 3B). In Caenorhabditis the median intron size is 51 bp for introns that have been lost versus 57 bp for introns that have been retained (P < 0.001, Mann–Whitney U test). For Drosophila, we found a median of 62 and 66 bp for lost and retained introns, respectively (P < 0.001, Mann–Whitney U test).
Fig. 3.—
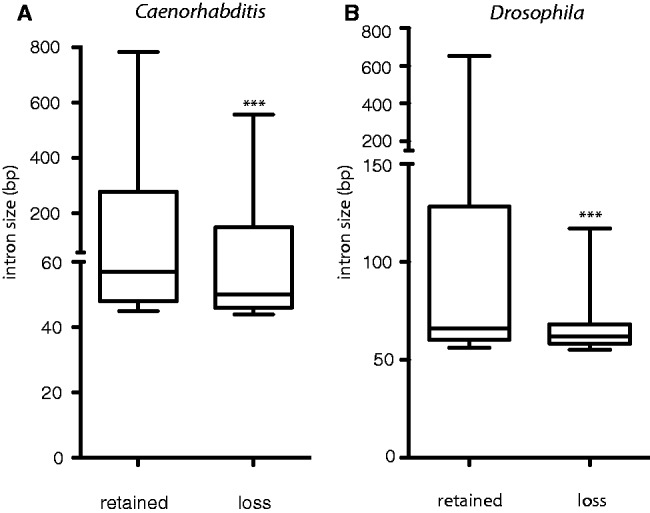
Preferential loss of small introns. A boxplot of the sizes of introns that were either 100% retained or found to be lost in at least one (A) Caenorhabditis or (B) Drosophila species. For the lost introns, we plotted the size of the introns that were retained at identical positions in neighboring species, excluding initial introns that possibly contain indispensable regulatory elements in the often larger introns. The median of introns that are lost was significantly smaller than that of retained introns for all Caenorhabditis (***P < 0.001) and Drosophila species (***P < 0.001, Mann–Whitney U test). For C. elegans: n = 97,220 for retained introns; n = 10,465 for lost intron. For Drosophila: n = 142,967 for retained introns; n = 3,274 lost introns.
Germline Expressed Genes Experience Increased Intron Loss
We next questioned whether each gene is equally susceptible to losing one or more of its introns. One feature of a gene is its transcriptional status. Using a published data set of germline expressed genes in C. elegans (Wang et al. 2009), we asked whether expression of a gene within the cells that pass on the genetic information to the next generation is of relevance. We found that approximately 47% of genes that suffered from the loss of an intron are transcribed in germ cells (fig. 4A). This is a significantly higher percentage than was found for genes that did not suffer from intron loss, which was approximately 38% (lost: 211 out of 450 genes vs. retained: 2,555 out of 6,916 genes; P < 0.001, χ2 test). A similar analysis was performed for Drosophila using a data set retrieved from FlyAtlas (Chintapalli et al. 2007). This set contains all genes that are moderately expressed in both the ovary and the testis of the adult fly (6,141 out of 13,558). Also here, we found that germline gene expression increases the probability of intron loss (fig. 4B), augmenting earlier work reporting elevated rates of intron loss for Drosophila (Yenerall et al. 2011) and mammals (Coulombe-Huntington and Majewski 2007a) for germline expressed genes. These observations are in perfect agreement with a DSB repair model of intron loss, as the more open chromatin structure of transcribed genes, as well as the activity of the transcription factories, are known to induce higher levels of DSBs in active genes (Ju et al. 2006; Lin et al. 2009; Haffner et al. 2011).
Fig. 4.—
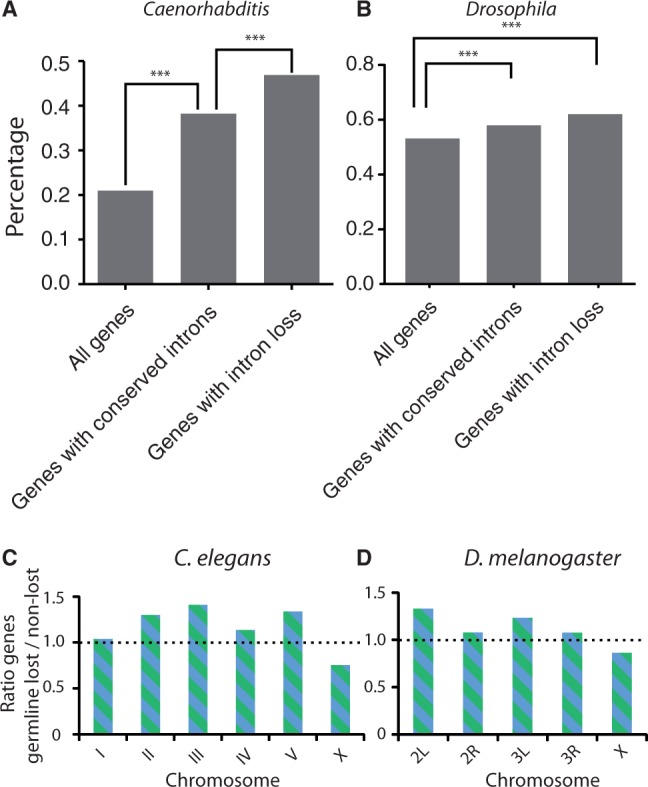
Increased likelihood of intron loss in germline-expressed genes in (A) Caenorhabditis elegans and (B) Drosophila melanogaster. Our criteria for conserved introns, selecting on highly conserved surrounding exons, enriches for germline-expressed genes (P < 0.001, χ2 test). Germline expression was highly overrepresented in the class of genes with associated intron loss (P < 0.001, χ2 test). ***P < 0.001. (C) Distribution of germline-expressed genes across the autosomes and the X-chromosome in C. elegans. For each chromosome, the ratio between germline-expressing genes that have lost at least one intron and genes that contain only retained introns is plotted, [(D as in C)] but now for D. melanogaster. We find the same outcome as for C. elegans: introns located in germline-expressing genes on X are less prone to be lost compared with introns located on the autosomes.
X-Chromosome Germline Expressed Genes Are Less Prone to Intron Loss
The C. elegans as well as the D. melanogaster genomes have been assembled into complete chromosomes. The constructed genomes allow us to plot the distribution of conserved and lost introns over the individual chromosomes. Using the reconstructed chromosomes, we asked whether the transcriptional status of genes influences the likelihood of losing an intron on each chromosome in a similar fashion. If intron loss were to be independent of their genomic location, a comparable distribution of lost and retained germline-expressed introns would be expected on each chromosome, and thus a ratio higher than one for lost/retained introns for all chromosomes. However, this is not what we observe the following: although this ratio is more than 1 for all autosomes, we found a clear decreased ratio (<1) on the X-chromosome in both C. elegans and D. melanogaster (fig. 4C and D).
Discussion
Recent studies have suggested DSB repair as being responsible for intron gains (Li et al. 2009), leading to the suggestion that similar mechanisms might work for intron loss (Farlow et al. 2011; Fawcett et al. 2012). Using a comparative analysis of five Caenorhabditis and eight Drosophila species, we now show that the degree of microhomology at the exon–intron junction dictates the rate of intron loss in nematodes and flies, which supports a prominent role for error-prone DSB repair in changing the intron landscape. We call this phenomenon microhomology-mediated intron loss (MMIL).
Previously, nonhomologous end-joining (NHEJ) has been suggested as a possible DNA repair mechanism for intron loss (Farlow et al. 2011; Yenerall et al. 2011; Fawcett et al. 2012). Although NHEJ can make use of a few nucleotides of microhomology to repair breaks (Lieber et al. 2010), we disfavor this pathway to account for MMIL, mostly because this pathway plays little or no role in C. elegans germ cells (Clejan et al. 2006). Alternative error-prone DNA repair pathways, which have been shown to contribute to inheritable genome alteration in C. elegans (Robert and Bessereau 2007), are known to be independent of the canonical NHEJ proteins CKU-70 and CKU-80 (Haber 2008; McVey and Lee 2008). The DSB repair mechanisms microhomology-mediated end-joining and single-stranded annealing use patches of (micro-) homology at either side of the break site to anneal to repair the DNA. Microhomology-mediated end-joining, although still rather ill defined, has been described as the pathway that uses only a few homologous nucleotides to establish contact between the two ends of the break. In our study, we have restricted the analysis to only four positions because, apart from the splice donor and acceptor site, intronic sequences experience little selection pressure and can freely mutate without apparent consequences. The degree of microhomology at the exon/intron border may thus very well have been more pronounced at the time the intron was lost. On an evolutionary time scale, DNA that is not under selective pressure will greatly vary between species that have relatively rapid turnover; it is estimated that each neutral base has been mutated 2–3 times since the divergence of C. elegans and C. briggsae (Stein et al. 2003). We thus also restricted our analysis to regions of genes that were highly conserved: Introns were included in our data set only if 15 amino acids on both sides of the intron were at least 50% identical across all species. We also performed a more restrictive analysis using 100% identity in 6 amino acids on both sides, giving similar outcomes (data not included). For the same reason, we omitted all wobble bases from our analysis, as also these are likely under less selective pressure after intron loss has occurred. It is thus more plausible that these bases in the current genome are different than at the moment the intron was lost. Although this filter sharpens the analysis and outcomes, it is not essential, as without it, an earlier notion of elevated homology at the exon–intron border was previously spotted for Drosophila (Coulombe-Huntington and Majewski 2007b).
We found MMIL to better fit the presented data than RTMIL, which has been suggested to account for precise intron loss in other species, such as mammals and flies (Coulombe-Huntington and Majewski 2007a; Yenerall et al. 2011). We did observe a slight bias for preferential intron retention at the 5′ side of a locus; however, we consider it more likely that this effect is attributed to the retention of the first intron due to selection pressure on regulatory elements which are frequently located in the most 5′ intron (Lynch and Kewalramani 2003). Indeed, the 5′ conservation is no longer significant upon exclusion of the first intron (supplementary fig. S1C and D, Supplementary Material online). Although the presence of microhomology is the quintessential feature to propose a MMIL model, two other observations are also in favor. First, the projection that homologous sequences are preferably used when they are in close proximity to a break can explain why smaller introns are more frequently found to be lost than larger introns, in accordance with previous findings in Drosophila (Yenerall et al. 2011). Interesting in this respect is that C. elegans genes that are expressed at higher levels tend to have shorter introns, which can increase the rate of intron loss if an intronic DSB occurs. We cannot, however, exclude other reasons for why smaller introns are more frequently lost over larger ones. Second, we found that genes that are germline-expressed are more susceptible to intron loss than those which are silent. This relationship could be explained by the notion that gene expression itself is a known inducer of DNA DSBs, which may ultimately lead to intron loss. The notion of enhanced intron loss in germline-expressed genes is in fact supportive of both the MMIL model as well as the RTMIL model. A difference between both models, however, is that RTMIL fully depends on transcriptional activity of the host gene in germ cells, whereas this dependency is far less strict for MMIL. RTMIL can thus not easily explain loss of introns in genes that are exclusively expressed in somatic tissue.
Surprisingly, we found that the preferential loss of introns from germline-expressed genes, while observed for all autosomes, is not seen for genes located on the X-chromosome. This is observed for both worms and flies. The C. elegans X-chromosome is silenced in early meiotic prophase in oogenic germ cells, and oocyte-enriched genes on the X-chromosomes are, on average, expressed at levels significantly lower than oocyte-enriched genes on autosomes (Kelly et al. 2002). In fact, transcription of several X-linked oocyte genes was found to be restricted to very late meiotic prophase I, a stage where DSBs are exclusively repaired via homologous recombination. This error-free repair pathway may thus protect X-linked genes from (intron) deletions at transcription-induced DSBs. Although mechanisms of sex-chromosome inactivation have been observed for nematodes, flies, and mammals (Namekawa and Lee, 2009; Meiklejohn et al. 2011), it is currently unknown whether they protect the sex chromosomes from mutations such as deletion of intronic sequences.
In summary, we here provide evidence that the presence of microhomology at the intron–exon junction is predictive for introns to be lost given enough time. We propose that the underlying mechanism for this MMIL phenomenon is microhomology-driven DNA DSB repair as this process is known to generate intron-size deletions, it explains why smaller introns are preferentially lost over larger ones, and it is in line with the observation that intron loss is more frequently found in actively transcribed genes, which are more susceptible to DNA damage. DNA repair may thus provide biological systems with the possibility to insert potential regulatory elements within encoding sequences as well as the means to remove them (fig. 3D), even in a very precise manner, from genes that are under strong evolutionary pressure.
Supplementary Material
Supplementary figures S1–S3 and tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
M.T. and R.S. wrote the paper. M.T. designed the study. R.S. wrote the Perl scripts and analyzed the data with M.T. This work was supported by the European Research Council Starting Grant (203379, “DSBrepair”) to M.T. The authors thank Jane van Heteren and Evelina Papaioannou for critically reading of the manuscript and members of the Tijsterman Lab for discussions.
Literature Cited
- Bradnam KR, Korf I. Longer first introns are a general property of eukaryotic gene structure. PLoS One. 2008;3:e3093. doi: 10.1371/journal.pone.0003093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JA. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- Clejan I, Boerckel J, Ahmed S. Developmental modulation of nonhomologous end joining in Caenorhabditis elegans. Genetics. 2006;173:1301–1317. doi: 10.1534/genetics.106.058628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan A, Wolfe KH. Origins of recently gained introns in Caenorhabditis. Proc Natl Acad Sci U S A. 2004;101:11362–11367. doi: 10.1073/pnas.0308192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbourne JK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331:555–561. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe-Huntington J, Majewski J. Characterization of intron loss events in mammals. Genome Res. 2007a;17:23–32. doi: 10.1101/gr.5703406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe-Huntington J, Majewski J. Intron loss and gain in Drosophila. Mol Biol Evol. 2007b;24:2842–2850. doi: 10.1093/molbev/msm235. [DOI] [PubMed] [Google Scholar]
- Croll D, McDonald BA. Intron gains and losses in the evolution of Fusarium and Cryptococcus fungi. Genome Biol Evol. 2012;4:1148–1161. doi: 10.1093/gbe/evs091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decottignies A. Microhomology-mediated end joining in fission yeast is repressed by pku70 and relies on genes involved in homologous recombination. Genetics. 2007;176:1403–1415. doi: 10.1534/genetics.107.071621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow A, Meduri E, Dolezal M, Hua L, Schlotterer C. Nonsense-mediated decay enables intron gain in Drosophila. PLoS Genet. 2010;6:e1000819. doi: 10.1371/journal.pgen.1000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow A, Meduri E, Schlotterer C. DNA double-strand break repair and the evolution of intron density. Trends Genet. 2011;27:1–6. doi: 10.1016/j.tig.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JA, Rouze P, Van de Peer Y. Higher intron loss rate in Arabidopsis thaliana than A. lyrata is consistent with stronger selection for a smaller genome. Mol Biol Evol. 2012;29:849–859. doi: 10.1093/molbev/msr254. [DOI] [PubMed] [Google Scholar]
- Fedorov A, Merican AF, Gilbert W. Large-scale comparison of intron positions among animal, plant, and fungal genes. Proc Natl Acad Sci U S A. 2002;99:16128–16133. doi: 10.1073/pnas.242624899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiber AL, Rangarajan J, Vaughn JC. The evolution of single-copy Drosophila nuclear 4f-rnp genes: spliceosomal intron losses create polymorphic alleles. J Mol Evol. 2002;55:401–413. doi: 10.1007/s00239-002-2336-y. [DOI] [PubMed] [Google Scholar]
- Haber JE. Alternative endings. Proc Natl Acad Sci U S A. 2008;105:405–406. doi: 10.1073/pnas.0711334105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Charlesworth B, Halligan DL, Andolfatto P. Patterns of intron sequence evolution in Drosophila are dependent upon length and GC content. Genome Biol. 2005;6:R67. doi: 10.1186/gb-2005-6-8-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner MC, De Marzo AM, Meeker AK, Nelson WG, Yegnasubramanian S. Transcription-induced DNA double strand breaks: both oncogenic force and potential therapeutic target? Clin Cancer Res. 2011;17:3858–3864. doi: 10.1158/1078-0432.CCR-10-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SH, So GM, Chow KL. Postembryonic expression of Caenorhabditis elegans mab-21 and its requirement in sensory ray differentiation. Dev Dyn. 2001;221:422–430. doi: 10.1002/dvdy.1161. [DOI] [PubMed] [Google Scholar]
- Ju BG, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- Kelly WG, et al. X-chromosome silencing in the germline of C. elegans. Development. 2002;129:479–492. doi: 10.1242/dev.129.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zahler AM. Conservation, regulation, synteny, and introns in a large-scale C. briggsae-C. elegans genomic alignment. Genome Res. 2000;10:1115–1125. doi: 10.1101/gr.10.8.1115. [DOI] [PubMed] [Google Scholar]
- Kersey PJ, et al. Ensembl Genomes: an integrative resource for genome-scale data from non-vertebrate species. Nucleic Acids Res. 2012;40:D91–D97. doi: 10.1093/nar/gkr895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Tucker AE, Sung W, Thomas WK, Lynch M. Extensive, recent intron gains in Daphnia populations. Science. 2009;326:1260–1262. doi: 10.1126/science.1179302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, Gu J, Lu H, Shimazaki N, Tsai AG. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell Biochem. 2010;50:279–296. doi: 10.1007/978-90-481-3471-7_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Kewalramani A. Messenger RNA surveillance and the evolutionary proliferation of introns. Mol Biol Evol. 2003;20:563–571. doi: 10.1093/molbev/msg068. [DOI] [PubMed] [Google Scholar]
- McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Landeen EL, Cook JM, Kingan SB, Presgraves DC. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol. 2011;9:e1001126. doi: 10.1371/journal.pbio.1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Lee JT. XY and ZW: is meiotic sex chromosome inactivation the rule in evolution? PLoS Genet. 2009;5:e1000493. doi: 10.1371/journal.pgen.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen CB, Friedman B, Birren B, Burge CB, Galagan JE. Patterns of intron gain and loss in fungi. PLoS Biol. 2004;2:e422. doi: 10.1371/journal.pbio.0020422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu DK, Hou WR, Li SW. mRNA-mediated intron losses: evidence from extraordinarily large exons. Mol Biol Evol. 2005;22:1475–1481. doi: 10.1093/molbev/msi138. [DOI] [PubMed] [Google Scholar]
- Pontier DB, Tijsterman M. A robust network of double-strand break repair pathways governs genome integrity during C. elegans development. Curr Biol. 2009;19:1384–1388. doi: 10.1016/j.cub.2009.06.045. [DOI] [PubMed] [Google Scholar]
- Robert V, Bessereau JL. Targeted engineering of the Caenorhabditis elegans genome following Mos1-triggered chromosomal breaks. EMBO J. 2007;26:170–183. doi: 10.1038/sj.emboj.7601463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, Wolf YI, Sorokin AV, Mirkin BG, Koonin EV. Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr Biol. 2003;13:1512–1517. doi: 10.1016/s0960-9822(03)00558-x. [DOI] [PubMed] [Google Scholar]
- Roy SW. Intron-rich ancestors. Trends Genet. 2006;22:468–471. doi: 10.1016/j.tig.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Roy SW, Gilbert W. The pattern of intron loss. Proc Natl Acad Sci U S A. 2005;102:713–718. doi: 10.1073/pnas.0408274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SW, Hartl DL. Very little intron loss/gain in Plasmodium: intron loss/gain mutation rates and intron number. Genome Res. 2006;16:750–756. doi: 10.1101/gr.4845406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, Dietrich FS, Roy SW. Comparative genomic analysis of fungal genomes reveals intron-rich ancestors. Genome Biol. 2007;8:R223. doi: 10.1186/gb-2007-8-10-r223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, et al. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 2003;1:E45. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. Identification of genes expressed in the hermaphrodite germ line of C. elegans using SAGE. BMC Genomics. 2009;10:213. doi: 10.1186/1471-2164-10-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenerall P, Krupa B, Zhou L. Mechanisms of intron gain and loss in Drosophila. BMC Evol Biol. 2011;11:364. doi: 10.1186/1471-2148-11-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LY, Yang YF, Niu DK. Evaluation of models of the mechanisms underlying intron loss and gain in Aspergillus fungi. J Mol Evol. 2010;71:364–373. doi: 10.1007/s00239-010-9391-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.