

Abstract
We have compared genomes of Alteromonas macleodii “deep ecotype” isolates from two deep Mediterranean sites and two surface samples from the Aegean and the English Channel. A total of nine different genomes were analyzed. They belong to five clonal frames (CFs) that differ among them by approximately 30,000 single-nucleotide polymorphisms (SNPs) over their core genomes. Two of the CFs contain three strains each with nearly identical genomes (∼100 SNPs over the core genome). One of the CFs had representatives that were isolated from samples taken more than 1,000 km away, 2,500 m deeper, and 5 years apart. These data mark the longest proven persistence of a CF in nature (outside of clinical settings). We have found evidence for frequent recombination events between or within CFs and even with the distantly related A. macleodii surface ecotype. The different CFs had different flexible genomic islands. They can be classified into two groups; one type is additive, that is, containing different numbers of gene cassettes, and is very variable in short time periods (they often varied even within a single CF). The other type was more stable and produced the complete replacement of a genomic fragment by another with different genes. Although this type was more conserved within each CF, we found examples of recombination among distantly related CFs including English Channel and Mediterranean isolates.
Keywords: Alteromonas macleodii, SNPs, microevolution, recombination, horizontal gene transfer
Introduction
The process of genome variation in prokaryotic cells in nature remains largely unknown. Cells reproducing clonally can suffer mutations and intragenomic recombination both, legitimate (mediated by recA and among similar DNA sequences) or illegitimate due to insertion and deletion of mobile genetic elements that does not require sequence homology. Prokaryotes do not have meiosis or sexual reproduction but have widespread recombination phenomena that can take place between individuals of very different genomic background (horizontal gene transfer). The development of high-throughput sequencing technologies has opened a new window for studying the variation of bacterial genomes through time. Experimental evolution in the laboratory indicate that genomes of bacteria can remain nearly unaltered through large numbers of generations (Conrad et al. 2009; Charusanti et al. 2010; Kishimoto et al. 2010). A suggestive figure is that of Barrick et al. (2009) in which only 29 single-nucleotide polymorphisms (SNPs) were found in a long-term adaptive evolution study of Escherichia coli evolved in glucose minimal medium after 20,000 generations. However, in all these cases, the situation departs largely from nature. First, the level of complexity that contributes to evolution in natural environments, such as the interaction with other populations that might act as donors of genetic material, the changes in nutrient availability, and physicochemical conditions, cannot be reproduced in experiments in controlled environments. Despite these limitations, laboratories experiments have significantly improved knowledge about the mechanisms underlying adaptive evolution in bacteria (for reviews see Conrad et al. [2011]).
Understanding of bacterial genome evolution in natural environments is more limited and has been largely restricted to pathogens (Morelli et al. 2010; Nübel et al. 2010; Mutreja et al. 2011; Reeves et al. 2011). There is information about the variation of pathogenic strains throughout epidemic outbreaks from host to host, but this again is a very special situation that, although extremely important for human health, does not help much in understanding the evolution of free living bacteria. One way to approach this topic is the study of isolates from similar environments that have space-time continuity, so that the cells represented by the isolated strains can be considered representatives of the same population sampled at different times and locations. A report using this approach for Vibrio cyclitrophicus isolates from different communities associated to different size particles (Vergin et al. 2007; Shapiro et al. 2012) indicated that the microbe was highly recombinogenic, although recombination was more frequent among close genomes.
Alteromonas macleodii is a gamma-proteobacterium commonly found in temperate waters around the world (Sass et al. 2001; López-López et al. 2005). Mesocosm studies (Schäfer et al. 2006) and metatranscriptomic data (McCarren et al. 2010; Shi et al. 2012) have provided new insights into the relevance of this microbe as an opportunistic strategist when nutrient availability increases in oligotrophic conditions. Recently, we have used a metagenomic fosmid library and two A. macleodii strain genomes (AltDE and AltDE1), obtained from the same seawater sample, to analyze the genomic diversity present at this single time/space point (Gonzaga et al. 2012). The two strains had a relatively conserved core genome (98.6% average nucleotide identity [ANI]) but differed widely in the gene content of several flexible genomic islands (fGIs) located at equivalent genomic locations. Many of these fGIs were involved in the synthesis of cell surface structures, such as the flagellum or the lipopolysaccharide O-chain and might change phage sensitivity, as found in other cases of fGIs (Rodriguez-Valera et al. 2009; Avrani et al. 2011). The aim of this study was to analyze the genomic diversity of another set of isolates belonging to the deep ecotype clade from different Mediterranean locations and isolation times (plus the single Atlantic isolate available). The genomes have been fully sequenced and assembled, and the results provide a snapshot of how variation happens at the microdiversity level in this marine bacterium in nature. Remarkably, we have found two nearly identical genomes separated by more than 1,000 km, 2,500 m depth, and 5 years, the two closest marine isolates obtained from different and distant samples found till now.
Materials and Methods
Sample Collection and Sequencing
Details of isolation and origin of the A. macleodii strains sequenced in this study have been described in supplementary figure S1, Supplementary Material online. Briefly, A. macleodii MED64 comes from waters of the Aegean Sea near Lebanon (Pinhassi and Berman 2003), A. macleodii U4, U7, U8, U12, UM4b, UM7, and UM8 were isolated from the Ionian Sea at the Urania Basin (West of Crete) from three different depths (3,455, 3,475, and 3,500 m) (Sass et al. 2001). Finally, A. macleodii 615 comes from the L4 long-term coastal monitoring station in the Western English Channel (Southward et al. 2004).
DNA was extracted by phenol–chloroform as described in Neumann et al. (1992) and checked for quality on a 1% agarose gel. The quantity was measured using Quant-iT PicoGreen dsDNA Reagent (Invitrogen). The genomes were sequenced using the IlluminaHiSeq 2000 (100-bp paired-end read) sequencing platform (Macrogen, Korea). The generated reads were trimmed and assembled de novo using VELVET, version 0.7.63 (Zerbino and Birney 2008). Combination of Geneious Pro 5.0.1 (with default parameters) using previously assembled genomes AltDE and AltDE1 as a reference (Gonzaga et al. 2012) and oligonucleotides designed from the sequence of the ends of assembled contigs were used to obtain one single closed contig.
Gene Prediction and Annotation
Gene prediction of the assembled contigs was done using the ISGA pipeline (http://isga.cgb.indiana.edu/, last accessed June 18, 2013). The predicted protein sequences were compared using BlastP to the National Center for Biotechnology Information (NCBI) nr protein database (e value: 10−5). Open reading frames (ORFs) smaller than 100 bp and without significant homology to other proteins were not considered. BioEdit was used to manipulate the sequences (Hall 1999). GC content was calculated using the EMBOSS tool geecee (Rice et al. 2000).For comparative analyses, reciprocal BlastN and TBlastXs searches between the genomes were carried out, leading to the identification of regions of similarity, insertions, and rearrangements. To allow the interactive visualization of genomic fragment comparisons, Artemis v.12 (Rutherford et al. 2000) and Artemis Comparison Tool ACTv.9 (Carver et al. 2005) were used to compare the genomes. ANI was calculated as defined before (Konstantinidis and Tiedje 2005), using a minimum cutoff of 50% identity and 70% of the length of the query gene. Sequences were aligned using MUSCLE version 3.6 (Edgar 2004) and ClustalW (Thompson et al. 1994) and edited manually as necessary. The CGView application (Stothard and Wishart 2005) was used to plot the circular representations of the CF plasmids.
SNP Analysis
To further investigate the differences among A. macleodii strains, nucmer program in the MUMmer3+ package (Kurtz et al. 2004) was used to identify the indels and the SNPs between small regions of the genome such as genomic islands (GIs). The program uses exact matching, clustering, and alignment extension strategies to create a dot plot based on the number of identical alignments between genomes. SNPs between whole genomes were identified using SNPsFinder (Song et al. 2005).
dNdS Analysis
The ratio of nonsynonymous (dN) to synonymous (dS) changes is one of the most widely methods used to quantify selection pressures acting on protein-coding regions. A low ratio (dN/dS < 1) indicates purifying selection, whereas a high ratio (dN/dS > 1) is a clear signal of diversifying selection. Orthologous protein sequence pairs were aligned using ClustalW and the protein alignments imposed upon the nucleotide sequences using the program pal2nal (Suyama et al. 2006). For each sequence pair, pairwise dN, dS, and dN/dS indices were estimated by maximum likelihood using the codeml program (Yang 1998).
Recombination
Multiple alignment of genomic sequences for A. macleodii strains was performed by using Mauve multiple alignment software (v2.3.1) (Darling et al. 2004). To detect potential recombination strains and possible recombination breakpoints (Martin et al. 2010), recombination detection methods implemented in the RDP4 (beta 16) software were used. To be considered as a reliable recombination event, the highest multiple-comparison corrected P-value cutoff was set at 106 for at least four different methods embedded in the RDP program (RDP, GENECONV, SiScan, BootScan, MaxChi, Chimaera, and 3Seq).
For estimating mutation and recombination rates, the ClonalFrame software v1.2 was used. ClonalFrame is a Bayesian inference method that reconstructs clonal relationships between the isolates in a sample. Three independent runs of ClonalFrame were performed each consisting of 100,000 Markov chain Monte Carlo iterations. To assess the relative contribution of recombination and mutation, r/m and ρ/θ statistics were used. ρ/θ is the proportion of rates at which recombination and mutation occur. It is therefore a measure of how often recombination events happen relative to mutations. r/m is the ratio of probabilities that a given site is altered through recombination and mutation and is therefore a measure of how important the effect of recombination was in the diversification of the sample relative to mutation.
Recruitments
Recruitment plots of the genomes were carried out against some available marine metagenomes (Rusch et al. 2007; Coleman and Chisholm 2010; Quaiser et al. 2011). BlastN (Altschul et al. 1997) was carried out between each A. macleodii genome (615, AltDE, AltDE1, MED64, U4, U7, U8, UM4b, and UM7) and the environmental databases. A very restrictive cutoff of 99% of identity in 90% of the length of the environmental read was established to guaranty that only similarities at the level of nearly identical microbes were counted. The numbers of hits were normalized against the genomes and the database sizes. In the metagenome recruitment of AltDE, 70% of identity in 50% of the length of the metagenomic read was used as a cutoff to construct the plots.
Accession Numbers
The genome sequences have been deposited in GenBank under the following accession numbers: CP004849 for A. macleodii U4, CP004851 for A. macleodii U7, CP004852 for A. macleodii U8, CP004853 for A. macleodii UM7, CP004855 for A. macleodii UM4b, CP004848 for A. macleodii MED64, and CP004846 for A. macleodii 615. U4, UM7, and 615 plasmid sequences have been also deposited at NCBI under the accession numbers CP004850, CP004854, and CP004847
Results
Diversity within the Deep Ecotype Core Genome
Two genomes of isolates belonging to the deep ecotype clade of A. macleodii, AltDE and AltDE1, obtained from 1,000 m deep in the South Adriatic have been already described (Ivars-Martínez, Martin-Cuadrado, et al. 2008; Gonzaga et al. 2012). We report here the genome sequence of seven new strains of A. macleodii isolated from different locations throughout the Mediterranean and one from the English Channel (Atlantic Ocean) (supplementary fig. S1, Supplementary Material online). They all belong to the “deep ecotype” clade (López-López et al. 2005; Ivars-Martínez, D'Auria, et al. 2008). This clade is quite divergent from the A. macleodii “surface clade” (López-Pérez et al. 2012) and, in spite of the high similarity of the 16S rRNA gene (>98%), could actually belong to a separate species (ANI below 85% over the core genome) (Ivars-Martínez, Martin-Cuadrado, et al. 2008) (supplementary fig. S2, Supplementary Material online). Although it seems contradictory, some representatives of the deep clade have been actually isolated from surface waters. We already proposed (Ivars-Martínez, Martin-Cuadrado, et al. 2008) that the “deep” clade is a group of strains adapted to live on larger fast-sinking particles and that this explains their frequent isolation from deep Mediterranean waters, although it can be found in surface samples as well. Most of the genomes reported here (strains designation starting by U) belong to isolates that were obtained from a much deeper sample (∼3,500 m) in the Urania Basin (Sass et al. 2001) located approximately 1,000 km away from the South Adriatic sampling site where AltDE and AltDE1 were obtained. Besides, the U isolates were retrieved 5 years earlier than the Adriatic ones and come from three separate samples taken at approximately 20-m intervals along the water column (see Materials and Methods). The two additional genomes belong to strains isolated from surface samples, MED64 was isolated from coastal waters off Israel (Eastern Aegean) (Pinhassi and Berman 2003), and 615 from the L4 long-term coastal monitoring station off Plymouth (English Channel) (Southward et al. 2004). In spite of the different locations and environmental conditions of the sampling sites, the isolates form a highly homogeneous clade with average nucleotide identities over 98% (table 1). There were two pairs of genomes (U8-U12 and UM7-UM8) that were identical, both pairs were retrieved from the same sample and can be considered resequencing of the same strain. The remaining genomes had a gradient of similarities that is illustrated in figure 1 and table 1. The genomes have synteny over most of the core genome that includes 2,614 genes and can be classified into five clonal frames (CFs). We use the term CF to describe bacterial lineages of common ancestry and apparent clonal (asexual) descent but in which replacement of genome fragments by recombination, selection, and drift by neutral genetic mutations has occurred (Milkman and Bridges 1990). Two of them, CF1 and CF2, comprised three different strain genomes each, and the remaining three were represented by one single genome each.
Table 1.
General Features of “Deep Ecotype” Alteromonas macleodii Genomes
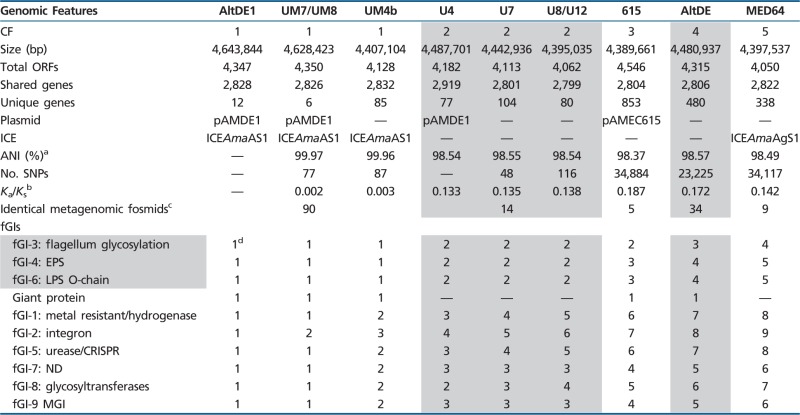 |
Note.—ND, not determined; EPS, exoplipopolysaccharide; LPS, lipopolysaccharide. Shading in column 1 has been used to highlight the replacement fGIs. For the number of SNPs, AltDE1 was used as a reference genome in CF1, CF3, CF4, and CF5; for CF2, U4 was the reference genome.
aANI (Konstantinidis and Tiedje 2005) to AltDE1 homologous genes.
bRatio of the number of nonsynonymous substitutions per nonsynonymous site (Ka) to the number of synonymous substitutions per synonymous site (Ks).
cNumber of identical fosmids found in an Adriatic metagenome (Gonzaga et al. 2012).
dNumbers identify the different versions (different gene content) of the fGI.
Fig. 1.—

Alignment of Alteromonas macleodii deep ecotype available genomes. They are arranged by numbers of SNPs along their core genomes starting from the bottom genome of MED64. Numbers of SNPs are indicated to the right and the amount corresponds to the comparison of the two strains connected by the arrow. Vertical green lines indicate SNPs among the strains within a CF, whereas white ones mark genes with ratio dN/dS > 1 between pairs of strains belonging to different CFs. Colored rectangles to the left delineate the genomes belonging to the same CF. Arrows show location of lysogenic phages inserted within the genomes, same color indicate same phage. ICEs are highlighted with blue rectangles. Some fGIs identified in the comparison between AltDE and AltDE1 (Gonzaga et al. 2012) have been highlighted in purple, identified by the inferred function (on top).
Strains belonging to the same CF diverged between 48 and 116 SNPs over the core genome, whereas those belonging to different CFs differed by between 25,576 and 31,564 SNPs, indicating that they must have diverged much earlier. Most SNPs were evenly distributed throughout the genomes and originated synonymous replacements. However, when comparing different CFs, some hotspots with high dN/dS values could be identified (fig. 1). These hotspots were located at different genes in the different strains, and we could not discern any obvious pattern from the types of genes or their location. The dN/dS values were higher (∼0.1) among CFs than within CFs (0.002), what seems contradictory (Rocha et al. 2006). However, the ClonalFrame analysis (see later) indicates that recombination is also much more common in close relatives, so many of the synonymous replacements found within the same CF can be due to recombination rather than to point mutations.
There have been reports of frequent recombination among A. macleodii strains, some even spanning the two clades (López-López et al. 2005; Ivars-Martínez, D'Auria, et al. 2008). Besides, in the previous work comparing only two strains and metagenomic fosmids (Gonzaga et al. 2012), there was evidence indicating that recombination happened mostly in close proximity to the fGIs (Gonzaga et al. 2012). However, the access to complete genomes allows for a more reliable and comprehensive assessment. We could generate genome alignments of more than 1.5 kb for 537 locally collinear blocks (65,8% of the core genome) shared by the 13 A. macleodii strains sequenced presently, including strains belonging to the surface clade. In total, 143 recombination events were identified (supplementary fig. S3, Supplementary Material online) that appeared spread along the core genome. There were some chromosomal recombination hotspots, but they do not appear to be more frequent near the fGIs. Two examples of the maximum-likelihood (ML) trees generated using these regions are shown in supplementary figure S3, Supplementary Material online.
To confirm the high level of recombination detected among CFs, we generated ML trees of different genomic regions including the alignable parts of the fGIs and compared them to the consensus tree generated by all the alignable regions in the genomes (core). The results shown in figure 2 illustrate that the topology varies depending on the regions selected, near fGIs, or in the core. This confirms that recombination events often break the clonal structure of the population as has been described for genomes of V. cyclitrophicus (Shapiro et al. 2012). Furthermore, recombination events often broke the line between surface and deep isolates. To assess the relative effect of recombination and mutation, we have used ClonalFrame software v1.2 to estimate the frequency of recombination relative to mutation (ρ/θ) and the weight of recombination on diversification relative to mutation (r/m) (Didelot and Falush 2007). The mean estimate including all the surface and deep clade representatives of ρ/θ and r/m ratios were 0.06 and 0.45, respectively. These results indicate that in spite of the frequent recombination, mutation is much more frequent than recombination. Similar ρ/θ and r/m ratios were estimated for the strains within the surface clade and among the members of the CF1 (supplementary table S1, Supplementary Material online). However, within representatives of the deep clade and the strains within CF2, the latter all coming from the same location, ratios of recombination-associated replacements were much higher (r/m = 5.13 and 8.61, respectively). These results suggest that, although recombination was less frequent than mutation, the weight of recombination for the total numbers of nucleotide replacements was quite significant, confirming previous observations (López-López et al. 2005; Ivars-Martínez, D'Auria, et al. 2008).
Fig. 2.—

ML trees of the nine Alteromonas macleodii deep ecotype representatives. Type strain A. macleodii ATCC 27126 was used as an outgroup to root the tree. Members of different CFs within the deep clade are labeled with color-coded dots. Trees below the genome were calculated based on aligned randomly selected core genome regions. The trees above the genome correspond to the alignable parts of some fGIs. The nucleotide lengths of the alignments are indicated between brackets for each tree. In the box is the consensus tree based on 2.8 Mbp of aligned core genome. AltDE1 was used as a reference genome to locate the position of the sequence used to generate the trees.
Plasmids, Conjugative Elements, and Phages
The plasmids and lysogenic phages were very variable genomic features. The 300-kb conjugative plasmid pAMDE1 previously described by Gonzaga et al. (2012) in the Adriatic Sea isolate AltDE1 was also found with identical sequence in the Urania isolate UM7, also from CF1, and in U4 that belongs to CF2. However, in the third representative of the CF1 (UM4b), it was lost. The perfect conservation of the plasmid sequence indicates recent transfer. An important feature of this plasmid was the presence of a hybrid polyketide and nonribosomal peptide (NRPS-PKS) cluster of 65 kb. This cluster is flanked by IS elements, suggesting that it could be a mobile genetic element (Gonzaga et al. 2012). Interestingly, within the genome of strains U7 and U8 of CF2, the plasmid was not present but an insertion of the same NRPS-PKS cluster was found in the chromosome (supplementary fig. S4, Supplementary Material online). The insertion was located next to the single Phe-tRNA, an insertion target producing high variability in all the known strains of A. macleodii, including those of the surface clade (López-Pérez et al. 2012). A completely different plasmid pAMEC615 was found in 615. The plasmid was confirmed by polymerase chain reaction (PCR) as a circular replicon of approximately 200 kb (supplementary fig. S4, Supplementary Material online). A fragment of 21 kb flanked by transposases in this plasmid is identical to a GI identified in the chromosome of A. macleodii 673, a surface clade strain obtained from the same water sample (López-Pérez et al. 2012). This GI was next to a tRNA and is probably involved in benzoate degradation (supplementary fig. S4, Supplementary Material online). Another 19-kb region (mostly hypothetical proteins) in this plasmid was nearly identical to a region in the chromosome of Alteromonas sp. SN2, an isolate from intertidal sediments in Korea that is only distantly related to A. macleodii. Altogether, these results reflect the frequent exchange of plasmids or plasmid fragments encompassing a taxon at the genus level and of global distribution.
A similar dynamic distribution was found for the integrative and conjugative elements (ICE). ICEs are also mobile genetic elements transferable by conjugation, but, unlike plasmids, they are always integrated into the chromosome. All the genomes of CF1 contain the same ICEAmaAS1 (supplementary fig. S5, Supplementary Material online) already reported in AltDE1 (Gonzaga et al. 2012). However, although the one in UM7 was identical to that of AltDE1, the one in UM4b has an insertion of three new genes located in a “hotspot” (Hs) (Beaber et al. 2002). A different ICE, which also belongs to the SXT/R391 family (Wozniak et al. 2009) was found in strain MED64. Synteny of the core ICE genes was well preserved (supplementary fig. S5, Supplementary Material online), but the MED64 Hs’ regions were more similar to Hs’ found in different V. cholera species. The SXT/R391 ICE family shares the same chromosomal integration site, the 5′-end of prfC, which encodes peptide chain release factor 3. However, in strain U4 of CF2, a 15-kb ICE-like region, similar to the ICEAmaAS1, was found at a different genomic location (supplementary fig. S5, Supplementary Material online). This ICE-like region has only 4 of the 14 tra genes found in the A. macleodii ICEs and a part of the Hs2 region found in ICEAmaAS1. Overall, the ICE is one of the most dynamic regions in the chromosome with frequent changes within CFs.
The strains AltDE1 and UM7 have both a lysogenic phage inserted at the same position in the genome. This insertion was positively identified as a lysogenic phage thanks to the presence of a CRISPR spacer with identical sequence in AltDE (Gonzaga et al. 2012). The presence of this phage in the two strains separated by site and time of isolation indicates that phages can stay in the lysogenic state for a long time in nature. On the other hand, UM4b also in CF1 was devoid of this lysogenic phage. In the three CF2 strains, a different phage, similar to E. coli prophage CP4-57 (Kirby et al. 1994), was found inserted at a tRNA-Leu (supplementary fig. S6A, Supplementary Material online). The att site could be detected due to the similarity to the site-specific integration of similar prophage found in Haemophilus infuenzae (Hauser and Scocca 1992; Wang et al. 2009) (supplementary fig. S6B, Supplementary Material online). MED64, the only representative of CF5 contained yet a third different phage that showed clear similarities to the lambdoid E. coli HK97 phage (Juhala et al. 2000), no obvious attachment site or insertion target was found (supplementary fig. S6C, Supplementary Material online). None of the two new phages detected here showed homology to any AltDE CRISPR spacers.
Flexible Genomic Islands
The main differences found among CFs were the presence of different fGIs. Recently, A. macleodii fGIs were defined as regions detected when comparing closely related strains that have similar location and inferred function but contain different genes (Gonzaga et al. 2012). The availability of more than one genome for some CFs allows the distinction of two types of fGIs that have probably different mechanisms of diversification.
The most obvious fGIs have a pattern of variation that could be described as “complete replacement.” In this kind, a cluster of genes is replaced by another totally different set, albeit coding for a similar or related function. In general, there is very little similarity, if any, among the genes present in the equivalent replacement fGI found in different CFs, like if the region had been completely replaced. In our case, the genes of this type of fGIs encoded the synthesis of exposed structures of the cell such as the lipopolysaccharide O-chain, the exopolysaccharide, and the flagellum (fGI3, 4 and 6) (table 1). Basically in all these cases, the fGIs present in different CFs have sets of genes that produce different sugar skeletons that form or decorate the exposed structure. As previously discussed (Rodriguez-Valera et al. 2009; Gonzaga et al. 2012; Rodriguez-Valera and Ussery 2012), this fGIs could generate different phage recognition targets in the population diluting the predation pressure of these viruses. These fGIs were very well conserved within CFs but were always different for different CFs (table 1). However, we found some interesting exceptions that might help understanding the mechanisms of variation at play in these genomic regions. One of them was found affecting the flagellum glycosylation fGI of strain 615 and the three strains in CF2. This fGI is located in the middle of the large flagellum gene cluster (Gonzaga et al. 2012) and contains flagellar structural genes that are exposed to the environment and genes involved in flagellin glycosylation (fig. 3). Flagellin glycosylation has been described in several bacterial species and is an essential modification, allowing both flagellar assembly and function (Logan 2006). This fGI was different for each CF except CF2 and CF3 for which the sequence found in this region was identical (fig. 3). It is remarkable that CF3 is represented by the single isolate 615 obtained from the English Channel, whereas all the members of CF2 come from the Urania Basin. The genomes of 615 and CF2 strains differ by an average of 11 SNPs per kb. However, only one SNP was found in this region of 17 kb. Contrastingly, sequence analysis of genes flgH and flgI located before the 5′-end of the island showed a large accumulation of SNPs, even though they code for the essential L- and P-rings of the flagellar basal body (fig. 3). A similar case of shared gene clusters (or rather in this case of a large gene) by different CFs was found for the giant protein (supplementary fig. S7, Supplementary Material online). Although this gene cannot be defined as an fGI because it was not present in all the strains, this GI was found in CF1, CF3, and CF4. It contains mostly a single gene coding for a “giant protein” of 6,573 aa. Most of these giant proteins have been identified in nonpathogenic environmental bacteria as large cell-surface glycoproteins (Reva and Tümmler 2008). The large protein sequence contains several VBCS repeats found in other giant proteins and, although their function is not well understood, they are believed to function in protein–protein and protein–carbohydrate interactions. SNPs analysis revealed that 615 have the most divergent version of these giant proteins. However, between CF1 and AltDE, most of the giant protein gene is identical except for the region that contains the VBCS repeats (supplementary fig. S7, Supplementary Material online). Like in the case of the flagellum glycosylation fGI, a large number of synonymous SNPs were found at the 5′-boundary of the island (fig. 3 and supplementary fig. S7, Supplementary Material online). This unusually high numbers of concentrated SNPs could be due to either positive selection or recombination. However, the highly significant enrichment of synonymous SNPs (fig. 3) suggests that the increase in SNP frequencies result from recombination events with divergent genomes. On the other hand, the nearly absolute sequence conservation along the swapped regions indicates that they have been exchanged quite recently, before a significant number of SNPs could accumulate.
Fig. 3.—

Comparison of the gene cluster involved in the biosynthesis and assembly of flagellar components, with the flagellum glycosylation island fGI found in different CFs. The plots above the 615 cluster indicate number of SNPs in a 100-bp window when compared with the three CF2 strains (all identical over this fGI). The average number of SNPs in the genome is indicated by the white line. The region enlarged in the red box indicates the rate of nonsynonymous (dN), synonymous (dS) substitutions and the dN/dS ratios.
The other kind of fGI, that could be called “additive,” contain variable numbers of gene cassettes in the different strains giving rise to very variable sizes but part of the fGI remains conserved. Most of the variability found within each CF was derived from changes that affected additive fGIs (table 1). Typically, these changes involve sequential insertions at a specific site such as tRNA gene as in the case of the fGI1, metal-resistant/hydrogenase island that has been already described (Gonzaga et al. 2012), or specific mobile elements such as an integron (fGI2, see later) or a mobilizable GI (MGI) fGI9. As an example of this kind of island (fig. 4), fGI9 is a typical MGI, conjugative mobile elements that utilize the conjugation machinery of ICEs, or plasmids for their transfer (Daccord et al. 2013). Interestingly, the size of this fGI in strains having ICEs (CF1 and CF5) was over twice as large as in the others. Although within CF2, this fGI was conserved, in CF1, a complete set of genes related to the Entner-Doudoroff pathway flanking by transposases was identified in AltDE1 and UM7 but not in UM4b. Finally, some of the additive fGIs (5, 7, and 8), although always rich in transposable elements, could not be characterized at the level of the mechanisms providing variability.
Fig. 4.—

fGI9 or MGI. (A) Comparison of gene content of the MGI in the strain genomes. The attachment sites, left (attL) and right (attR), are highlighted in red, the integrase is highlighted in yellow, and the red arrow indicates the position of the conserved sequence that mimics the origin of transfer (oriT). Predicted ORFs with the same color are involved in the same function. (B) Alignment of the attachments sites attL and attR.
Presence of the CFs at the Different Sites
The most similar genomes by SNPs, aside from the identical ones mentioned before, were U4 and U7 both in CF2. Interestingly, a similar degree of identity was found between AltDE1 (from the South Adriatic) and UM7 (from Urania), in spite of the time and space distance of the isolation of these two strains. Actually, when considering the fGIs, the pair from the South Adriatic and the Ionian AltDE1 and UM7 was the most similar (table 1). The finding of two nearly identical clones isolated from different locations and at different times has not been reported yet outside of clinical settings (Reeves et al. 2011). The major difference between AltDE1 and UM7 was found in fGI2 (Gonzaga et al. 2012). This variable region was the only fGI that was different in all the strains sequenced (table 1), and its high variability was due to the presence of an integron. Integrons are mobile DNA elements, and their core structure consists of a gene that codes for an integrase, IntI, and a proximal primary recombination sequence called attI site that allow bacteria to capture and express gene cassettes (Mazel 2006; Boucher et al. 2007). Multiple alignments of fGI2 showed that the integrase was identical for all the strains sequenced and possessed an identifiable single attl site, typical of class 1 integrons (Partridge et al. 2000) (data not shown). Inclusion of different cassettes makes the length variable among the strains, going from 5 kb in MED64 (that contains only one cassette) to 27 kb in AltDE. The function of most of these cassettes is unknown. Strains AltDE1 two cassettes and UM7 single cassette are completely different (supplementary table S2, Supplementary Material online), illustrating the highly dynamic nature of this genomic region. Aside from the SNPs, the only other difference between these two strains was found in fGI8 (containing glycosyltransferases). In CF1, four different thrombospondin type 3 repeat family protein genes have been found in this fGI. Thrombospondins are multimeric multidomain glycoproteins that function at the cell surface (Lawler and Hynes 1986). UM7 and UM4b have lost an internal repeat (459 nucleotides) in the first thrombospondin-like protein compared with AltDE1 (data not shown). This could be attributed to intragenomic recombination or duplication-related errors due to the repetitive nature of this region. Summarizing, the two differences in gene content found between the closest pair of isolates are 1) the acquisition of different cassettes by an integron and 2) a deletion of approximately 450 nucleotides due likely to an intragenomic recombination event.
Another way to gauge the presence of a CF at different sites is by comparing the genomes with metagenomes where the microbes are abundant (Gonzaga et al. 2012). Unfortunately, the presence of A. macleodii in most metagenomic data sets such as the Global Ocean Survey (Rusch et al. 2007) is too small to allow for a precise estimation of the presence of the different CFs detected here at different locations. There are, however, two data sets that are rich enough in biomass of this microbe to assess the presence of the different CFs. One is a large metagenomic fosmid library built with biomass retrieved from the same sample from which AltDE and AltDE1 were isolated (Gonzaga et al. 2012). From this fosmid library (38,704), 245 fosmids could be assigned to environmental A. macleodii by similarity to the genomes available, and 161 were fully sequenced. The availability of the new genomes allows to assign the fosmids to the five CFs described here (table 1). Although most of the fosmids are more similar to the two strains isolated from the same location, there are significant numbers of them that seem more related to CF 2 or to the CFs represented by 615 and MED64. This could be taken as an indication of the presence of all the CFs at this location although in very different proportions. One direct pyrosequencing metagenome carried out with a sample from the deepest Mediterranean basin Matapan-Varilov (Smedile et al. 2012) contained large amounts of A. macleodii reads and recruitment analysis indicated that most of the biomass here corresponded to clones highly related to the CF represented by AltDE because even the fGIs of this strain recruited at high similarity (supplementary fig. S8, Supplementary Material online).
Discussion
The genomes described here have been sequenced at high coverage, and the assembly has been confirmed by PCR providing a very high quality and reliability of data. Furthermore, for isolates UM7 and U8, the independent sequencing of identical strains (UM8 and U12, respectively) provided an even more robust confirmation of the genome data, very important when comparing highly similar genomes. This allows a reliable description of the gradient of similarity, revealing the way these microbes change in the short to medium time scale in a marine planktonic habitat. It is difficult to get an accurate time frame for the divergence of the most recent common ancestor (MRCA) of the isolates studied here, but recent work using genomes of isolates from clonal expansion of pathogenic bacteria provide some relevant numbers. For example, uropathogenic E. coli isolates obtained within a single household showed 1.1 SNPs per genome and year (Reeves et al. 2011). However, probably the most relevant estimation for our case is that of Mutreja et al. (2011) that analyzed the genomes of 113 monophyletic 7th pandemic V. cholera isolates. These authors calculated a ratio of 3.3 SNPs per genome and year. Although these are pathogenic clones isolated from human patients, V. cholera survive in natural waters, is a close phylogenetic relative of A. macleodii, and has probably a similar lifestyle. If we accept this value for the rate of change of the core genome of A. macleodii in the marine environment (an admittedly risky extrapolation), then the CFs diverged all at a similar timeframe between 7,000 and 10,000 years ago. This figure is quite meaningful because the Mediterranean Sea and even the global ocean has not changed much since the end of the last glacial period about 10,000 years ago (Annan and Hargreaves 2013). Since then, temperature wise at least, conditions have remained quite constant. Therefore, this would have been a reasonable starting point for the CFs of the A. macleodii deep clade to radiate.
Strains belonging to the same CF diverged between 48 and 116 SNPs over the core genome. These are remarkably small figures for free living independent isolates. A recent report for two Listeria genomes obtained from two food packaging facilities 6 years apart had 18 SNPs difference (Holch et al. 2013). In a widespread study of methicillin-resistant Staphylococcus aureus, the most similar strains were different at 14 SNPs but were obtained only 11 weeks apart (Harris et al. 2010). The L2 El Tor V. cholera isolates obtained over 53 years mentioned earlier (Mutreja et al. 2011) had between 50 and 250 SNPs. Interestingly, the two most similar genomes found here were isolated from different locations. Both samples came from the Eastern Mediterranean, and the deep Adriatic and Ionian are communicated by currents. However, the main connection flows in the opposite direction of the collection date (Menna and Poulain 2010), that is, from the deep Adriatic taken in 2003 to the deep Ionian taken in 1998. Furthermore, the samples were also taken in the opposite direction of the sinking flow (first the 3,500 m and later the 1,000 m). All this oceanographic parameters indicate that the water masses of origin of both isolates are not mixed rapidly, and the MRCA must be at least decades old. Just the isolation date, 5 years apart, gives a minimal framework for the endurance of this clonal lineage. Using the 3.3 SNPs/year described earlier, the MRCA of AltDE1 and UM7 (87 SNPs) would be 26 years older than the strains. Actually, even the most hypervariable regions are quite well conserved between these two genomes, including all the fGIs and even the lambda-like lysogenic phage, providing one of the largest examples of persistence in nature of a phage in a lysogenic state. The main differences found between these two strains were due to the insertion of different gene cassettes in an integron, what indicates that this could be the most rapidly changing type of mobile genetic element. Incidentally, the presence of totally different genes at this site prove also that they do not belong to a single laboratory clone, that is, that they are real and independent natural isolates and not the results of laboratory contamination. On the other hand, the other genome within CF1, UM4b, with likely a few decades older MRCA had no phage inserted and many more differences affecting some of the additive fGIs. Actually five out of seven fGIs of this type identified had already changed (table 1).
Recently, it has been proposed for V. cyclitrophicus strains (Shapiro et al. 2012) that the main driver of divergence is the higher rate of recombination taking place among strains that share a similar habitat (Shapiro et al. 2012). Along these lines, we have found a higher impact of recombination within the CFs than between them. Furthermore, for isolates from the same location, such as those of CF2, the impact of recombination was even higher. It seems clear that these groups of gammaproteobacteria are very recombinogenic, and the abundance of conjugative elements such as plasmids, ICEs, MGIs, or lysogenic phages provide plenty of mechanisms to exchange genome fragments between cells. However, in Alteromonas, there are also many examples of significant genomic exchange between the surface and deep clades and even with other species such as Alteromonas sp. SN2.
Prominently, the degree of nucleotide divergence seems to have little effect on the recombination events that precede the replacement of fGIs. Particularly, the presence of identical versions of the flagellum glycosylation fGI3 and the giant protein GI in very different genomic backgrounds indicates that in these cases recombination can take place between distant relatives. A similar complete replacement of a very large protein by homologous recombination has been described in V. cholera where also a high density of SNPs was followed by this gene cluster (Mutreja et al. 2011). In our case, the identity of the fGIs was so high that the exchange must have happened very recently and indicates that replacement fGIs have a fast turnover. Also the low values of dN/dS ratio of the high variability site upstream from these fGIs indicate that they are frequently subjected to recombination. In these cases, rare recombination events maybe favored by the strong selective pressures to evade phage predation. The change of any of these exposed structures would make the clones resistant to some of the phages preying upon them. It is remarkable that a very similar pattern of replacement of fGIs has been found in 113 genomes of V. cholera (Mutreja et al. 2011) in which also within a background of very little core variation there was a replacement of the O-chain polysaccharide gene cluster and the giant protein gene described before.
The availability of complete and fully assembled genomes of closely related strains opens a window into the dynamics of variation of prokaryotic genomes. Studies of free-living microbes such as Alteromonas are particularly relevant because they provide information that applies to ecologically relevant microbes and help also in understanding the more complex cases of pathogenic bacteria that have reservoirs in natural habitats. The different rates and mechanisms of variation of the core and flexible genomes illustrate how prokaryotic cells balance the needs for change and conservation. The presence of multiple concurrent CFs of A. macleodii has already been explored (Gonzaga et al. 2012). Here, we have proven the presence of some of them at different locations, expanding the model (Rodriguez-Valera et al. 2009; Rodriguez-Valera and Ussery 2012) and refining the description of the patterns of variation among the different CFs.
Supplementary Material
Supplementary figures S1–S8 and tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by projects MAGYK (BIO2008-02444), MICROGEN (Programa CONSOLIDER-INGENIO 2010 CDS2009-00006), CGL2009-12651-C02-01 from the Spanish Ministerio de Ciencia e Innovación, DIMEGEN (PROMETEO/2010/089), ACOMP/2009/155 from the GeneralitatValenciana, and MaCuMBA from European Community (EC) (Ref. FP7-KBBE-2012-6-311975) and by FEDER funds.
Literature Cited
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annan JD, Hargreaves JC. A new global reconstruction of temperature changes at the Last Glacial Maximum. Clim Past. 2013;9(1):367–376. [Google Scholar]
- Avrani S, Wurtzel O, Sharon I, Sorek R, Lindell D. Genomic island variability facilitates Prochlorococcus-virus coexistence. Nature. 2011;474(7353):604–608. doi: 10.1038/nature10172. [DOI] [PubMed] [Google Scholar]
- Barrick JE, et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461(7268):1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- Beaber JW, Burrus V, Hochhut B, Waldor MK. Comparison of SXT and R391, two conjugative integrating elements: definition of a genetic backbone for the mobilization of resistance determinants. Cell Mol Life Sci. 2002;59(12):2065–2070. doi: 10.1007/s000180200006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher Y, Labbate M, Koenig JE, Stokes HW. Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbiol. 2007;15(7):301–309. doi: 10.1016/j.tim.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Carver TJ, et al. ACT: the Artemis comparison tool. Bioinformatics. 2005;21(16):3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Charusanti P, et al. Genetic basis of growth adaptation of Escherichia coli after deletion of pgi, a major metabolic gene. PLoS Genet. 2010;6(11):e1001186. doi: 10.1371/journal.pgen.1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Chisholm SW. Ecosystem-specific selection pressures revealed through comparative population genomics. Proc Natl Acad Sci U S A. 2010;107:18634–18639. doi: 10.1073/pnas.1009480107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, et al. Whole-genome resequencing of Escherichia coli K-12 MG1655 undergoing short-term laboratory evolution in lactate minimal media reveals flexible selection of adaptive mutations. Genome Biol. 2009;10(10):R118. doi: 10.1186/gb-2009-10-10-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad TM, Lewis NE, Palsson BO. Microbial laboratory evolution in the era of genome-scale science. Mol Syst Biol. 2011;7:509. doi: 10.1038/msb.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daccord A, Ceccarelli D, Rodrigue S, Burrus V. Comparative analysis of mobilizable genomic islands. J Bacteriol. 2013;195(3):606–614. doi: 10.1128/JB.01985-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175(3):1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5(1):113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga A, et al. Polyclonality of concurrent natural populations of Alteromonas macleodii. Genome Biol Evol. 2012;4(12):1360–1374. doi: 10.1093/gbe/evs112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Harris SR, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327(5964):469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser MA, Scocca JJ. Site-specific integration of the Haemophilus influenzae bacteriophage HP1: location of the boundaries of the phage attachment site. J Bacteriol. 1992;174(20):6674–6677. doi: 10.1128/jb.174.20.6674-6677.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holch A, et al. Genome sequencing identifies two nearly unchanged strains of persistent Listeria monocytogenes isolated in two different fish processing plants sampled six years apart. Appl Environ Microbiol. 2013;79:2944–2951. doi: 10.1128/AEM.03715-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivars-Martínez E, D`Auria G, et al. Biogeography of the ubiquitous marine bacterium Alteromonas macleodii determined by multilocus sequence analysis. Mol Ecol. 2008;17(18):4092–4106. doi: 10.1111/j.1365-294x.2008.03883.x. [DOI] [PubMed] [Google Scholar]
- Ivars-Martínez E, Martin-Cuadrado AB, et al. Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2008;2(12):1194–1212. doi: 10.1038/ismej.2008.74. [DOI] [PubMed] [Google Scholar]
- Juhala RJ, et al. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J Mol Biol. 2000;299(1):27–51. doi: 10.1006/jmbi.2000.3729. [DOI] [PubMed] [Google Scholar]
- Kirby JE, Trempy JE, Gottesman S. Excision of a P4-like cryptic prophage leads to Alp protease expression in Escherichia coli. J Bacteriol. 1994;176(7):2068–2081. doi: 10.1128/jb.176.7.2068-2081.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T, et al. Transition from positive to neutral in mutation fixation along with continuing rising fitness in thermal adaptive evolution. PLoS Genet. 2010;6(10):e1001164. doi: 10.1371/journal.pgen.1001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102(7):2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler J, Hynes RO. The structure of human thrombospondin, an adhesive glycoprotein with multiple calcium-binding sites and homologies with several different proteins. J Cell Biol. 1986;103(5):1635–1648. doi: 10.1083/jcb.103.5.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan SM. Flagellar glycosylation—a new component of the motility repertoire? Microbiology. 2006;152(5):1249–1262. doi: 10.1099/mic.0.28735-0. [DOI] [PubMed] [Google Scholar]
- López-López A, Bartual SG, Stal L, Onyshchenko O, Rodríguez-Valera F. Genetic analysis of housekeeping genes reveals a deep-sea ecotype of Alteromonas macleodii in the Mediterranean Sea. Environ Microbiol. 2005;7(5):649–659. doi: 10.1111/j.1462-2920.2005.00733.x. [DOI] [PubMed] [Google Scholar]
- López-Pérez M, et al. Genomes of surface isolates of Alteromonas macleodii: the life of a widespread marine opportunistic copiotroph. Sci Rep. 2012;2:696. doi: 10.1038/srep00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26(19):2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazel D. Integrons: agents of bacterial evolution. Nat Rev Microbiol. 2006;4(8):608–620. doi: 10.1038/nrmicro1462. [DOI] [PubMed] [Google Scholar]
- McCarren J, et al. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci U S A. 2010;107(38):16420–16427. doi: 10.1073/pnas.1010732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menna M, Poulain PM. Mediterranean intermediate circulation estimated from Argo data in 2003-2010. Ocean Sci. 2010;6(1):331–343. [Google Scholar]
- Milkman R, Bridges MM. Molecular evolution of the Escherichia coli chromosome. III. Clonal frames. Genetics. 1990;126(3):505–517. doi: 10.1093/genetics/126.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli G, et al. Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010;6(7):e1001036. doi: 10.1371/journal.pgen.1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja A, et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477(7365):462–465. doi: 10.1038/nature10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann BR, Pospiech A, Schairer HU. Rapid isolation of genomic DNA from Gram-negative bacteria. Trends Genet. 1992;8(10):332–333. doi: 10.1016/0168-9525(92)90269-a. [DOI] [PubMed] [Google Scholar]
- Nübel U, et al. A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog. 2010;6(4):e1000855. doi: 10.1371/journal.ppat.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge SR, et al. Definition of the attI1 site of class 1 integrons. Microbiology. 2000;146(11):2855–2864. doi: 10.1099/00221287-146-11-2855. [DOI] [PubMed] [Google Scholar]
- Pinhassi J, Berman T. Differential growth response of colony-forming α- and γ-proteobacteria in dilution culture and nutrient addition experiments from Lake Kinneret (Israel), the Eastern Mediterranean Sea, and the Gulf of Eilat. Appl Environ Microbiol. 2003;69(1):199–211. doi: 10.1128/AEM.69.1.199-211.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaiser A, Zivanovic Y, Moreira D, Lopez-Garcia P. Comparative metagenomics of bathypelagic plankton and bottom sediment from the Sea of Marmara. ISME J. 2011;5(2):285–304. doi: 10.1038/ismej.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PR, et al. Rates of mutation and host transmission for an Escherichia coli clone over 3 years. PLoS One. 2011;6(10):e26907. doi: 10.1371/journal.pone.0026907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva O, Tümmler B. Think big—giant genes in bacteria. Environ Microbiol. 2008;10(3):768–777. doi: 10.1111/j.1462-2920.2007.01500.x. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rocha EPC, et al. Comparisons of dN/dS are time dependent for closely related bacterial genomes. J Theor Biol. 2006;239(2):226–235. doi: 10.1016/j.jtbi.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Valera F, Ussery D. Is the pan-genome also a pan-selectome? F1000 Res. 2012;1:16. doi: 10.12688/f1000research.1-16.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Valera F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7(11):828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- Rusch DB, et al. The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5(3):e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Sass AM, Sass H, Coolen MJL, Cypionka H, Overmann J. Microbial communities in the chemocline of a hypersaline deep-sea basin (Urania Basin, Mediterranean Sea) Appl Environ Microbiol. 2001;67(12):5392–5402. doi: 10.1128/AEM.67.12.5392-5402.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer H, et al. Microbial community dynamics in Mediterranean nutrient-enriched seawater mesocosms: changes in the genetic diversity of bacterial populations. FEMS Microbiol Ecol. 2006;34(3):243–253. doi: 10.1111/j.1574-6941.2001.tb00775.x. [DOI] [PubMed] [Google Scholar]
- Shapiro BJ, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336(6077):48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, McCarren J, DeLong EF. Transcriptional responses of surface water marine microbial assemblages to deep-sea water amendment. Environ Microbiol. 2012;14(1):191–206. doi: 10.1111/j.1462-2920.2011.02598.x. [DOI] [PubMed] [Google Scholar]
- Smedile F, et al. Metagenomic analysis of hadopelagic microbial assemblages thriving at the deepest part of Mediterranean Sea, Matapan-Vavilov Deep. Environ Microbiol. 2012;15(1):167–182. doi: 10.1111/j.1462-2920.2012.02827.x. [DOI] [PubMed] [Google Scholar]
- Song J, Xu Y, White S, Miller KWP, Wolinsky M. SNPsFinder, a web-based application for genome-wide discovery of single nucleotide polymorphisms in microbial genomes. Bioinformatics. 2005;21(9):2083–2084. doi: 10.1093/bioinformatics/bti176. [DOI] [PubMed] [Google Scholar]
- Southward AJ, et al. Long-term oceanographic and ecological research in the Western English Channel. Adv Mar Biol. 2004;47:1–105. doi: 10.1016/S0065-2881(04)47001-1. [DOI] [PubMed] [Google Scholar]
- Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2005;21(4):537–539. doi: 10.1093/bioinformatics/bti054. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34(2 Suppl):W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergin KL, et al. High intraspecific recombination rate in a native population of Candidatus Pelagibacter ubique (SAR11) Environ Microbiol. 2007;9(10):2430–2440. doi: 10.1111/j.1462-2920.2007.01361.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Kim Y, Wood TK. Control and benefits of CP4-57 prophage excision in Escherichia coli biofilms. ISME J. 2009;3(10):1164–1179. doi: 10.1038/ismej.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak RAF, et al. Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. 2009;5(12):e1000786. doi: 10.1371/journal.pgen.1000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15(5):568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.