

Abstract
Chagas disease is a deadly infection caused by the protozoan parasite Trypanosoma cruzi. Afflicting approximately 8 million people in Latin America, Chagas disease is now becoming a serious global health problem proliferating beyond the traditional geographical borders, mainly because of human and vector migration. Because the disease is endemic in low-resource areas, industrial drug development has been lethargic. The chronic form remains incurable, there are no vaccines, and 2 existing drugs for the acute form are toxic and have low efficacy. Here we report the efficacy of a small molecule, VNI, including evidence of its effectiveness against chronic Chagas disease. VNI is a potent experimental inhibitor of T. cruzi sterol 14α-demethylase. Nontoxic and highly selective, VNI displays promising pharmacokinetics and administered orally to mice at 25 mg/kg for 30 days cures, with 100% cure rate and 100% survival, the acute and chronic T. cruzi infection.
Keywords: Chagas disease, Trypanosoma cruzi, sterol biosynthesis, sterol 14alpha-demethylase (CYP51), inhibition, drug discovery, VNI
Chagas disease is a vector-borne zoonosis. In areas of endemicity, Trypanosoma cruzi is mainly transmitted by blood-feeding insects (triatomines; also known as kissing bugs) through their feces and contaminated objects, including crops, food, and drink, with hundreds of different mammalian species forming an inexhaustible reservoir [1]. The area where kissing bug species have been reported spans from the southern parts of South America up to the Great Lakes in the United States [2].
Clinical Chagas disease is classified into acute and chronic phases. The acute phase begins when T. cruzi infiltrates the body and starts multiplying within different organs and tissues. The symptoms during this phase can be very mild, nonspecific, and fever like (and often require no medical attention) or severe and sometimes deadly (myocarditis and meningoencephalitis have been reported, with an estimated fatality rate of 0.5% [3]), depending on factors such as the parasite burden and host immunocompetence. The acute phase lasts 4–8 weeks, after which the symptoms cease, and the infection enters the lifelong chronic phase. Intracellular T. cruzi amastigotes remain in infected tissues, especially in cardiac and skeletal muscle, but the parasitemia levels become undetectable by microscopy. Clinical symptoms of chronic Chagas disease develop decades later in up to 30% of patients. Therefore, most people remain unaware of their infection and can unwillingly transmit the parasite via blood, tissue, and organ donations and from mother to child. As an example, the estimated number of babies born with congenital Chagas disease each year in the United States, where approximately 300 000 infected persons live [4], ranges from 58 to 582 [5]. The chronic form of Chagas disease is a life-threatening illness that most severely affects the heart and gastrointestinal tract, accounting for nearly 5 times as many disability-adjusted life-years lost as malaria [2]. In the United States, undiagnosed Chagas disease is responsible for 30 000–45 000 cases of cardiomyopathy [4].
For more than a century since the discovery of T. cruzi, Chagas disease has remained highly neglected. The only 2 drugs used to treat acute-phase disease, benznidazole (since 1960s) and nifurtimox (since 1970s), are impractical because of low efficiency and frequent side effects; neither is approved by the Food and Drug Administration [3]. Treatment of the chronic form of the disease is still mainly limited to the management of symptoms (eg, antiarrhythmic medications, steroids, and pacemakers). Even heart transplantation cannot ensure a cure, because the parasites hidden within other organs will eventually reinvade it. Despite the well-recognized limitations of current therapeutic options and the general acceptance that specific treatment should be offered to all patients [4, 6], Chagas disease still remains outside the commercial interests of pharmaceutical companies, because treatment is considered unprofitable. In academic settings, the development of specific chemotherapeutic approaches was stalled for decades because of the hypothesis that chronic Chagas disease had an autoimmune, rather than an infectious, etiology [7]. Eventually, a clear correlation between T. cruzi and the inflammatory processes was made [8], leading to a consensus that elimination of the parasite from the human body is necessary for cure.
Sterols are essential components of eukaryotic cells. They play key roles in controlling the fluidity and permeability of plasma membranes, as well as in cell development and division. Some eukaryotic phyla (eg, insects) or species (eg, bloodstream forms of Trypanosoma brucei) acquired an ability to scavenge sterols for their membranes from exogenous sources (eg, diet). Others (eg, T. cruzi) must synthesize sterols endogenously. In all cases, sterol deficiency is lethal. Inhibition of sterol biosynthesis is used for treatment of human fungal infections. The most efficient clinical antifungals (azoles) target the fungal sterol 14α-demethylase (CYP51), a cytochrome P450 monooxygenase that catalyses oxidative removal of the 14α-methyl group from cyclized sterol precursors [9]. By inhibiting CYP51, azoles block the sterol biosynthetic pathway in the parasites and cause membrane damage, owing to the lack of ergosterol and the accumulation of cytotoxic methylated sterols [10]. Five azole drugs that are currently used for treatment of systemic fungal infections represent modifications of 2 basic inhibitory scaffolds: ketoconazole (I) and fluconazole (II; Figure 1A). Several derivatives of these 2 scaffolds are under development [11]. Some of these antifungals were found to be effective against T. cruzi [7, 12–14], with the 1,2,4-triazole posaconazole (scaffold I; Merck) and ravuconazole (scaffold II; Eisai; Figure 1A) entering clinical trials for Chagas disease [6]. However, ravuconazole has only suppressive rather than curative effects, and it is unable to eradicate T. cruzi in murine and canine models of Chagas disease [13, 15], whereas posaconazole, which resulted in cure in a patient with chronic Chagas disease [16], is too expensive for widespread use in countries where the disease is endemic [4, 16–18]. The cost of posaconazole, estimated to be well over $1000/patient, derives in part from its long (>17 steps for the longest linear sequence) and low-yielding (<1% overall) synthesis [18]. Other drugs with the same mechanism of action, improved safety profile, and lower potential cost are urgently needed [4, 6].
Figure 1.

Sterol 14α-demethylase (CYP51) inhibitory scaffolds of the antifungal drugs posaconazole and ravuconazole (derivatives of ketoconazole and fluconazole, respectively; A), and VNI (B). The numbers represent the comparative binding and inhibition parameters for Trypanosoma cruzi CYP51. Data denote the apparent dissociation constant (Kd), inhibition of the enzyme activity in vitro at equimolar ratio inhibitor/P450 (1-hour reaction; I), and median inhibitory concentrations (IC50) for inhibition of human CYP3A4, the cytochrome P450 that metabolized >90% of drugs in the human body [45]. B, VNI bound in the CYP51 active site [3gw9]. Shown is a slice through the surface of the binding cavity. VNI, the heme, and Y103 are depicted as stick models; other residues located within 4 Å are shown as lines. Water molecules are presented as red spheres. The H-bonds are displayed as red dashed lines. The substrate access channel entrance is marked with an arrow. Key features of VNI include an imidazole ring that directly coordinates to the enzyme heme iron (N3), a 2,4-dichlorinated β-phenyl ring that is projected into the deepest portion of the CYP51-substrate binding cavity, and a 3-ring linear polycycle positioned along the substrate access channel and connected to the rest of the inhibitor molecule through a carboxamide fragment with a chiral (+) amine carbon (R). The carbonyl oxygen of this fragment forms a hydrogen bond with the catalytic water molecule, thus connecting the inhibitor with the enzyme I-helix and, presumably, disrupting the CYP51 proton delivery route. The amide nitrogen repositions the side chain of Y103, which, in the enzymatically active protein, is H-bonded with the heme propionate. In total, VNI-CYP51 interactions include iron coordination, van der Waals contacts with 16 amino acid residues, and a H-bond network that disrupts 2 functionally essential protein regions.
VNI, (R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadi-azol-2-yl)benzamide, represents a novel CYP51 inhibitory scaffold, the first one to come not from antifungal programs but from studying the inhibition of T. cruzi sterol 14α-demethylase activity in the reconstituted reaction in vitro [19]. Later, VNI was structurally characterized in complex with the target enzyme [20, 21]. Structural data suggest that the potency of VNI to inhibit trypanosomal CYP51s (which have only approximately 25% amino acid identity to the fungal CYP51 orthologs) is likely to be enhanced by the hydrogen bond interactions, particularly between the VNI carboxamide fragment and the heme/substrate binding area of the enzyme (Figure 1). Moreover, the inhibitory effect of VNI is highly selective: by acting as a stoichiometric, functionally irreversible inhibitor of protozoan sterol 14α-demethylases, it has a moderate inhibitory effect on fungal (Candida albicans) CYP51 and does not inhibit the human CYP51 ortholog [21]. VNI exhibits no toxicity to mammalian cells [19], and doses of up to 400 mg/kg do not cause signs of acute toxicity in mice in vivo [22]. Unlike posaconazole or fluconazole, this scaffold does not induce T. cruzi CYP51 gene expression and does not require an increase in the dose to maintain its antiparasitic efficiency over time [20], suggesting that it may have a lower propensity to induce resistance.
METHODS
Cellular Infection Assays
The highly invasive trypomastigote clone 20A of the Tulahuen strain of T. cruzi, which was shown to infect >98% of exposed cardiomyocytes [23], was used to evaluate the anti–T. cruzi efficacy of inhibitors in cellular infection assays. T. cruzi trypomastigotes expressing green fluorescent protein GFP were generated as described elsewhere [20, 21]. Trypomastigotes were then used to infect cardiomyocyte monolayers in 96-well tissue culture plates and in 8-well LabTech tissue culture chambers in triplicate at a ratio of 10 parasites per cell. Plates were cultured in Dulbecco's modified Eagle's medium (DMEM) free of phenol red and supplemented with 10% fetal bovine serum (FBS) at 37°C in an atmosphere of 5% CO2 for 24 hours. Unbound trypomastigotes were removed by washing with DMEM free of phenol red. Infected monolayers were exposed to several concentrations of the inhibitors VNI, posaconazole, or ravuconazole (range, 1–22 nM), dissolved in dimethyl sulfoxide (DMSO)/DMEM free of phenol red in triplicate, and cocultured in DMEM plus 10% FBS for 48 hours to observe parasite multiplication. Seventy-two hours after infection, the cardiomyocyte monolayers were washed with phosphate-buffered saline, and the infection was quantified using a Synergy HT fluorometer (Biotek Instruments) [20]. For fluorescence microscopy of the drug-associated inhibition of T. cruzi multiplication within cardiomyocytes, infection assays were performed in 8-well LabTech tissue culture chambers in triplicate under the same conditions described above and elsewhere [24]. A total of 72 hours after infection, the monolayers were fixed with 2.5% paraformaldehyde and stained with 4′,6-diamidino-2-phenylindole, to visualize DNA, and with Alexa fluor 546 phalloidin (Invitrogen), to visualize cardiomyocyte actin myofibrils, which stain red.
Efficacy Studies in Animal Models of Chagas Disease
In the model of acute infection [25], female BALB/c mice (Jackson Laboratory; age, 8 weeks; weight, 25 g; 5 mice/group) were injected intraperitoneally with 1 × 105 cells of bloodstream T. cruzi, obtained from the peak of parasitemia in BALB/c mice infected with the Tulahuen strain 20A clone. The Tulahuen strain of T. cruzi is widely used to evaluate the efficacy of drugs in BALB/c mice [26–29]; the model of T. cruzi Tulahuen 20A clone infection in BALB/c mice has been documented elsewhere [30]. The parasites were resuspended in DMEM to obtain a trypanosome concentration of 1 × 106 parasites/mL. Each mouse was injected with 0.1 mL of the T. cruzi suspension. In these conditions, parasitemia reaches its maximum on day 14 after infection, causing death in 100% of untreated animals. VNI treatment was started 24 hours after infection. A 5% stock solution of VNI in DMSO was dissolved in sterile 5% Arabic gum, and 25 mg/kg was administered twice per day for 30 days by oral gavage. The control mouse group received only the vehicle. Parasitemia was monitored by placing 5 µL of tail blood under a coverslip and counting 100 high-powered fields, as described elsewhere [25]. Animals reaching the peak of parasitemia were euthanized because previous studies showed that animals do not survive for >24 hours after the peak [25]. The day on which mice were euthanized was recorded. The organs (heart, spleen, and liver), skeletal muscle tissue, and blood were analyzed for the presence of parasites by means of real-time polymerase chain reaction (qPCR), as indicated below. Three in vivo independent experiments were performed using the same protocol, and they yielded similar results.
In the model of chronic infection, female BALB/c mice (Jackson Laboratory; age, 8 weeks; weight, 25 g; 9 mice/group) were injected intraperitoneally with a less potent inoculum (50 bloodstream forms) of T. cruzi Tulahuen strain 20A clone, which produces a slowly developing parasitemia with a low peak level. Infection is effectively controlled by most infected animals (60%) that survive this initial parasitemic phase. Ninety days after infection, when no circulating parasites were found, VNI (25 mg/kg) or vehicle was orally administered twice daily for 30 days. Between 50 and 70 days after conclusion of VNI treatment, mice in both groups received 6 injections of cyclophosphamide (100 mg/kg) to induce immunosuppression, as described elsewhere [26], to reactivate the infection (Figure 3B). Parasitemia was evaluated as described for the model of acute infection. When parasitemia in the control group (ie, mice receiving only the drug vehicle) reached maximum levels, the blood, tissues, and organs of each mouse from the control and VNI-treated groups were collected to determine the presence of T. cruzi by real-time PCR analysis, as described below. Three in vivo independent experiments were performed using the same protocol and provided similar results. Animal studies were conducted under the National Institutes of Health guidelines on the humane use and care of laboratory animals for biomedical research (Guide for the Care and Use of Laboratory Animals. NIH Publication No.85-23. Revised 1985). Protocols were approved by the Meharry Medical College Institutional Animal Care and Use Committee.
Figure 3.
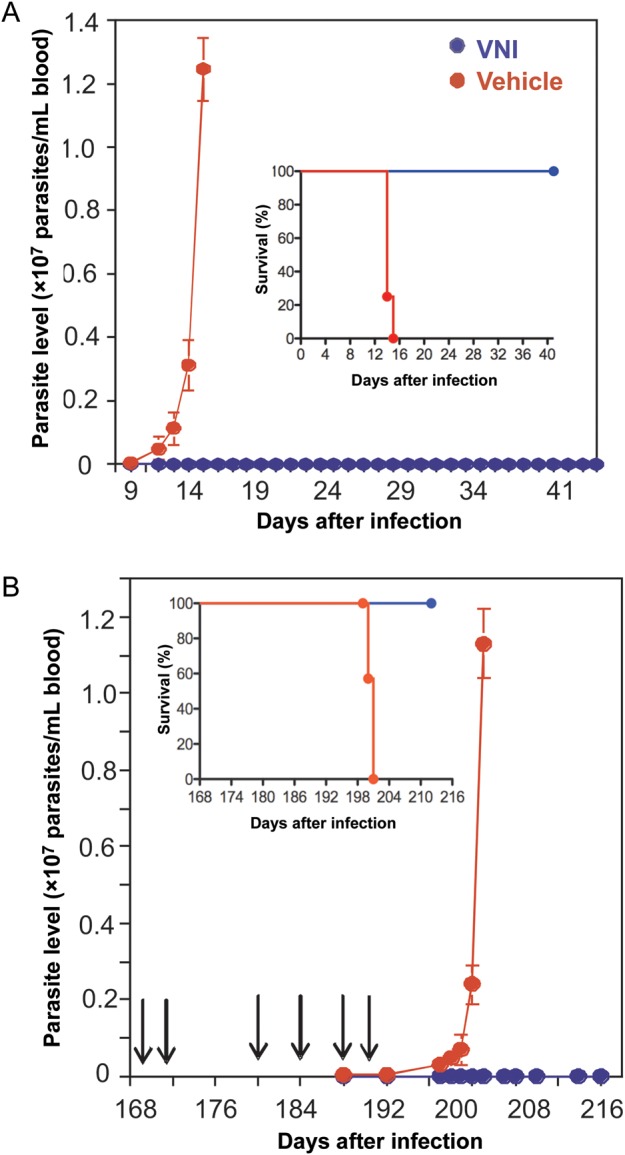
VNI clears Trypanosoma cruzi infection in vivo. A, Acute model of Chagas disease. Mice (5 per group) were intraperitoneally infected with a lethal dose of T. cruzi trypomastigotes (1 × 105 organisms), followed by oral treatment, 1 day after infection, with either VNI (25 mg/kg) or with drug vehicle twice per day for 30 days. Inset, Kaplan-Meier survival plot. B, Chronic model of Chagas disease. Mice (9 per group) were intraperitoneally infected with T. cruzi blood trypomastigotes (50 organisms) to induce chronicity. Ninety days after infection, oral treatment with VNI (25 mg/kg) or drug vehicle was administered twice daily for 30 days, after which mice were immunosuppressed by 6 injections of cyclophosphamide (CY; arrows) to reactivate infection. Inset, Kaplan-Meier survival plot.
Real-Time PCR for Detection of Tissue and Blood Parasites
Genomic DNA was purified from the collected organs, tissues, and blood. Parasite genomic DNA was purified from epimastigotes. A standard curve in the range of 50 pg to 50 ng for the detection of T. cruzi DNA by real-time PCR was developed using the T. cruzi 195-bp repeat satellite DNA–specific primers (GenBank accession no. AY520044) that amplified a T. cruzi 188-bp sequence, using the primers TCZ1 5′-CGA GCT CTT GCC CAC ACG GGT GCT-3′cand TCZ2 5′-CCT CCA AGC AGC GGA TAG TTC AGG-3′ [27, 31]. The PCR reactions were performed as previously described [32, 33], with minor modifications to detect the presence of parasite DNA in the genomic DNA purified from animal tissues. Samples from each mouse group were tested in triplicate. The mean of mice samples per group and their standard errors are presented.
Pharmacokinetics
The pharmacokinetics analysis was based on the VNI-specific absorption maximum (291 nm; ∑291 = 36 mM−1 cm−1) and quantification, using 10 μM ketoconazole as an internal standard (∑259 = 6 mM−1 cm−1). VNI was orally administered to a group of 6 BALB/c mice in a single dose (25 mg/kg). Blood samples were collected over time from the mouse tail, using a StatSampler blood collector (Iris Sample Processing, Westwood, MA) as described by the manufacturer, and plasma was collected by centrifugation at 500 g for 5 minutes. VNI was extracted from plasma, using the procedure described by Kraus et al [34]. The samples were centrifuged for 10 minutes at 12 000 g, and the supernatant was dried and then dissolved in a 1:1 ratio of acetonitrile to water. The VNI concentration in plasma was monitored using reverse-phase high-performance liquid chromatography equipped with the dual-wavelength UV 2489 detector (Waters) set at 291 and 259 nm and a Symmetry C18 (3.5 μm) 4.6 × 75-mm column. The injection volume was 100 μL. The mobile phase was 56% 0.01 M potassium phosphate (pH 7.4) and 44% acetonitrile (vol/vol), with an isocratic flow rate of 1.0 mL/minutes. VNI and ketoconazole were eluted after 6.5 and 3.9 minutes, respectively. This procedure afforded the detection limit of 0.4 μM (0.2 μg/mL) VNI in 10 μL of mouse plasma.
Drugs and Analytical Methods
Synthesis of VNI and its enantiomer as a racemic mixture, followed by chiral separation of R and S enantiomers, was performed as described elsewhere [22]. We recently developed a short, enantioselective, gram-scale synthesis of VNI [35]. Posaconazole and ravuconazole were purchased from Santa Cruz Biotechnology. Reconstitution of CYP51 activity was performed as previously reported [36]. The influence of VNI, ketoconazole, posaconazole, and ravuconazole on the activity of CYP3A4 was compared using a BD Biosciences screening kit, in accordance with the manufacturer instructions; fluorescence was measured using the Molecular Devices SpectraMax M5 fluorometer. A standard high-throughput screening thallium assay for detection of human ether-a-go-go–related gene (hERG) activity was performed at the Vanderbilt University High-Throughput Screening Facility to test for the possible influence of VNI on hERG channel (Supplementary Figure S1).
RESULTS AND DISCUSSION
Antiparasitic Effect of VNI in T. cruzi Amastigotes
T. cruzi has a complex life cycle that includes insect and mammalian stages. During the initial, acute phase of infection, the nonreplicative bloodstream trypomastigotes invade different mammalian cell types, where they transform into replicative intracellular amastigotes and multiply within the cytoplasm. Cellular experiments (Figure 2) showed that the potency of VNI in eradicating T. cruzi amastigotes from infected cardiomyocytes was higher than the potencies of either posaconazole or ravuconazole (50% maximal effective concentration [EC50], 1.3 nM vs 5 nM). Since the EC50 of VNI for mammalian cells (HL60) is >50 μM, this gives a selectivity index of >50 000, which exceeds the minimal selectivity requirement [37] of at least 2500-fold. A 1-nM concentration represented a 35% maximal effective concentration for VNI and a 15% maximal concentration for posaconazole, whereas 1-nM concentration of ravuconazole displayed no inhibitory effect. A 7.5-nM solution of VNI cleared T. cruzi infection in cardiomyocytes, whereas posaconazole and ravuconazole concentrations of >30 nM were required to reach the same effect.
Figure 2.

VNI is more effective than posaconazole and ravuconazole in clearing Trypanosoma cruzi–infected cardiomyocytes. A, Comparative dose-dependent antiparasitic effects. Cardiomyocyte monolayers were infected with green fluorescent protein–expressing trypomastigotes for 24 hours and then treated with different concentrations of VNI, posaconazole (POS), ravuconazole (RAV), or dimethyl sulfoxide (DMSO). Parasite multiplication within monolayers was estimated by green fluorescent protein fluorescence (expressed in relative fluorescence units [RFU]) 72 hours after infection. Data represent the mean values ± SD of the results from triplicate samples. B, Fluorescence microscopic observation of T. cruzi multiplication inside cardiomyocytes treated with 7.5 nM of VNI, posaconazole, and ravuconazole 72 hours after infection. T. cruzi amastigotes are green, cardiomyocyte nuclei are blue, and cardiomyocyte actin myofibrils are red.
Curative Effect of VNI in the Acute and Chronic Stage of Chagas Disease in the Murine Model
These promising in vitro results encouraged us to start animal treatment using a relatively low oral dose of VNI (25 mg/kg); the reported doses for oral administration of posaconazole in the murine models ranges from 20 to 300 mg/kg [38–41]. In the acute model, the parasitological clearance was 100%, with 100% survival (Figure 3A), and there were no detectable toxic side effects. After VNI treatment, mice did not lose weight and appeared normal. In the chronic model, treatment with VNI started 90 days after infection, lasted for 30 days, and then was followed by 6 rounds of immunosuppressive therapy (Figure 3B). After immunosuppression, all untreated animals had high levels of parasitemia, whereas all VNI-treated mice were free of parasites and survived. In both the acute and chronic models of the disease, qPCR analysis confirmed parasitological clearance: blood and all tested tissues of the VNI treated animals were free of T. cruzi (Table 1).
Table 1.
Results of Real-Time Polymerase Chain Reaction for Detection of Trypanosoma cruzi Organs, Tissues, and Blood of Mice
| Infection Model, Mouse Group | Heart | Muscle | Liver | Spleen | Blood |
|---|---|---|---|---|---|
| Acute | |||||
| T. cruzi infected + vehicle | 5/5 (12.40 ± 0.13) | 5/5 (12.71 ± 0.57) | 5/5 (16.26 ± 0.18) | 5/5 (12.86 ± 0.20) | 5/5 (14.91 ± 0.90) |
| T. cruzi infected + VNI | 0/5 (28.52 ± 0.53) | 0/5 (28.82 ± 0.96) | 0/5 (28.44 ± 0.71) | 0/5 (28.82 ± 0.42) | 0/5 (29.52 ± 0.44) |
| Uninfected | 0/5 (28.63 ± 0.13) | 0/5 (28.56 ± 0.20) | 0/5 (28.74 ± 0.57) | 0/5 (29.36 ± 0.11) | 0/5 (28.74 ± 0.10) |
| Chronic | |||||
| T. cruzi infected + vehicle | 9/9 (16.64 ± 0.83) | 9/9 (12.90 ± 0.54) | 9/9 (15.02 ± 0.27) | 9/9 (17.33 ± 1.09) | 9/9 (14.59 ± 0.78) |
| T. cruzi infected + VNI | 0/9 (31.98 ± 0.81) | 0/9 (28.12 ± 0.84) | 0/9 (30.34 ± 1.74) | 0/9 (31.62 ± 0.11) | 0/9 (30.31 ± 1.62) |
| Uninfected | 0/9 (28.34 ± 0.66) | 0/9 (28.44 ± 0.70) | 0/9 (29.90 ± 0.55) | 0/9 (29.69 ± 0.61) | 0/9 (30.12 ± 0.57) |
Data are no. of T. cruzi–positive mice/no. in group (mean [±standard error] cycle threshold for parasite detection).
VNI Pharmacokinetics
The curative efficiency of VNI may be enhanced by its promising pharmacokinetic properties (Figure 4). When administered as a single dose by oral gavage (25 mg/kg, which corresponds to the dose used for treatment), VNI reached peak plasma levels of about 40 μM. This exceeded maximal concentrations reported for orally administered posaconazole by 2–9-fold, depending on the experimental details and formulations [38–41], whereas the reported plasma concentrations of ravuconazole did not exceed 4 μM [42–44]. As a starting point, the initial PK of VNI in mice meets perfectly the definition of an ideal target product profile [37]: it is orally available, remains in mouse plasma for >8 hours, reaches the plasma level that exceeds the 99% maximal effective concentration for T. cruzi within cardiomyocytes by 5000-fold, and has a selectivity index for mammalian(HL60)/protozoan(T. cruzi) cells of >50 000. We surmise that, after proceeding to clinical trials, the VNI oral bioavailability may be further improved by using alternative formulations, such as cyclodextrin solutions, which were proven to increase the bioavailability of posaconazole [40] and other azole compounds [34].
Figure 4.

Single-oral-dose profile of the pharmacokinetics of VNI in mouse plasma. VNI was administered by oral gavage at 25 mg/kg. Data represent mean values (±SD) from 6 mice per point. Inset, Log scale for VNI concentration. The arrow shows a possible point of separation between zero- and first-order kinetics.
To summarize, VNI cures the acute and chronic forms of Chagas disease in mice, with 100% survival and no observable side effects. Low cost (<$0.10/mg; [35]), oral bioavailability, favorable pharmacokinetics, and low toxicity make this compound an exceptional candidate for clinical trials. The efficacy of VNI provides additional compelling support for efficacious antiparasitic treatment of chronic Chagas disease, further validating CYP51 as a viable drug targeting T. cruzi, and it opens a new opportunity for therapeutic cure of patients. Although widespread searches for other new drugs that target T. cruzi are surely being pursued, there are millions of patients with this debilitating illness who need immediate therapy, and VNI or a derivative might fulfill this need.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the VICB Synthesis Core (Vanderbilt University), for synthesizing a portion of the VNI used to initiate this study; V. Furtak and C. Taylor, for technical assistance; and M. F. Lima (Meharry Medical College), for reading the manuscript.
Financial support. The work was supported by National Institutes of Health grant GM067871 (GIL, MRW), Vanderbilt Institute of Chemical Biology (VICB) Pilot project grant 2011 (GIL), and in part by GM084333 (JNJ), AI080580 and U54 MD007593 (FV), AI083925 (PNN) and AI007281 (CAJ).
Potential conflict of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Kirchhoff LV. Epidemiology of American trypanosomiasis (Chagas disease) Adv Parasitol. 2011;75:1–18. doi: 10.1016/B978-0-12-385863-4.00001-0. [DOI] [PubMed] [Google Scholar]
- 2.Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas’ disease in the United States. Clin Microb Rev. 2011;24:655–81. doi: 10.1128/CMR.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bern C. Antitrypanosomal therapy for chronic chagas’ disease. N Eng J Med. 2011;364:2527–34. doi: 10.1056/NEJMct1014204. [DOI] [PubMed] [Google Scholar]
- 4.Leslie M. Drug developers finally take aim at a neglected disease. Science. 2011;333:933–5. doi: 10.1126/science.333.6045.933. [DOI] [PubMed] [Google Scholar]
- 5.Yadon ZE, Schmunis GA. Congenital Chagas disease: estimating the potential risk in the United States. Am J Trop Med Hyg. 2009;81:927–33. doi: 10.4269/ajtmh.2009.09-0257. [DOI] [PubMed] [Google Scholar]
- 6.Clayton J. Chagas disease: pushing through the pipeline. Nature. 2010;465:S12–5. doi: 10.1038/nature09224. [DOI] [PubMed] [Google Scholar]
- 7.Urbina JA. Ergosterol biosynthesis and drug development for Chagas disease. Mem Inst Oswaldo Cruz. 2009;104(Suppl 1):311–8. doi: 10.1590/s0074-02762009000900041. [DOI] [PubMed] [Google Scholar]
- 8.Tarleton RL, Zhang L, Downs MO. “Autoimmune rejection” of neonatal heart transplants in experimental Chagas disease is a parasite-specific response to infected host tissue. Proc Natl Acad Sci U S A. 1997;94:3932–7. doi: 10.1073/pnas.94.8.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lepesheva GI, Villalta F, Waterman MR. Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51) Adv Parasitol. 2011;75:65–87. doi: 10.1016/B978-0-12-385863-4.00004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maertens JA. History of the development of azole derivatives. Clin Microbiol Infect. 2004;10:1–10. doi: 10.1111/j.1470-9465.2004.00841.x. [DOI] [PubMed] [Google Scholar]
- 11.Lepesheva GI, Waterman MR. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr Top Med Chem. 2011;11:2060–71. doi: 10.2174/156802611796575902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urbina JA, Lira R, Visbal G, Bartroli J. In vitro antiproliferative effects and mechanism of action of the new triazole derivative UR-9825 against the protozoan parasite Trypanosoma (Schizotrypanum) cruzi. Antimicrob Agents Chemother. 2000;44:2498–502. doi: 10.1128/aac.44.9.2498-2502.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urbina JA, Payares G, Sanoja C, Lira R, Romanha AJ. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int J Antimicrob Agents. 2003;21:27–38. doi: 10.1016/s0924-8579(02)00273-x. [DOI] [PubMed] [Google Scholar]
- 14.Molina J, Martins-Filho O, Brener Z, Romanha AJ, Loebenberg D, Urbina JA. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob Agents Chemother. 2000;44:150–5. doi: 10.1128/aac.44.1.150-155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diniz Lde F, Caldas IS, Guedes PM, et al. Effects of ravuconazole treatment on parasite load and immune response in dogs experimentally infected with Trypanosoma cruzi. Antimicrob Agents Chemother. 2010;54:2979–86. doi: 10.1128/AAC.01742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinazo MJ, Espinosa G, Gallego M, Lopez-Chejade PL, Urbina JA, Gascon J. Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am J Trop Med Hyg. 2010;82:583–7. doi: 10.4269/ajtmh.2010.09-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buckner FS, Urbina JA. Recent developments in sterol 14-demethylase inhibitors for Chagas disease. Int J Parasitol Drugs Drug Resist. 2012 Dec;2:236–242. [DOI] [PMC free article] [PubMed]
- 18.Schiller DS, Fung HB. Posaconazole: an extended-spectrum triazole antifungal agent. Clin Ther. 2007;29:1862–86. doi: 10.1016/j.clinthera.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Lepesheva GI, Ott RD, Hargrove TY, et al. Sterol 14 alpha-demethylase as a potential target for antitrypanosomal therapy: Enzyme inhibition and parasite cell growth. Chem Biol. 2007;14:1283–93. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lepesheva GI, Hargrove TY, Anderson S, et al. Structural insights into inhibition of sterol 14 alpha-demethylase in the human pathogen Trypanosoma cruzi. J Biol Chem. 2010;285:25582–90. doi: 10.1074/jbc.M110.133215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lepesheva GI, Park HW, Hargrove TY, et al. Crystal structures of Trypanosoma brucei sterol 14 alpha-demethylase and implications for selective treatment of human infections. J Biol Chem. 2010;285:1773–80. doi: 10.1074/jbc.M109.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hargrove TY, Kim K, de Nazaré Correia Soeiro M, et al. CYP51 structures and structure-based development of novel, pathogen-specific inhibitory scaffolds. Int J Parasitol Drugs Drug Resist. 2012;2:178–86. doi: 10.1016/j.ijpddr.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lima MF, Villalta F. Trypanosoma cruzi trypomastigote clones differentially express a parasite cell adhesion molecule. Mol Biochem Parasitol. 1989;33:159–70. doi: 10.1016/0166-6851(89)90030-3. [DOI] [PubMed] [Google Scholar]
- 24.Augustine SAJ, Kleshchenko YY, Nde PN, et al. Molecular cloning of a trypanosoma cruzi cell surface casein kinase II substrate, Tc-1, involved in cellular infection. Infect Immun. 2006;74:3922–3929. doi: 10.1128/IAI.00045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villalta F, Kierszenbaum F. Immunization against a challenge with insect vector, metacyclic forms of Trypanosoma cruzi simulating a natural infection. Am J Trop Med Hyg. 1983;32:273–6. doi: 10.4269/ajtmh.1983.32.273. [DOI] [PubMed] [Google Scholar]
- 26.Hayes MM, Kierszenbaum F. Experimental Chagas’ disease: kinetics of lymphocyte responses and immunological control of the transition from acute to chronic Trypanosoma cruzi infection. Infect Immun. 1981;31:1117–24. doi: 10.1128/iai.31.3.1117-1124.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Combs TP, Nagajyothi , Mukherjee S, et al. The adipocyte as an important target cell for Trypanosoma cruzi infection. J Biol Chem. 2005;280:24085–94. doi: 10.1074/jbc.M412802200. [DOI] [PubMed] [Google Scholar]
- 28.Cencig S, Coltel N, Truyens C, Carlier Y. Parasitic loads in tissues of mice infected with Trypanosoma cruzi and treated with AmBisome. PLoS Negl Trop Dis. 2011;5:e1216. doi: 10.1371/journal.pntd.0001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knubel CP, Martínez FF, Fretes RE, et al. Indoleamine 2,3-dioxigenase (IDO) is critical for host resistance against Trypanosoma cruzi. FASEB J. 2010;24:2689–701. doi: 10.1096/fj.09-150920. [DOI] [PubMed] [Google Scholar]
- 30.Villalta F, Smith CM, Burns JM, Chaudhuri G, Lima MF. Fab’ fragments of a mAb to a member of family 2 of trans-sialidases of Trypanosoma cruzi block Trypanosome invasion of host cells and neutralize infection by passive immunizationa. An N Y Acad Sci. 1996;797:242–5. doi: 10.1111/j.1749-6632.1996.tb52965.x. [DOI] [PubMed] [Google Scholar]
- 31.Williams JT, Mubiru JN, Schlabritz-Loutsevitch NE, et al. Polymerase chain reaction detection of Trypanosoma cruzi in Macaca fascicularis using archived tissues. Am J Trop Med Hyg. 2009;81:228–34. [PMC free article] [PubMed] [Google Scholar]
- 32.Nde PN, Simmons KJ, Kleshchenko YY, Pratap S, Lima MF, Villalta F. Silencing of the laminin γ-1 gene blocks Trypanosoma cruzi Infection. Infect Immun. 2006;74:1643–8. doi: 10.1128/IAI.74.3.1643-1648.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simmons KJ, Nde PN, Kleshchenko YY, Lima MF, Villalta F. Stable RNA interference of host thrombospondin-1 blocks Trypanosoma cruzi infection. FEBS Letters. 2006;580:2365–70. doi: 10.1016/j.febslet.2006.03.054. [DOI] [PubMed] [Google Scholar]
- 34.Kraus JM, Verlinde CL, Karimi M, Lepesheva GI, Gelb MH, Buckner FS. Rational modification of a candidate cancer drug for use against Chagas disease. J Med Chem. 2009;52:1639–47. doi: 10.1021/jm801313t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobish MC, Villalta F, Waterman MR, Lepesheva GI, Johnston JN. Organocatalytic, enantioselective synthesis of VNI: a robust therapeutic development platform for Chagas, a neglected tropical disease. Org Lett. 2012;14:6322–6325. doi: 10.1021/ol303092v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lepesheva GI, Zaitseva NG, Nes WD, et al. CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B’ helix defines substrate preferences of sterol 14alpha-demethylase. J Biol Chem. 2006;281:3577–85. doi: 10.1074/jbc.M510317200. [DOI] [PubMed] [Google Scholar]
- 37.Buckner FS. Experimental chemotherapy and approaches to drug discovery for Trypanosoma cruzi infection. Adv Parasitol. 2011;75:89–119. doi: 10.1016/B978-0-12-385863-4.00005-8. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez MM, Pastor FJ, Calvo E, Salas V, Sutton DA, Guarro J. Correlation of in vitro activity, serum levels, and in vivo efficacy of posaconazole against Rhizopus microsporus in a murine disseminated infection. Antimicrob Agents Chemother. 2009;53:5022–5. doi: 10.1128/AAC.01026-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Theuretzbacher U, Clancy CJ, Nguyen MH, Derendorf H. Pharmacokinetic/pharma-codynamic profile of posaconazole. Clin Pharmacokin. 2010;49:379–96. doi: 10.2165/11319340-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 40.Nomeir AA, Kumari P, Hilbert MJ, et al. Pharmacokinetics of SCH 56592, a new azole broad-spectrum antifungal agent, in mice, rats, rabbits, dogs, and cynomolgus monkeys. Antimicrob Agents Chemother. 2000;44:727–31. doi: 10.1128/aac.44.3.727-731.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calvo E, Pastor FJ, Rodríguez MM, Mayayo E, Salas V, Guarro J. Murine model of a disseminated infection by the novel fungus fonsecaea monophora and successful treatment with posaconazole. Antimicrob Agents Chemother. 2010;54:919–23. doi: 10.1128/AAC.01284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueda Y, Matiskella JD, Golik J, et al. Phosphonooxymethyl prodrugs of the broad spectrum antifungal azole, ravuconazole: Synthesis and biological properties. Bioorg Med Chem Lett. 2003;13:3669–72. doi: 10.1016/j.bmcl.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 43.Hata K, Kimura J, Miki H, Toyosawa T, Nakamura T, Katsu K. In vitro and in vivo antifungal activities of ER-30346, a novel oral triazole with a broad antifungal spectrum. Antimicrob Agents Chemother. 1996;40:2237–42. doi: 10.1128/aac.40.10.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andes D, Marchillo K, Stamstad T, Conklin R. In vivo pharmacodynamics of a new triazole, ravuconazole, in a murine candidiasis model. Antimicrob Agents Chemother. 2003;47:1193–9. doi: 10.1128/AAC.47.4.1193-1199.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guengerich FP. Human cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: structure, mechanism, and biochemistry. New York: Plenum Publishing; 2005. pp. 377–530. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.