

Abstract
African sleeping sickness, caused by the protozoan parasite Trypanosoma brucei, is universally fatal if untreated, and current drugs are limited by severe toxicities and difficult administration. New antitrypanosomals are greatly needed. Heat shock protein 90 (Hsp90) is a conserved and ubiquitously expressed molecular chaperone essential for stress responses and cellular signaling. We investigated Hsp90 inhibitors for their antitrypanosomal activity. Geldanamycin and radicicol had nanomolar potency in vitro against bloodstream-form T. brucei; novobiocin had micromolar activity. In structure-activity studies of geldanamycin analogs, 17-AAG and 17-DMAG were most selective against T. brucei as compared to mammalian cells. 17-AAG treatment sensitized trypanosomes to heat shock and caused severe morphological abnormalities and cell cycle disruption. Both oral and parenteral 17-DMAG cured mice of a normally lethal infection of T. brucei. These promising results support the use of inhibitors to study Hsp90 function in trypanosomes and to expand current clinical development of Hsp90 inhibitors to include T. brucei.
Keywords: Trypanosoma brucei, human African trypanosomiasis, Hsp90, inhibitor, drug, kinetoplast, cell cycle, geldanamycin, 17-AAG, 17-DMAG
African sleeping sickness (human African trypanosomiasis; HAT) is caused by the protozoan kinetoplastid parasite Trypanosoma brucei [1]. Kinetoplastids, including the human pathogens Trypanosoma cruzi and Leishmania species, are early diverging eukaryotes named for the kinetoplast, the characteristic dense granule of DNA at the base of the flagellum that comprises the mitochondrial genome. T. brucei is transmitted by the tsetse fly and causes endemic disease in both livestock (nagana) and humans (HAT) in sub-Saharan Africa. The parasite propagates extracellularly and moves throughout the body, including the central nervous system during the late stage of disease. In humans, this infection is fatal unless treated. Current therapies require repeated parenteral dosing and have formidable toxicities, exemplified by the 5%–10% lethality caused by the commonly used drug melarsoprol. New antitrypanosomal drugs are long overdue.
Heat shock protein 90 (Hsp90) is a phylogenetically conserved, abundant, and essential molecular chaperone [2–7]. In mammalian cells it functions as a homodimer complexed with regulatory cochaperones. Hsp90 stabilizes substrate (ie, client) proteins, enabling their proper activities. Over 200 clients have been identified, with many acting in signal transduction pathways, stress responses, and cell cycle regulation [4]. Thus, Hsp90 is a critical node coordinating many networks necessary for survival. This combinatorial function and a disproportionate dependence on Hsp90 activity in malignant cells have established it as a cancer chemotherapy target [5–8]. Dozens of Hsp90 inhibitors have entered the drug development pathway, including derivatives of the natural products geldanamycin (eg, 17-AAG [17-N-allylamino-17-demothoxygeldanamycin] and 17-DMAG [17-dimethylaminoethylamino-17-demethoxygeldanamycin]) and radicicol, as well as novel structural scaffolds identified in screens. 17-AAG has been in phase 3 trials [6]. Geldanamycin treatment leads to proteosomal degradation of Hsp90 client proteins, cell cycle arrest, and heat shock response in mammalian cells [9]. Damage to multiple proteins and pathways may be the basis for reported synergism between Hsp90 inhibitors and several chemotherapeutic agents [10].
The kinetoplastid homologue of Hsp90 is Hsp83, and there are 10 tandem copies in the T. brucei genome [11]. Hsp83 influences the temperature-sensitive differentiation of insect to mammalian-form Leishmania organisms and T. cruzi [12, 13], and Hsp90 inhibitors arrest the growth of several kinetoplastids in vitro and have activity against Trypanosoma evansi in mice [12–16]. However, little is known about the role of Hsp83 in cellular metabolism and its clients and cochaperones remain to be identified. Initial studies have found that Hsp83 interacts with the deacetylase Sir2 in Leishmania organisms and with protein phosphatase 5 in T. brucei [15, 17]. Although the trypanosome genome lacks evidence of steroid hormone receptors and tyrosine kinases that are prominent Hsp90 clients in mammalian cells, homologues of other clients are recognizable, including serine/threonine kinases and cell cycle regulators [18, 19].
The cell cycle of kinetoplastids differs dramatically from that of host cells. In trypanosomes, the replication of mitochondrial DNA (the kinetoplast) occurs once, and its S, G2, and M phases precede those of nuclear DNA (Figure 1A) [20–22]. The Golgi, basal bodies, and flagellum are also replicated in an ordered fashion and then precisely positioned so cytokinesis can occur via cleavage furrow ingression. The complex temporal requirements of these processes, coupled with a lack of tight checkpoints characteristic in higher eukaryotes [23], make trypanosomes particularly vulnerable to cell cycle disaster [24].
Figure 1.

The cell cycle of Trypanosoma brucei is disrupted by 17-AAG. A, In trypanosomes both the nuclear and the mitochondrial genomes can be visualized by light microscopy, and their replication is temporally linked but not strictly synchronous. Cells progress from having 1 nucleus and kinetoplast (1N1K), to having 1 nucleus and 2 kinetoplasts (1N2K), and finally to having 2 nuclei and kinetoplasts (2N2K) before the cells divide by cleavage furrow ingression to yield 2 daughter cells (adapted from [20, 22]). G1, gap 1; SK/N, synthesis kinetoplast/nuclear DNA; G2, gap 2; D, kinetoplast division; M, nuclear mitosis; P, positioning of organelles, C, cytokinesis. B, Trypanosomes were treated with solvent, 50 nM 17-AAG, or 100 nM 17-AAG for the indicated times and then examined by flow cytometry. Since the kinetoplast composes only 5% of the total cellular DNA [32], this method focuses on the nuclear genome. DMSO, dimethyl sulfoxide; 2C, nonreplicating diploid cells with 2 copies of nuclear genomic DNA; 4C, cells late in replication with 4 copies of genomic DNA; 6C or 8C, abnormal cells with 6 or 8 copies of the nuclear genome.
We evaluated structurally diverse Hsp90 inhibitors, including some already studied in humans, for their activity against bloodstream-form trypanosomes in vitro, explored structure-activity relationships for the geldanamycin scaffold, assessed the effect of inhibitors on the cell cycle, tested 17-AAG in combination with other drugs, and demonstrated the efficacy of orally dosed 17-DMAG against T. brucei infection in mice.
MATERIALS AND METHODS
Cell Culture, Reagents, and Cytotoxicity Assay
All studies used bloodstream T. brucei brucei (MiTat 1.2 strain 427) maintained continuously in exponential growth (104–105 cells/mL) at 37°C and 5% CO2 in phenol red–free HMI-9, 10% fetal bovine serum (Invitrogen), and 10% Serum Plus (SAFC Biosciences). Motile parasites were counted by a hemocytometer. L1210 murine leukemia cells (ATCC CCL-219) were maintained in phenol red–free Roswell Park Memorial Institute 1640 medium (Sigma–Aldrich) and 15% fetal bovine serum. Stock solutions of compounds were aliquoted and stored at −20°C, as follows: radicicol (Sigma–Aldrich) and ansamycins (including geldanamycin, 17-AAG, and 17-DMAG; The NCI/DTP Open Chemical Repository [available at: http://dtp.cancer.gov]) in sterile dimethyl sulfoxide (DMSO; Hybri-Max, Sigma–Aldrich); novobiocin (Sigma–Aldrich), eflornithine (NCI/DTP), and pentamidine (Lymphomed) in H2O; and melarsoprol (Centers for Disease Control and Prevention) in 1,2-propanediol (Sigma–Aldrich). Final DMSO percentage of ≤0.5% had no effect in the cytotoxicity assay. Cytotoxicity was assayed by a colorimetric 96-well plate acid-phosphatase method [25]. Cells were exposed to compound for 24 hours (T. brucei seeding concentration, 1 × 105 cells/mL) or 48 hours (L1210 seeding concentration, 7 × 104 cells/mL), reflecting respective doubling-times of 6 and 11 hours. Dose-response curves and half-maximal effective concentrations (EC50 values) were obtained (Microsoft Excel and DeltaGraph Pro v3.5).
Flow Cytometry
For each time point, 3 × 106 cells from treated cultures (seeded at 2 × 105 cells/mL) were pelleted at 1000 × g for 10 minutes; washed twice with glucose- and sucrose-supplemented phosphate-buffered saline (vPBS) [26]; fixed by dropwise addition of ice-cold 70% EtOH in PBS; and stored at 4°C. Cells were pelleted at 3000 × g for 10 minutes, washed with PBS, resuspended in 500 µL 10 μg/mL RNaseA in PBS, and incubated at 37°C for 45 minutes. After addition of propidium iodide (Sigma–Aldrich) to achieve a final concentration of 20 μg/mL, 10 000 cells were analyzed on FACSCalibur (BD Biosciences), gated on FL2-A versus FL2-W to exclude doublets, and the G1 peak of controls was centered at 200 units FL2-A (CellQuest Pro v5.2, BD Biosciences).
DAPI Staining and Microscopy
For each time point, 2 × 106 cells from treated cultures seeded at 2 × 105 cells/mL were pelleted at 1000 × g for 10 minutes, washed with vPBS, fixed with 3% paraformaldehyde in PBS for 10 minutes on ice, diluted 5-fold in vPBS, pelleted at 3000 × g for 10 minutes, washed with vPBS, applied to polylysine-coated slides for 1 hour, permeabilized with 0.1% Triton X-100 (Sigma–Aldrich) in PBS for 10 minutes, washed thrice with PBS, and mounted using Fluoroshield with DAPI (Sigma–Aldrich) [26]. Slides were imaged on a Zeiss Axioskop with a Retiga Exi charge-coupled-device camera (QImaging), using iVision v4.0.13 (Biovision Technologies). Images were merged and adjusted for brightness and contrast only (ImageJ v1.45s, Rasband). For quantitation, images of ≥100 cells/condition were randomized then scored by 3 blinded independent observers with respect to the number of nuclei and kinetoplasts and the appearance by fluorescence and/or phase.
Animal Studies
Protocols were approved by the Johns Hopkins Institutional Animal Care and Use Committee. Drug solutions were prepared immediately before use in 5% glucose vehicle. Six-week-old female CD1 mice were infected with T. brucei (MiTat 1.2 strain 427; 5 × 104) intraperitoneally on day 0. On day 1, after confirmation of parasitemia, animals were divided into groups of 3 (groups 1–4) or 4 (group 5) and then treated once daily with vehicle (200 µL intraperitoneally for 3 days; group 1), 3.5 mg/kg Berenil (Sigma–Aldrich; 200 µL intraperitoneally for 3 days; group 2), 150 mg/kg 17-DMAG (400 µL intraperitoneally once; group 3), 30 mg/kg 17-DMAG (200 µL intraperitoneally for 5 days; group 4), or 50 mg/kg 17-DMAG (200 µL by oral gavage for 5 days; group 5). Mice were weighed prior to infection and daily during the first week; parasitemia was monitored in tail snip blood samples for 30 days. Mice with >5 × 108 trypanosomes/mL or in evident distress were euthanized.
RESULTS
In Vitro Antitrypanosomal Activity of Parent Compounds
Initial studies were designed to determine whether structurally diverse Hsp90 inhibitors had cytotoxic activity against African trypanosomes in vitro. Geldanamycin and radicicol both bind to the N-terminal adenosine triphosphate (ATP)–binding pocket, which has a rare Bergerat-fold, of Hsp90 and inhibit ATPase activity [27, 28]. Novobiocin binds the C-terminal domain [29]. Geldanamycin and radicicol had potent nanomolar EC50 values against T. brucei (Table 1), in agreement with reported 50% growth inhibition at 40 nM geldanamycin [15]. Novobiocin had micromolar activity. For comparison, clinically used antitrypanosomals pentamidine, melarsoprol, and eflornithine had EC50 values of 7 nM, 10 nM, and 50 μM, respectively, in this assay. Geldanamycin and radicicol values fall well within this range.
Table 1.
Activity of Heat Shock Protein 90 Inhibitors Against Trypanosomes In Vitro
| Compound | EC50 (nM)a |
|---|---|
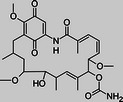
Geldanamycin |
12 ± 4 |
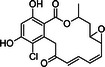
Radicicol |
70 ± 14 |
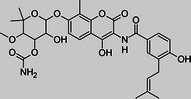
Novobiocin |
180 000 ± 21 000 |
Abbreviation: EC50, half maximal effective concentration
a Values are mean ± SD of at least 3 independent experiments. r2 values for dose-response curves all exceeded 0.99; within an assay, the coefficients of variation for quadruplicate determinations were all ≤10% and averaged 1.8%.
Structure Activity Relationships on the Ansamycin Scaffold
On the basis of the impressive potency of geldanamycin, 12 related ansamycins were analyzed for structure-activity relationships (Tables 2 and 3). For some measure of therapeutic index, compounds were also evaluated against L1210 mammalian cells. Disruption of the geldanamycin quinone (compound 255108; Table 2) reduced activity severely against trypanosomes and moderately against L1210. Sterically demanding hydrazone moieties at R4 (210753, 255112, and 265482) substantially decreased activity against both cell types. Modifications at C17 were well tolerated. Replacement of the methoxy at R1 with a hydroxyl group (255104) decreased potency by 10-fold against both cell types, but incorporation of a chloroethylamino group (320877) had little to no effect on potency. Importantly, however, other amino moieties at R1 enhanced selective toxicity against trypanosomes: a primary amine (255109) or allyl amino group (330507) resulted in a 30–50-fold increase in selectivity, and a dimethylaminoethylamino group (707545) increased selectivity 300-fold. An even more substantial change, a large, rigid bromo-benzoxazine bridging positions R1 and R2 (255105), maintained good activity against both cell types, suggesting that this position is not constrained in the drug and target interaction. Modifications in more remote ansamycin ring substituents and reduction of the quinone (330500; Table 3) impaired potency and selectivity, while opening the macrocycle (265481) resulted in complete loss of activity.
Table 2.
Structure Activity Relationships of Ansamycins
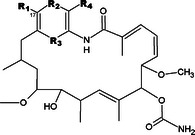 | |||||||
|---|---|---|---|---|---|---|---|
| Ansamycin Scaffold | |||||||
| NSC Number | Modification |
EC50 (nM)a |
|||||
| R1 | R2 | R3 | R4 | T. brucei | L1210 | Selectivityb | |
| 122750 Geldanamycin | OCH3 | CO | CO | H | 13 | 12 | 1 |
| 255108 | OCH3 | H | 38 000 | 7100 | 0.2c | ||
| 210753 | OCH3 | CO | CO | 2800 | 940 | 0.3c | |
| 255112 | OCH3 | CO | CO | 13 000 | 9600 | 0.7 | |
| 265482 | OCH3 | CO | CO |
|
3400 | 5800 | 2 |
| 255104 | OH | CO | CO | H | 130 | 180 | 1 |
| 320877 | CO | CO | H | 17 | 21 | 1 | |
| 255109 | NH2 | CO | CO | H | 6.0 | 180 | 30c |
| 330507 17-AAG | CO | CO | H | 38 | 1800 | 50c | |
| 707545 17-DMAG | CO | CO | H | 3.1 | 1000 | 300c | |
| 255105 | 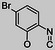 |
CO | H | 27 | 23 | 0.9 | |
Abbreviations: EC50, half maximal effective concentration; T. brucei, Trypanosoma brucei.
a Values are mean of at least 3 independent determinations. Coefficients of variation for EC50 values were all ≤33%. r2 values for dose-response curves all exceeded 0.98; within an assay, the coefficients of variation for quadruplicate determinations were ≤10% and averaged 2.1%.
b Defined as the ratio of the L1210 EC50 to the T. brucei EC50; large numbers are favorable.
c P < .05, by a 2-tailed t test, for comparison of T. brucei and L1210 values.
Table 3.
Structure Activity Relationships on the Ansamycin Scaffold
| NSC Number | Structure | EC50 (nM)a |
||
|---|---|---|---|---|
| T. brucei | L1210 | Selectivityb | ||
| 330500 Macbecin II | 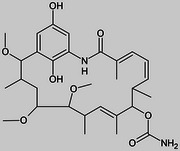 |
240 | 47 | 0.2c |
| 265481 | 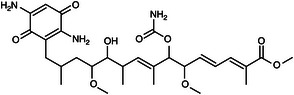 |
>80 000 | >80 000 | NA |
Abbreviations: EC50, half maximal effective concentration; NA, not applicable; T. brucei, Trypanosoma brucei.
a Values are mean of at least 3 independent determinations. Coefficients of variation for EC50 values were all ≤35%. r2 values for dose-response curves all exceeded 0.98; within an assay, the coefficients of variation for quadruplicate determinations were ≤10% and averaged 2.6%.
b Defined as the ratio of the L1210 EC50 to the T. brucei EC50; large numbers are favorable.
c P < .05, by a 2-tailed t test.
The general trends of these structure activity relationships correlate with crystal structure data of ansamycins complexed with Hsp90, where C17 modifications are exposed to solvent and outside the Bergerat-fold ATP-binding pocket [30]. However, the series also reveals differences in the susceptibility of mammalian and trypanosome cells. Many factors may account for this, including phylogenetic differences in the target enzymes. This possibility is supported by the several-log difference in ATPase activity of kinetoplastid Hsp83, compared with mammalian Hsp90 [31]. Compounds with the most favorable selectivity were 17-AG (255109), 17-AAG (330507), and 17-DMAG (707545); the latter was also most potent against trypanosomes. As 17-AAG is the most widely studied, we chose to characterize its antitrypanosomal effect more fully.
17-AAG Affects Growth Rate, Mitosis, and Cytokinesis
Previous reports indicate that, for insect-form T. cruzi or Leishmania donovani, 24 hours geldanamycin treatment arrests the cell cycle at either G1 or G2 [12–14]. We found that, within 5 hours, 50 or 100 nM 17-AAG (EC80 or EC99, respectively) arrested growth of bloodstream T. brucei, and cell numbers declined thereafter (Supplementary Figure 1A). Cell cycle progression was inhibited, and this was examined by flow cytometry and microscopy of DAPI-stained cells, across time and concentration.
On the basis of nuclear DNA content, as measured by flow cytometry, control cultures in mid-log growth were 38% G1 (2C DNA content for the diploid trypanosome), 21% S, and 35% G2/M (4C DNA content). This distribution was maintained until the culture reached stationary phase at 24 hours (Figure 1B). Treatment with 50 nM 17-AAG caused a transient increase in the G1 peak at 5 hours, which decreased progressively thereafter. At 10 hours the G2/M (or 4C) peak was increased, indicating a problem with mitosis and/or cytokinesis. At 24 hours, the 4C peak remained significant, but substantial 6C and 8C peaks were also present. These abnormal forms are possible in bloodstream trypanosomes because of inadequate or missing checkpoints to stop rereplication of DNA when cytokinesis is delayed or inhibited [23]. With 100 nM 17-AAG, the 5-hour increase in G1 and 10-hour increase in G2/M (or 4C) were also seen, but at 24 hours the dominant population was 4C, along with an 8C but no 6C peak. With both concentrations, the percentage of cells in S phase declined (quantitations appear in Supplementary Figure 1B–D).
To obtain further insight into this cell cycle disruption, we examined trypanosomes by fluorescence microscopy. Unlike flow cytometry, which reflects nuclear DNA content, visual analysis of DAPI-stained cells assays both nuclear and kinetoplast DNA, and phase contrast provides concomitant analysis of cytokinesis. Control cells can be divided into 3 populations: 1N1K cells, which have a single nucleus and kinetoplast (68% of the total); 1N2K cells, in which mitochondrial DNA has divided but nuclear mitosis has not occurred (21%); and 2N2K cells, in which the kinetoplast and nucleus have both divided (8%) (Figure 2Ai–iii and 2B). These proportions are in good agreement with flow cytometry: G1 plus S (38% + 21%) are a subset of 1N1K (68%), and 1N2K plus 2N2K (21% + 8%) are a subset of G2/M (35%) (Figure 1A). Likewise consistent with flow cytometry, treatment with 50 nM 17-AAG reduced 1N cells and increased 2N cells and forms containing greater than 2N (Figure 2B). Simultaneous accounting for both nuclei and kinetoplasts revealed the vivid loss of coordinate replication of these genomes. Flawed nuclear mitosis was evidenced by cells with abnormally high numbers of kinetoplasts but only 1 or 2 giant nuclei (Figure 2Ci and 2Cii). Conversely, defective kinetoplast segregation, with unchecked nuclear mitosis, produced cells with multiple nuclei and a single large kinetoplast, demonstrated numerically by the increase in 2N1K cells (Figure 2B, 2Ciii, and 2Civ). Grouping cells on the basis of the ratio of nuclei to kinetoplasts established kinetoplast segregation as more defective than nuclear mitosis: 29% had more nuclei than kinetoplasts (Figure 2D). Of cells that retained a normal census of nuclei and kinetoplasts, many still had evident dysregulation, including 2N2K cells whose kinetoplasts had begun to rereplicate prior to cytokinesis (Figure 2Cv). Phase contrast confirmed the cytokinesis inhibition suggested by 6C and 8C flow cytometry peaks. At 24 hours, cells with multiple partially ingressed cleavage furrows indicated repeated failed attempts at cell division (Figure 2Ci and 2Ciii), and some cells were arrested at abscission (Figure 2Cvi).
Figure 2.

Analyses of trypanosome nuclear and kinetoplast content by DAPI staining and fluorescence microscopy. A, Examples, as indicated, of normal 1 nucleus and kinetoplast (1N1K) cells, 1 nucleus and 2 kinetoplast (1N2K) cells, and 2 nuclei and kinetoplast (2N2K) cells in control cultures. Bar, 1 µm. B, Trypanosomes were treated with 50 nM 17-AAG or solvent for the indicated times and then scored for nuclear and kinetoplast content on the basis of DAPI staining. Values are the means ± SD of counts made in a blinded fashion by 3 observers. The insert shows nuclear content of other cells across time. DMSO, dimethyl sulfoxide; X, ≥5; Z, not identifiable. C, Examples of aberrant cells after 24 hours of treatment with 50 nM 17-AAG. White arrowhead, rereplicating kinetoplast. D, Analysis of data from panel B to determine the percentage of cells with a number of nuclei equal to, less than, or greater than the number of kinetoplasts.
Interestingly, abnormalities from 100 nM 17-AAG were substantially less varied. Although morphologically abnormal (Supplementary Figure 1E) and with an increased 2N population (including aberrant 2N1K; Supplementary Figure 1F), few trypanosomes had incomplete cleavage furrows or >2 nuclei or kinetoplasts. In conjunction with flow cytometry results, this suggests strong mitosis inhibition at 24 hours: although 52% of cells had 4C nuclear DNA content, only 28% were 2N, and, conversely, just 8% had 2C DNA content, but 66% were 1N.
To test whether structurally different Hsp90 inhibitors caused cell cycle disruption, trypanosomes were treated with 350 nM radicicol (EC99). Patterns comparable to those described above were seen by microscopy and flow cytometry (Supplementary Figure 1G and 1H), indicating the effects are likely attributable to Hsp83 inhibition.
The multiple abnormalities caused by Hsp90 inhibitors suggest that Hsp83 deficiency affects several cell cycle proteins. At submaximal inhibition, the lack of effective checkpoints results in abnormal nuclear and kinetoplast content and abortive cytokinesis. With more stringent inhibition of Hsp83, the regulated ability of cells to initiate DNA segregation and cytokinesis is more severely impaired, which limits the variety of abnormalities. Overall, and in contrast to previous reports involving the insect forms of other kinetoplastids [12–14], these results indicate time- and concentration-dependent disruption of both G1 and G2 phases (Figure 1).
Additional Effects of 17-AAG
Heat shock chaperones were initially recognized as proteins whose levels increased after heat stress. Hsp90, among other chaperones, helps maintain protein quality control. We hypothesized that if 17-AAG inhibits Hsp83, then 17-AAG might sensitize trypanosomes to heat shock (Figure 3). In control cells, a heat pulse for one hour caused an arrest in trypanosome cell growth for 10 hours. In a dose-dependent fashion, 17-AAG sensitized trypanosomes to heat shock. Cells shocked in 30 nM 17-AAG (which is ordinarily an EC30) continuously declined in number and did not recover. Other antitrypanosomals with different mechanisms of action, pentamidine and melarsoprol, sensitized trypanosomes to heat shock, but neither was as potent as 17-AAG. Eflornithine had no effect (Supplementary Figure 2).
Figure 3.

Trypanosoma brucei treated with 17-AAG are more sensitive to heat shock. Starting at −1.5 hours, cells at 37°C were treated with 17-AAG or solvent as indicated. The temperature for control cells remained at 37°C for the duration of the experiment. At −1 hour, tubes containing cells to be heat shocked (HS) were transferred to a 41°C water bath and then returned to a 37°C environment at 0 hours for the remainder of the experiment. Motile cells were counted by hemocytometer to determine parasite density. Values are the mean ± SD of 3 determinations.
Somewhat surprisingly, in 2 assays we found that trypanosomes differed from mammalian cells in response to Hsp90 inhibitors: we saw no decrease in argonaute protein levels (a mammalian Hsp90 client [33]) and no synergy with other drugs, including etoposide, melarsoprol, pentamidine, or novobiocin (Supplementary Figure 3).
Activity of 17-DMAG in T. brucei–Infected Mice
The promising in vitro cytotoxicity of Hsp90 inhibitors supported evaluation in a mouse model of T. brucei infection. 17-DMAG was chosen because of its superior water solubility, potency, and selectivity (Table 2). In this acute model, parasites rapidly proliferated in vehicle controls and caused death within 4 days (Figure 4). The veterinary antitrypanosomal diminazen (Berenil) cured all mice. Three dosing strategies with 17-DMAG provided initial clearance of parasitemia and prolonged survival relative to controls. A single high parenteral dose of 150 mg/kg led to dose-limiting toxicity and day 10 recurrence of parasitemia in the surviving mouse. When this same total dose was divided over 5 days, parasitemia dropped below the lower limit of detection by day 3, and all mice were cured (ie, lacked detectable parasitemia) for 30 days (Figure 4). Of considerable importance, oral 17-DMAG (50 mg/kg daily for 5 days) caused prompt reduction in parasitemia and cured all but 1 mouse, which died on day 6 from gavage-induced trauma and/or drug toxicity. The remaining mice in this group appeared healthy and remained parasite free for 30 days (Figure 4). Weight change as an index of health was consistent with these outcomes. On day 0, the average weight was 21.6 g. Between days 0 and 7, average weight changes were +2.2 g (Berenil group), −0.75 g (17-DMAG, 150 mg/kg intraperitoneally once), +3.5 g (17-DMAG, 30 mg/kg intraperitoneally for 5 days), and +3.2 g (17-DMAG, 50 mg/kg by oral gavage for 5 days).
Figure 4.

17-DMAG has antitrypanosomal activity in vivo. Mice were infected with Trypanosoma brucei on day 0. Starting on day 1, they were treated once daily with vehicle or drug as indicated and then monitored for up to 30 days for survival (A) and parasitemia (average value for group; B) by visual analysis and hemocytometer counts of trypanosomes in tail snip blood samples. ip, intraperitoneally; og, by oral gavage; ×1, once; ×5, for 5 days.
DISCUSSION
We evaluated Hsp90 inhibitors against bloodstream-form T. brucei in vitro and in vivo. Geldanamycin has potent activity in vitro, and orally dosed 17-DMAG cures mice of infection. Is this promising antitrypanosomal activity due to Hsp83 inhibition? Multiple lines of evidence from our laboratory and reported by others suggest that it is. Genetic studies have shown that T. cruzi Hsp83 complements an Hsp90 knockout yeast strain [34]. In L. donovani, geldanamycin selects Hsp83 from a cosmid library screen, and overexpression of Hsp83 decreases sensitivity to geldanamycin [12]. Biochemical evidence includes geldanamycin-mediated retrieval of Hsp83 from T. evansi lysates [16]. With T. brucei, we find that the structurally dissimilar Hsp90 inhibitors 17-AAG and radicicol cause the same cell cycle dysregulation; that despite important differences, major structural requirements on the ansamycin scaffold are similar for antitrypanosomal and anti-tumor activities; and that 17-AAG sensitizes trypanosomes to heat shock. The antitrypanosomal effects we have shown validate Hsp83 as a drug target in bloodstream-form African trypanosomes.
Less clear are the downstream consequences of Hsp83 inhibition: why does Hsp83 deficiency kill trypanosomes? In higher eukaryotes, the toxicity of Hsp90 inhibitors is attributed to subsequent functional loss of client proteins, which include key cell cycle regulators. We find vivid evidence of cell cycle dysregulation in trypanosomes treated with Hsp90 inhibitors. Many gene products essential for orderly progression through the trypanosome cell cycle have been identified via RNA interference silencing, including kinases (eg, cyclin-related, Aurora, Polo-like [35–37]) and kinase regulators (eg, cyclins, kinase activator MOB1, and kinase receptor TRACK [38–40]), several of whose mammalian homologs are recognized Hsp90 clients [41–44]. Individual knockdown of these proteins in kinetoplastids generates a cell cycle defect, such as abnormal numbers of kinetoplasts or nuclei and arrested or misplaced cleavage furrows. Unlike these discrete phenotypes, however, with Hsp90 inhibitors we see multiple abnormalities in the same cell and pleomorphic abnormalities in the population (Figure 2C). These complex and profound disruptions of nuclear and kinetoplast segregation and cytokinesis support the notion that Hsp83, like Hsp90 in higher eukaryotes, is an important node in the midst of numerous regulatory pathways.
In the development of any antiparasitic drug, a critical consideration is therapeutic index. Our findings that trypanosomes and L1210 tumor cells differ in susceptibility to Hsp90 inhibitors and, more importantly, that 17-DMAG cures mice of T. brucei infection provide convincing evidence that Hsp90 inhibitors can be selectively toxic for trypanosomes. Given the 61% protein sequence identity between Hsp90 and Hsp83, it is interesting to speculate about the basis for this discrimination. Precedent for selective toxicity to Hsp90 inhibitors exists in the greater susceptibility (up to 200-fold) of tumor cells as compared to nontransformed cells [8, 45]. Cancer cells are thought to be unusually dependent on Hsp90 to chaperone upregulated oncogene clients and mutant proteins and to counter the toxic metabolic environment commonly present in tumors [5–8]. Many factors could heighten susceptibility of trypanosomes to Hsp90 inhibitors, including the previously suggested differences in inhibitor binding affinity at the ATPase site, or less stringent checkpoints lowering the threshold for cell cycle disruption. However, the unique cellular pathways in these ancient eukaryotes suggest other downstream processes, not present in host cells, that may be unusually dependent on Hsp83. These include the heat shock–sensitive requisite trans-splicing of a 5′-leader sequence onto all messenger RNAs [46] (the disruption of which would globally cripple protein synthesis) or the production of essential variable surface glycoprotein for the cell surface coat (which requires chaperone activity) [47, 48].
Extensive studies of Hsp90 inhibitors in humans provide a strong starting point for their possible use in African trypanosomiasis. In phase 1 trials of 17-DMAG, maximum tolerated doses generated maximum blood concentrations of 600–2700 nM [49, 50], well in excess of the 3 nM EC50 against trypanosomes in vitro (Table 2). Distinctly different susceptibilities and doubling times of trypanosomes and mammalian cells suggest that doses and prolonged regimens suitable for cancer therapy may be inappropriate for trypanosomiasis; indeed, our mouse study indicates that limited regimens may suffice. Furthermore, there are 16 other Hsp90 inhibitors currently in clinical trials, several of which have better safety profiles than 17-DMAG and 7 of which are administered orally [7].
We have demonstrated that Hsp90 inhibitors in vitro cause potent growth inhibition of T. brucei and vivid morphological abnormalities and that oral dosing cures trypanosomiasis in mice. In the laboratory, these agents provide a valuable tool for studying the role of Hsp83 in kinetoplastids. More importantly, good clinical promise is afforded by the numerous Hsp90 inhibitors now in human trials. Our results support further investigation into the repurposing of Hsp90 inhibitors as antitrypanosomal agents.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Rahul Bakshi, Elizabeth Nenortas, and Calvin Tiengwe, for technical support; Elizabetta Ullu, for the generous gift of Ago1 antibody; Rahul Bakshi, Paul Englund, Rob Jensen, and Elizabeth Nenortas, for comments on the manuscript; and Caren Freel Meyers, for comments on the SAR section. Instrument support was provided by the Becton Dickinson Immune Function Laboratory of the Flow Cytometry Core, Johns Hopkins Bloomberg School of Public Health. This work would not have been possible without the provision of compounds by the Open Chemical Repository of the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (available at: http://dtp.cancer.gov).
Financial support. This work was supported by the National Institutes of Health (grants R01AI028855-16 and R01A1095453 to T. S.) and Fulbright New Zealand (Fulbright, Ministry of Research Science and Technology, Graduate Award to K. M.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–59. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- 2.Taipale M, Jarosz DF, Lindquist S. Hsp90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–28. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 3.Kaplan KB, Li R. A prescription for “stress”—the role of Hsp90 in genome stability and cellular adaptation. Trends Cell Biol. 2012;22:576–83. doi: 10.1016/j.tcb.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta. 2012;1823:624–35. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic Hsp90 complex in cancer. Nat Rev Cancer. 2010;10:537–49. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitesell L, Lin NU. Hsp90 as a platform for the assembly of more effective cancer chemotherapy. Biochim Biophys Acta. 2012;1823:756–66. doi: 10.1016/j.bbamcr.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Travers J, Sharp S, Workman P. Hsp90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today. 2012;17:242–52. doi: 10.1016/j.drudis.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 8.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 9.Matts RL, Brandt GE, Lu Y, et al. A systematic protocol for the characterization of Hsp90 modulators. Bioorg Med Chem. 2011;19:684–92. doi: 10.1016/j.bmc.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu X, Xiao L, Wang L, Ruden DM. Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem Pharmacol. 2012;83:995–1004. doi: 10.1016/j.bcp.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mottram JC, Murphy WJ, Agabian N. A transcriptional analysis of the Trypanosoma brucei Hsp83 gene cluster. Mol Biochem Parasitol. 1989;37:115–27. doi: 10.1016/0166-6851(89)90108-4. [DOI] [PubMed] [Google Scholar]
- 12.Wiesgigl M, Clos J. Heat shock protein 90 homeostasis controls stage differentiation in Leishmania donovani. Mol Biol Cell. 2001;12:3307–16. doi: 10.1091/mbc.12.11.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graefe SEB, Wiesgigl M, Gaworski I, Macdonald A, Clos J. Inhibition of Hsp90 in Trypanosoma cruzi induces a stress response but no stage differentiation. Eukaryot Cell. 2002;1:936–43. doi: 10.1128/EC.1.6.936-943.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Zhou Y, Yao C, et al. Apoptosis caused by Hsp90 inhibitor geldanamycin in Leishmania donovani during promastigote-to-amastigote transformation stage. Parasitol Res. 2009;105:1539–48. doi: 10.1007/s00436-009-1582-y. [DOI] [PubMed] [Google Scholar]
- 15.Jones C, Anderson S, Singha UK, Chaudhuri M. Protein phosphatase 5 is required for Hsp90 function during proteotoxic stresses in Trypanosoma brucei. Parasitol Res. 2008;102:835–44. doi: 10.1007/s00436-007-0817-z. [DOI] [PubMed] [Google Scholar]
- 16.Pallavi R, Roy N, Nageshan RK, et al. Heat shock protein 90 as a drug target against protozoan infections: biochemical characterization of Hsp90 from Plasmodium falciparum and Trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J Biol Chem. 2010;285:37964–75. doi: 10.1074/jbc.M110.155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adriano MA, Vergnes B, Poncet J, et al. Proof of interaction between Leishmania Sir2RP1 deacetylase and chaperone Hsp83. Parasitol Res. 2007;100:811–8. doi: 10.1007/s00436-006-0352-3. [DOI] [PubMed] [Google Scholar]
- 18.Parsons M, Worthey EA, Ward PN, Mottram JC. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics. 2005;6:127. doi: 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nett IR, Martin DM, Miranda-Saavedra D, et al. The phosphoproteome of bloodstream form Trypanosoma brucei, causative agent of African sleeping sickness. Mol Cell Proteomics. 2009;8:1527–38. doi: 10.1074/mcp.M800556-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodward R, Gull K. Timing of nuclear and kinetoplast DNA replication and early morphological events in the cell cycle of Trypanosoma brucei. J Cell Sci. 1990;95(Pt 1):49–57. doi: 10.1242/jcs.95.1.49. [DOI] [PubMed] [Google Scholar]
- 21.Jensen RE, Englund PT. Network news: the replication of kinetoplast DNA. Annu Rev Microbiol. 2012;66:473–91. doi: 10.1146/annurev-micro-092611-150057. [DOI] [PubMed] [Google Scholar]
- 22.McKean PG. Coordination of cell cycle and cytokinesis in Trypanosoma brucei. Curr Opin Microbiol. 2003;6:600–7. doi: 10.1016/j.mib.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Hammarton TC. Cell cycle regulation in Trypanosoma brucei. Mol Biochem Parasitol. 2007;153:1–8. doi: 10.1016/j.molbiopara.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z. Regulation of the cell division cycle in Trypanosoma brucei. Eukaryot Cell. 2012;11:1180–90. doi: 10.1128/EC.00145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bodley AL, McGarry MW, Shapiro TA. Drug cytotoxicity assay for African trypanosomes and Leishmania species. J Infect Dis. 1995;172:1157–9. doi: 10.1093/infdis/172.4.1157. [DOI] [PubMed] [Google Scholar]
- 26.Field MC, Allen CL, Dhir V, et al. New approaches to the microscopic imaging of Trypanosoma brucei. Microsc Microanal. 2004;10:621–36. doi: 10.1017/S1431927604040942. [DOI] [PubMed] [Google Scholar]
- 27.Grenert JP, Sullivan WP, Fadden P, et al. The amino-terminal domain of heat shock protein 90 (Hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates Hsp90 conformation. J Biol Chem. 1997;272:23843–50. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- 28.Schulte TW, Akinaga S, Soga S, et al. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones. 1998;3:100–8. doi: 10.1379/1466-1268(1998)003<0100:arbttn>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. 2000;275:37181–6. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 30.Johnson VA, Singh EK, Nazarova LA, Alexander LD, McAlpine SR. Macrocyclic inhibitors of Hsp90. Curr Top Med Chem. 2010;10:1380–402. doi: 10.2174/156802610792232088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nadeau K, Sullivan MA, Bradley M, Engman DM, Walsh CT. 83-kilodalton heat shock proteins of trypanosomes are potent peptide-stimulated ATPases. Protein Sci. 1992;1:970–9. doi: 10.1002/pro.5560010802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borst P, van der Ploeg M, van Hoek JF, Tas J, James J. On the DNA content and ploidy of trypanosomes. Mol Biochem Parasitol. 1982;6:13–23. doi: 10.1016/0166-6851(82)90049-4. [DOI] [PubMed] [Google Scholar]
- 33.Pare JM, Tahbaz N, Lopez-Orozco J, LaPointe P, Lasko P, Hobman TC. Hsp90 regulates the function of argonaute 2 and its recruitment to stress granules and P-bodies. Mol Biol Cell. 2009;20:3273–84. doi: 10.1091/mbc.E09-01-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmer G, Louvion JF, Tibbetts RS, Engman DM, Picard D. Trypanosoma cruzi heat shock protein 90 can functionally complement yeast. Mol Biochem Parasit. 1995;70:199–202. doi: 10.1016/0166-6851(95)00007-n. [DOI] [PubMed] [Google Scholar]
- 35.Tu X, Wang CC. The involvement of two cdc2-related kinases (CRKs) in Trypanosoma brucei cell cycle regulation and the distinctive stage-specific phenotypes caused by CRK3 depletion. J Biol Chem. 2004;279:20519–28. doi: 10.1074/jbc.M312862200. [DOI] [PubMed] [Google Scholar]
- 36.Tu X, Kumar P, Li Z, Wang CC. An aurora kinase homologue is involved in regulating both mitosis and cytokinesis in Trypanosoma brucei. J Biol Chem. 2006;281:9677–87. doi: 10.1074/jbc.M511504200. [DOI] [PubMed] [Google Scholar]
- 37.Hammarton TC, Kramer S, Tetley L, Boshart M, Mottram JC. Trypanosoma brucei Polo-like kinase is essential for basal body duplication, kDNA segregation and cytokinesis. Mol Microbiol. 2007;65:1229–48. doi: 10.1111/j.1365-2958.2007.05866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hammarton TC, Clark J, Douglas F, Boshart M, Mottram JC. Stage-specific differences in cell cycle control in Trypanosoma brucei revealed by RNA interference of a mitotic cyclin. J Biol Chem. 2003;278:22877–86. doi: 10.1074/jbc.M300813200. [DOI] [PubMed] [Google Scholar]
- 39.Hammarton TC, Lillico SG, Welburn SC, Mottram JC. Trypanosoma brucei MOB1 is required for accurate and efficient cytokinesis but not for exit from mitosis. Mol Microbiol. 2005;56:104–16. doi: 10.1111/j.1365-2958.2005.04542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rothberg KG, Burdette DL, Pfannstiel J, Jetton N, Singh R, Ruben L. The RACK1 homologue from Trypanosoma brucei is required for the onset and progression of cytokinesis. J Biol Chem. 2006;281:9781–90. doi: 10.1074/jbc.M600133200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lange BMH, Rebollo E, Herold A, Gonzalez C. Cdc37 is essential for chromosome segregation and cytokinesis in higher eukaryotes. Embo J. 2002;21:5364–74. doi: 10.1093/emboj/cdf531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caldas-Lopes E, Cerchietti L, Ahn JH, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A. 2009;106:8368–73. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simizu S, Osada H. Mutations in the Plk gene lead to instability of Plk protein in human tumour cell lines. Nat Cell Biol. 2000;2:852–4. doi: 10.1038/35041102. [DOI] [PubMed] [Google Scholar]
- 44.Basto R, Gergely F, Draviam VM, Ohkura H, Liley K, Raff JW. Hsp90 is required to localise cyclin B and Msps/ch-TOG to the mitotic spindle in Drosophila and humans. J Cell Sci. 2007;120:1278–87. doi: 10.1242/jcs.000604. [DOI] [PubMed] [Google Scholar]
- 45.Amolins MW, Blagg BS. Natural product inhibitors of Hsp90: potential leads for drug discovery. Mini Rev Med Chem. 2009;9:140–52. doi: 10.2174/138955709787316056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muhich ML, Boothroyd JC. Polycistronic transcripts in trypanosomes and their accumulation during heat-shock - evidence for a precursor role in messenger-RNA synthesis. Mol Cell Biol. 1988;8:3837–46. doi: 10.1128/mcb.8.9.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bangs JD, Brouch EM, Ransom DM, Roggy JL. A soluble secretory reporter system in Trypanosoma brucei. Studies on endoplasmic reticulum targeting. J Biol Chem. 1996;271:18387–93. doi: 10.1074/jbc.271.31.18387. [DOI] [PubMed] [Google Scholar]
- 48.Descoteaux A, Avila HA, Zhang K, Turco SJ, Beverley SM. Leishmania LPG3 encodes a GRP94 homolog required for phosphoglycan synthesis implicated in parasite virulence but not viability. Embo J. 2002;21:4458–69. doi: 10.1093/emboj/cdf447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramanathan RK, Egorin MJ, Erlichman C, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-dimethylaminoethylamino-17-demethoxygeldanamycin, an inhibitor of heat-shock protein 90, in patients with advanced solid tumors. J Clin Oncol. 2010;28:1520–6. doi: 10.1200/JCO.2009.25.0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pacey S, Wilson RH, Walton M, et al. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin Cancer Res. 2011;17:1561–70. doi: 10.1158/1078-0432.CCR-10-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.