

Abstract
The aliphatic side chain plays a pivotal role in determining the cannabinergic potency of tricyclic classical cannabinoids. We have synthesized a series of analogues in which the C3 position is substituted either directly or through a one-carbon atom linker with an adamantylamine or with an oxa- or an oxazaadamantane. The oxaadamantane pharmacophore in analogue 16 showed the best binding profile for both receptors.
Introduction
The discovery of the two cannabinoid receptors, CB11,2 and CB2,3 ushered in a new era in research into the chemistry and pharmacology of this class of compounds. Both receptors are membrane-bound and belong to the family of G-protein-coupled receptors (GPCRs). The structural analysis of CB1a and CB2 as well as the study of their interactions with their ligands are hampered by the lack of a crystal structure. Consequently, there is little direct evidence for the mode(s) of interaction between ligand and receptor.4 The recognition of CB1 as an important therapeutic target for, inter alia, glaucoma,5 pain,6 and appetite modulation,7 indicates a need for a better understanding of the specific interactions between the cannabinoid pharmacophore and the key amino acids associated with the CB1 binding site.
It has long been known that the aliphatic side chain is important for determining the cannabinergic potency of classical cannabinoids and also that the presence of a tert-alkyl appendage at C1′ potentiates receptor binding affinity.8 Nevertheless, it was surprising that cannabinoids bearing a pendant adamantyl group at C3 in place of the n-pentyl group that is found in naturally occurring materials, such as (–)- 3-(1-adamantyl)-Δ8-tetrahydrocannabinol (1, AM411), were tolerated in both CB1 and CB2 binding sites.9 Furthermore, considerable receptor subtype selectivity was observed depending on the relative orientation of the adamantyl group with respect to the tricyclic nucleus. The ability of the receptor to accommodate the steric bulk of the 1-adamantyl group revealed an unanticipated flexibility and furthermore suggested that introducing heteroatomic functionality on the adamantane structure could be used to interrogate the receptor. Changes in binding affinities of cannabinoids bearing heteroatom-substituted adamantyl residues might indicate the close proximity of polar amino acid residues within the binding pocket. We have reported the synthesis of cannabinoids 2 and 310 from known oxazaadamantane I.11–13 Oxazaadamantyl cannabinoids 4, 5, 6, and 7, azaadamantyl cannabinoids 8–15, and oxacannabinoids 16 and 17 were designed and prepared as ligands with which to interrogate the CB1 and CB2 receptors (Figure 1). The northern aliphatic hydroxyl group is known to be an important cannabimimetic pharmacophore and was introduced at C9 as an R or β hydroxyl group.
Figure 1.

Adamantyl and heteroadamantyl cannabinoids.
Chemistry
To prepare a diverse set of cannabinoid ligands without preparing each individual alkyl resorcinol, we designed an advanced common intermediate from which all analogues could be derived. Following an approach that had been developed in our group,10,13 we prepared bicyclic intermediate 18a via acid catalyzed condensation between phloroglucinol 19a and a mixture of diacetates 20 and 21 (Scheme 1). This key step had been developed by a team at the Eli Lilly company for the synthesis of nabilone.14 Although the condensation works beautifully in chloroform, the solvent that had been used in the nabilone synthesis, phloroglucinol is very sparingly soluble in chloroform. To overcome this problem, we had used dichloromethane/acetone for our earlier synthesis of 18a but were able to isolate only moderate (40%) yields of the product.10 Part of the difficulty is associated with the high reactivity of 19a that leads to condensation with a second molecule of 20 or 21. It also became apparent that the choice of solvent had a large effect on the yield. We had explored a variety of solvents, acids, and conditions in order to overcome this difficulty to no avail. It is a testament to the skilled efforts of the Lilly team who carefully optimized this process that chloroform appears to be uniquely suited for the reaction. Fortunately, we were able to devise a simple modification of the procedure to overcome the problems associated with the low solubility of phloroglucinol in chloroform. Exposure of phloroglucinolto trimethylsilyl chloride (TMSCl) and triethylamine (TEA) in dichloromethane (DCM) led to the persilylated derivative 22 (Scheme 1). This hydrolytically sensitive material was not purified but was used in the condensation with 20 and 21 in place of phloroglucinol. Masking the phenolic hydroxyl groups as trimethylsilyl ethers led to a large increase in solubility in chloroform. This allowed us to perform the reaction in a mixture of chloroform and acetone (4/1) with p-toluenesulfonic acid in slight excess. This led to a much cleaner reaction and more than doubling of the yield of 18a to approximately 70%. The phenolic trimethylsilyl groups were hydrolyzed during workup. The separation of 18a from unreacted phloroglucinol was difficult. Although the separation can be accomplished by means of careful flash column chromatography on silica gel, we found that it was more practical to peracetylate the mixture of 18a and 19a following workup. The chromatographic separation of acetates 18b and 19b was straightforward and pure 18a was recovered in 68% overall yield following hydrolytic cleavage of the acetoxy groups by KOH in methanol. The large improvement in the yield in the first step of the synthesis greatly increased the pace of progress.
Scheme 1.

Synthesis of Amides 4,5 and 8–11a
aReagents and conditions: (a) p-TsOH, CHCl3/acetone (4/1), 0 °C, 1 h; rt, 1 h; ca. 70%; (b) Me3SiCl, Et3N, CH2Cl2, 0 °C to rt; (c) CH2Cl2, DMAP (cat.), pyr, Ac2O, 0 °Cto rt, 12 h; (d) KOH, MeOH, 0 °C, 2 h; 68% overall from 20 and 21; (e)Me3SiOTf,MeNO2, 0 °C, 2.5 h, >95%; (f ) PhNTf2, Et3N, CH2Cl2, 0 °C to rt; 57% from 18a; (g) CsF, 26, DMF, rt, 12 h; 57%; (h) NaBH4, MeOH, 0 °C, 1 h; 97% 27 + 28, ca. 95/5; (i) L-Selectride, THF,−78°C, 2 h; rt, 1 h; 90%; ( j)MeOCH2Cl, (i-Pr)2NEt,CH2Cl2, 0 °C to rt, 2.5 h; 29, 93%; 30, 94%; (k) Zn(CN)2, Pd(PPh3)4 (cat.), 10 wt%PMHS, DMF, 60 °C, 8 h; 31, 96%; 32, 97%; (l) LiOH,MeOH/H2O(4/1), 70 °C, 3 d; 33, 91%; 34, 91%; (m) I, 1-adamantylamine or 2-adamantylamine, EDCI,DMAP,CH2Cl2, rt, 2 h; from 33: 35, 91%; 37, 90%; 39, 90%; from 34: 36, 88%; 38, 91%; 40, 91%; (k) n-BuSH, ZnBr2, CH2Cl2, 45°C; 4, 77%; 5, 89%; 8, 89%; 9, 92%; 10, 91%; 11, 90%.
An improvement was made in the yield for the cyclocondensation step that converts 18a to tricyclic intermediate 23 as well. In our earlier synthesis of 23, we had used SnCl4 as the Lewis acid. While on a small scale this procedure was practical, upon scale-up, the formation of emulsions during workup led to irreproducible yields of product. Trimethylsilyl triflate (TMSOTf) proved to be a much better choice of Lewis acid both in terms of the ease of workup as well as the yield (>95% vs 84%). Treatment of 23 with N-phenyltriflimide15 in dichloromethane led to 24 regioselectively and in 68% yield (57% overall from 18a). Intermediate 23 was also converted to nonaflate 25 in 57% yield through treatment with reagent 26 in the presence of CsF in N,N-dimethylformamide (DMF).
Because it was our goal to produce both diastereomers of the C9 alcohol, ketone 24 was reduced with NaBH4 in 97% yield to give a ca. 95/5 mixture of C9-β (equatorial) alcohol 27 and C9-α (axial) diastereomer 28. Alcohol 28 was the exclusive product of the reduction of ketone 24 with L-Selectride at −78°C.
The phenolic and aliphatic hydroxyl groups in 27 and 28 were simultaneously protected as the methoxymethyl ether, leading to 29 and 30 in 93% and 94% yield, respectively, setting the stage for palladium catalyzed cyanation. Exposure of the aryl triflates to catalytic Pd(PPh3)4 and Zn(CN)2 in DMF led to nitriles 31 and 32, however, the reaction was not always reproducible. Addition of 10 wt % polymethylhydrosiloxane (PMHS)16 to the reaction mixture as an oxygen scavenger resulted in a consistent and reproducible yield of greater than 95% for each of the diastereomers 31 and 32. Nitriles 31 and 32 were hydrolyzed in the next step with LiOH in aqueous methanol, yielding carboxylic acids 33 (91% yield) and 34 (91% yield), respectively. Condensation of 33 with I, 1-adamantylamine, or 2-adamantylamine using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and 4-dimethylaminopyridine (DMAP) led to amides 35 (91% yield), 37 (90% yield), and 39 (90% yield), respectively. Condensation of 34 with I, 1-adamantylamine, or 2-adamantylamine under the same conditions led to amides 36 (88% yield), 38 (91% yield), and 40 (91% yield), respectively. Cleavage of the methoxymethyl ether protecting groups with n-BuSH/ZnBr217 led to amides 4, 5, and 8–11 in yields ranging from 77% to 92%.
The next task was to prepare compounds 6 and 7 that incorporate a methylene spacer group between amine I and the tricyclic cannabinoid nucleus. The synthesis from carboxylates 33 and 34 is summarized in Scheme 2. Reduction of acids 33 and 34 with borane–tetrahydrofuran (THF) complex led to benzylic alcohols 41 and 42, each in 88% yield. Mesylation followed by immediate displacement of the mesylate by bromide gave the diastereomeric benzylic bromides. These were allowed to react in DMF with a slight excess of amine I at ambient temperature to give oxaza-adamantylamines 43 and 44 in 75% and 72% yield, respectively. Protecting group removal was carried out as in Scheme 1 by exposure to n-BuSH/ZnBr 172 in dichloromethane at 45 °C, leading to oxaza adamantylcannabinoids 6 and 7 in 81% and 73% yield, respectively. It should be noted that all attempts to remove the ether protecting groups from 43 and 44 by means of trimethylsilyl bromide (TMSBr) conditions that we found to be satisfactory in molecules bearing an aromatic nitrogen atom10 led to inferior yields of 6 and 7. This is quite likely due of the buffering effect of the more basic benzylic nitrogen atom in 6 and 7. In contrast, the conditions for methoxymethyl ether group cleavage developed by Rawal and co-workers were optimal in our system and should be given consideration for similar applications in synthesis.
Scheme 2.

Synthesis of Benzylamines 6 and 7a
aReagents and conditions: (a) BH3 · THF, THF, 0 °C; 0 °C to rt; 41, 88%; 42, 88%; (b) MsCl, Et3N, THF, 0 °C to rt; LiBr, THF, rt; (c) I, DMF, K2CO3, rt; 43, 75%; 44, 72%; (d) n-BuSH, ZnBr2, CH2Cl2, 45 °C; 6, 81%; 7, 73%.
We followed a slightly different strategy for the synthesis of compounds 12–15, all of which were prepared by means of reductive amination reactions (Scheme 3). Nitriles 31 and 32 were deprotected by treatment with ZnBr2 and n-BuSH. Temporary masking of the hydroxyl groups as triethylsilyl (TES) ethers was followed by reduction of the nitrile to the aldehyde and fluorodesilylation to produce 47 and 48, both in 61% overall yield for the three steps. Removal of the methoxymethyl groups in the presence of the aldehyde took place in poor yield, making the protecting group exchange necessary. Imine formation with 1- or 2-adamantylamine was followed by catalytic hydrogenation with 10% Pd/C in methanol under an atmosphere of hydrogen, resulting in the clean formation of compounds 12–15.
Scheme 3.

Synthesis of Benzylamines 12–15a
a Reagents and conditions: (a) n-BuSH, ZnBr2, CH2Cl2, rt; 45, 90%, 46, 76%; (b) Et3SiCl, (i-Pr)2NEt, CH2Cl2, 0 °C to rt; (c) DIBAL, CH2Cl2, PhMe, −78 °C; (d) TBAF, THF, rt; 47, 61% from 45; 48, 61% from 46; (e) PhH, 1- or 2-adamantylamine, 4 Å MS, reflux, Dean–Stark; (f) Pd/C, H2, MeOH, rt; 12, 81% from 47; 13, 78% from 48; 14, 60% from 47; 15, 84% from 48.
The synthesis of oxaadamantyl cannabinoids 16 and 17 proved to be a difficult challenge. Our first approach was an attempt to trap a cannabinoid-derived benzyne with an appropriate carbon nucleophile, a strategy that had served us well in the past.10 Ultimately we were unable to define effective reaction conditions and were forced to abandon this approach in favor of a cross coupling process. The synthesis of the required vinyl boronate is summarized in Scheme 4. Treatment of commercially available 1,3-adamantanediol with benzenesulfonyl chloride18 in a mixture of pyridine and benzene at 70 °C led to the Grob fragmentation product 49. Ozonolysis in the next step gave diketone 50 in 66% yield over the two steps. Selective protection of one of the two ketone carbonyl groups as the ethylene ketal formed 51 in 94% yield. Sequential treatment of 51 with lithium diisopropylamide (LDA) and N-phenyltriflimide followed by acid catalyzed exchange of the ketal led to vinyl triflate 52 in 82% yield over the two steps from 51. Suzuki coupling of 52 with bis-pinacolato diborane (PinB-BPin) led to keto boronate 53 in good yield.19,20
Scheme 4.
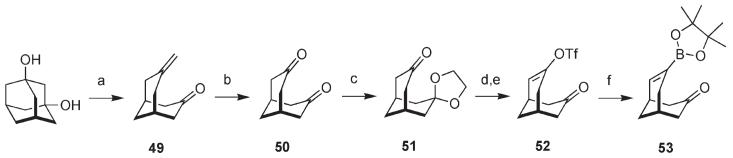
Synthesis of Ketone 53a
a Reagents and conditions: (a) PhSO2Cl, PhH, pyr, 70 °C; (b) O3, CH2Cl2, −78 °C; Me2S; 66% for two steps; (c) HO(CH2)2OH, PhH, TsOH (cat.), reflux, Dean–Stark; 94%; (d) LDA, THF, −78°C; PhNTf2, THF, warm to 0 °C; (e) acetone, TsOH (cat.), rt; 82% from 51; (f ) PdCl2(PPh3)2, PPh3, K2CO3, 1,4-dioxane, PinB-BPin, 70 °C; 73%.
Vinyl boronate 53 was coupled with aryl triflates 29 and 30 (Scheme 5). Treatment of 29 or 30 with 1,1′-bis(diphenylphosphino)ferrocene palladium(II) dichloride (PdCl2(dppf)), K2CO3, and a slight excess of 53 in a mixture of DMF/EtOH (4:1) at 70 °C led to products 54 and 55 in 87% and 67% yield, respectively. Because 53 is racemic whereas 29 and 30 are homochiral, products 54 and 55 are formed as diastereomeric mixtures. For the sake of simplicity, only one diastereomeric structure is shown in Scheme 5. Exposure of 54 and 55 to sodium borohydride unsurprisingly led exclusively to the endo alcohols 56 and 57 in 90% and 82% yield, respectively. Removal of the methoxymethyl protecting groups from 56 and 57 with ZnBr2 and n-BuSH induced cyclization of the seco-oxaadamantanes forming 16 and 17 in 95% and 79% yield, respectively.
Scheme 5.
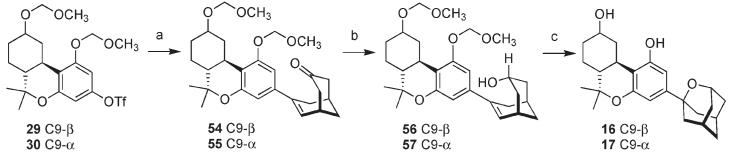
Synthesis of Oxaadamantane Cannabinoids 16 and 17a
a Reagents and conditions: (a) 53, PdCl2(dppf), K2CO3, DMF/EtOH (4/1), 70 °C; 54, 87%; 55, 67%; (b) NaBH4, MeOH, 0 °C; 56, 90%; 57, 82%; (c) n-BuSH, ZnBr2, CH2Cl2, rt; 16, 95%; 17, 79%.
Results and Discussion
Structure–Activity Relationships
Earlier work from our laboratory9 has shown that tetrahydrocannabinol analogues substituted at the 3-position of the phenolic ring exhibit moderate to high affinities for the CB1 and CB2 receptors where the 1-adamantyl analogue 1 exhibited preferential affinity for CB1 while its 2-adamantyl regioisomer (–)- 3-(2-adamantyl)-Δ8-tetrahydrocannabinol (AM744) had higher affinity for CB2. Our effort to introduce heteroatoms within the novel adamantyl cannabinoid structures involved the use of 9β- and 9α-hydroxy hexahydrocannabinols as prototypes for the novel ligands. This was motivated by earlier work in which we have demonstrated that the tricyclic component in this series of analogues is a very successful pharmacophore for the two known cannabinoid receptors.21
To probe the stereoelectronic requirements of the adamantyl group and also explore potential opportunities for improving the polar properties of the adamantyl cannabinergic ligands, we synthesized three groups of heteroadamantyl cannabinoids. The first includes analogues in which heteroatoms are incorporated into the adamantyl group either as a 2,6-oxazaadamantyl ring in which the ring nitrogen is directly attached to the 3-position of the tricyclic ring (2, 3) or alternatively as a 2-oxaadamantyl substituent (16, 17) (see 1 in Figure 1). In the second group, the heteroatom(s) are incorporated into the carbocyclic 1- or 2-adamantyl residue appended at C3 through carboxamido (8–11) or methylamino (12–15) groups. In the third group, the 2, 6-oxazaadamantyl ring is attached to the 3-position either through carbonyl (4, 5) or methylene groups (6, 7).
The SAR of all novel adamantyl analogues was examined by measuring their respective affinities for the CB1 and CB2 receptors (Table 1). All novel oxazacannabinoids exhibited reduced affinities for both receptors when compared with the earlier reported carbocyclic cannabinoids.9 The 9β-OH analogues exhibited more favorable affinities for both receptors when compared to their 9α-OH isomers. Compounds belonging to the second group (8–15) generally had low affinities for both receptors, although all analogues had significant CB2 selectivities over CB1. This is congruent with the earlier data suggesting that the CB2 receptors, either human or mouse, are capable of accommodating larger side chain substituents when compared to CB1. The data also suggest that mCB2 is capable of accommodating larger groups compared to hCB2.
Table 1.
Affinities (Ki) for CB1 and CB2 Cannabinoid Receptorsa
| Ki (μM)a | |||
|---|---|---|---|
| compd | rCB1 | mCB2 | hCB2 |
| l 9 a | 0.0068 | 0.052 | NA |
| 2 | 1.8 | 1.2 | 1.8 |
| 3 | 22.4 | 17 | 14.9 |
| 4 | no binding | 2.5 | 20 |
| 5 | no binding | 8.6 | 25 |
| 6 | 10 | 8.7 | 15 |
| 7 | 125 | 7.5 | 5 |
| 8 | 80 | 0.5 | 8.7 |
| 9 | 375 | 7.5 | 35 |
| 10 | 15 | 1.2 | 2.7 |
| 11 | 100 | 3.7 | 15 |
| 12 | 150 | 0.5 | 10 |
| 13 | 100 | 5 | 25 |
| 14 | 150 | 6.2 | 20 |
| 15 | no binding | 7.5 | 25 |
| 16 | 0.023 | 0.018 | 0.019 |
| 17 | 0.55 | 0.54 | 1.3 |
Affinities for CB1 and CB2 were determined using rat brain (CB1) or membranes from HEK293 cells expressing mouse or human CB2 and [3H]CP-55,940 as the radioligand following previously described procedures. 26Ki values for compounds 2, 3, 16, and 17 were obtained from one experiment (8 point) run in triplicate. Ki values for compounds 4–15, which were all in the micromolar range, were derived from a single experiment (2 points) run in triplicate.
Analogues from the third group (4–7) carrying an appended oxazaadamantyl ring exhibited a similar affinity trend as group I. Again, all analogues showed substantial CB2 vs CB1 selectivities where the mCB2 had somewhat more favorable affinities than hCB2. Also, at the CB2 receptors, 9β-OH analogues (12, 14) had slightly more favorable affinities compared to their 9α-OH analogues.
The first group of heterocannabinoids carries the structurally most compact 3-substituents. Their synthesis was motivated by work with analogues carrying a carbocyclic adamantyl group which exhibited interesting pharmacological profiles.
In earlier work, we synthesized a pair of 2,5-oxazacannabinoids (2, 3) in which the ring nitrogen is directly attached to the 3-phenolic position. Both of these analogues had low affinities for both CB1 and CB2.10 To further explore the basis for this somewhat surprising finding, we have now synthesized the respective 2-oxaadamantyl analogues in which the 1-adamantyl carbon is directly attached to the tricyclic cannabinoid 3-position. We were pleased to observe that both 9β-OH (16) and 9α-OH (17) isomers exhibited favorable affinities for both receptors. Expectedly the 9β-isomer had the higher affinity. This interesting new compound had a somewhat different pharmacological profile than its reported carbocyclic isomer 1. Unlike 1, which exhibited selectivity for CB1, 16 had only modest selectivity (2-fold) for CB2. The new oxaadamantyl analogues have more polar properties (clogP) than 1 and may serve as the basis for the design of novel heteroadamantyl analogues with improved physicochemical and pharmacological properties.
We have used molecular modeling to explain the observed high differences in affinities among the heteroadamantyl cannabinoids described here. Since among the series of analogues reported the only pharmacophoric variable is the 3-substituent, we focused our attention on the conformational and stereoelectronic properties of this moiety and examined the conformational space available for the C3 substituents in each of the analogues. To explore the energetically favorable conformations in each analogue, we used force field methods and retained all conformers within 6 kcal/mol from the global minimum. Representative examples for each of the three groups of analogues are shown in Figure 2. It is clear that the conformational space for the 3-substituents in the second and third groups covers a significantly larger volume than that of the first group. It can thus be argued that the low affinity of these hexahydrocannabinol analogues is attributable to steric factors as well as large desolvation penalties due to their polar linkers. Accordingly, these bulky substituents are unable to engage in an optimal interaction at the adamantyl side chain with its respective pharmacophoric site within the cannabinoid receptors. This is more accentuated in their interaction with CB1 compared to CB2. It should be pointed out that three of the ligands from the second family (8, 10, 12) exhibited the most favorable Ki values for mCB2 with 8 and 12 having the highest affinity (Ki = 0.5 μM). Conversely, all three analogues had 10–50-fold lower affinities for CB1. This observed preference of ligands carrying 3-substituents capable of assuming larger conformational space for the CB2 vs CB1 receptor is congruent with our earlier work with carbocyclic adamantyl analogues. In this work, we have argued that the CB2 selectivity of 2-adamantyl analogues exhibited a larger conformational space for this pharmacophore with the mCB2 being more accommodating than hCB2.
Figure 2.

Accessible conformers with 6 kcal/mol of the global energy minimum for 16 (green), 2 (cyan), 7 (orange), and 14 (magenta). Analogues are shown superimposed at their aromatic rings.
Our arguments for the low affinities of analogues for groups 2 and 3 cannot be easily applied tothe compounds belonging to the first group. A comparison of the pharmacophoric space for compounds 2 and 16 does not reveal striking differences in the accessible conformational volumes of the two analogues. To further explore potential differences in the manner in which the low affinity oxaza analogue (2) and the high-affinity oxa analogues (16, 17) interact with the two cannabinoid receptors, we calculated the energy barriers for rotation of these compounds around the Ph–N (2) or Ph–C (16, 17) respectively (Figure 3). Our calculations show that while the oxa analogues have a rotational barrier of ~ 6.3 kcal/mol, the oxaza analogue has a significantly higher barrier ~ of 12.3 kcal/mol. We would thus argue that while both analogues in question occupy similar conformational spaces, entropic advantages associated with a more facile rotation for the oxaadamantyl analogues may improve ligand binding affinity. Alternatively, it can be argued that in the 3-oxaza compounds (2 and 3) both N and O heteroatoms undergo unfavorable interactions within its allowable pharmacophoric space.
Figure 3.

Plot of the QM energy relative to the lowest energy structure calculated at the B3LYP/6–31G** level for 16 (dashed) and 2 (solid) during a coordinate scan around the C3–C1′ bond.
Methods
Along with entropic factors, the conformational space available to the analogues may offer insight into the steric factors required for CB1 and CB2 selectivity. To explore the permissible rotations of the bulky substituent, a conformational search was performed using optimized potentials for liquid simulations (OPLS) force field.23,24 The cannabinoid tricyclic moiety was held fixed and minimization on the remaining geometric parameters was performed.23 Conformers with greater than 0.5Å root-mean-square deviation (rmsd) with 6 kcal mol−1 of the global minimum were retained. All calculations were performed in Macromodel.25 The entropic factors involved for a ligand are also important, and compounds 2 and 16 were of particular interest and higher level calculations were performed at the B3LYP/6-31G** level. In these computationally more demanding calculations, a conformational scan was performed using dihedral drive around the C3–C1′ bond. The dihedral angle was restrained at a value between 0 and 360° in 10° steps, and minimization on the remaining geometric parameters was performed.
Summary
The 3-phenyl substituents of cannabinoid ligands are an essential pharmacophore capable of significantly modulating their affinities for the CB1 and CB2 cannabinoid receptors. In earlier work, we have shown that the 3-alkyl groups of classical cannabinoids can be successfully substituted with an adamantyl ring. The present work is aimed at further exploring this interesting pharmacophore. In addition to exploring the steric requirements of the adamantyl ring, we have sought to introduce heteroatoms within the ring or within the pendant fragment connecting the ring to the tricyclic component in order to modulate the hydrophobic physical properties of the ligands.
Our results indicate that optimal affinities are obtained with compounds in which the adamantyl pharmacophore is directly attached to the 3-position of the phenolic ring. The ligands’ affinities for both receptors are severely diminished if the adamantyl unit is attached through a linker, an observation attributable to the larger pharmacophoric space required by these compounds. The most successful ligands were the 2-oxaadamantyl analogues, suggesting that the oxygen atom in the adamantyl ring could be accommodated within the binding domain of the CB1 and CB2 receptors. However, the oxaza analogues in which the nitrogen atom is directly attached to the ring exhibited reduced affinities for both receptors. This is attributed to entropic factors because of restricted rotation around the N-Ph bond. The overall SAR of the new compounds follows trends congruent with earlier work in the classical cannabinoid field. This includes the observation that 9β-OH analogues have higher affinities compared to their 9α-stereoisomers. The work also confirmed earlier observations suggesting that the space for the adamantyl pharmacophore is most restricted at the CB1 receptor. Additionally, there appears to be a species difference with the mCB2 receptor being more accommodating than hCB2.
Experimental Section
Chemistry
1H NMR and 13C NMR spectra were recorded either at 300 MHz (1H) and 75 MHz (13C) or at 500 MHz (1H) and 126 MHz (13C). Chemical shifts are reported in parts per million (δ) and are referenced to the solvent, i.e. 7.26/77.0 for CDCl3. Multiplicities are indicated as br (broadened), s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sept (septet), or m (multiplet). Coupling constants (J) are reported in Hertz (Hz). Thin layer chromatography (TLC) was performed on glass plates 250 μm, particle size 5–17 μm, pore size 60Å. Flash column chromatography was performed on silica gel, 200–400 mesh, or premium silica gel, 60Å, 40–75 μm. All moisture sensitive reactions were performed under a static atmosphere of nitrogen or argon in oven-dried or flame-dried glassware. Purity and homogeneity of all materials was determined to be at least 95% from TLC, 1H NMR, 13C NMR, and HPLC. All optical rotations were measured on a JASCO digital polarimeter in a 0.1 dL cell.
Synthesis of 18b
To a suspension of phloroglucinol 19a (5.50 g, 43.7 mmol) in 300 mL of DCM at 0 °C was added TEA (24.3 mL, 174.6 mmol) followed by TMSCl (22.3 mL, 174.6 mmol). After 20 min, the cooling bath was removed and the mixture was allowed to stir at room temperature for an additional 2 h. The salts were removed via filtration and the filtrate was washed with ice cold water (3 ×), dried over Na2SO4, and concentrated affording persilylated phloroglucinol 22. Crude 22 was then dissolved in 440 mL of a 4:1 mixture of CHCl3:acetone and cooled to 0 °C. In a separate flask, diacetates 20 and 21 (4.42 g, 18.6 mmol; 5.2 g of 85% pure diacetates 20 and 21 were used) were dissolved in 150 mL of 4:1 CHCl3:acetone along with TsOH · H2O (4.57 g, 24.0 mmol). The TsOH · H2O and diacetates mixture was then added dropwise to persilylated phloroglucinol 22 at a rate of approximately 1 drop/s via an addition funnel. The reaction mixture was then slowly warmed to room temperature. Once the diacetates were shown to be consumed by TLC, the reaction was quenched with saturated NaHCO3 and stirred for 45 min. The organic layer was separated and dried over MgSO4 while the aqueous layer was back-extracted with EtOAc (6×) and dried over MgSO4. To the crude condensation product 18a and DMAP (100 mg, 0.82 mmol) in 200 mL of DCM at 0 °C was added pyridine (13.0 mL, 160.0 mmol) followed by Ac2O (15.1 mL, 160.0 mmol) and stirred for 12 h. The mixture was quenched with ice cold water, washed with 1 M HCl and brine, and dried over MgSO4. The crude product was then purified via flash column chromatography on silica gel eluting with 30% EtOAc/hexanes affording 18b (4.90 g, 68% yield over 2 steps) as a white solid.
1H NMR (CDCl3, 300 MHz): δ 6.83 (s, 2H), 3.63 (t, J = 8.4 Hz, 1H), 2.84–2.75 (m, 1H), 2.66–2.57 (m, 3H), 2.29–2.25 (m, 10H), 2.15–2.11 (m, 1H), 1.37 (s, 3H), 0.95 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 212.4, 168.5, 168.4, 149.5, 148.5, 125.0, 114.6, 57.1, 45.3, 42.0, 38.7, 30.6, 26.0, 24.5, 21.8, 20.9; mp: 138–140 °C. IR (neat, cm−1): 2947, 1769, 1709, 1608, 1427, 1371, 1185, 1122, 1031, 903. Mass spec calcd for C21H24O7, 388.1522; found, 388.1532. EI+(amu): 388 (M, 10), 346 (62), 304 (33), 303 (25), 262 (30), 262 (51), 244 (27), 219 (53), 207 (20), 194 (32), 177 (34), 152 (50), 83 (100); [α]23D −8.0 °(c 0.010, EtOAc).
Synthesis of 18a
To a solution of the triacetate 18b (4.90 g, 12.6 mmol) in 50 mL of MeOH at 0 °C was added KOH (2.48 g, 44.2 mmol) under N2. The reaction mixture was stirred at this temperature for an additional 2 h and then quenched with 1 N HCl. MeOH was removed under reduced pressure, and the residue was diluted with EtOAc, washed with brine, and dried over MgSO4. The crude product was carried on without further purification. An analytical sample could be purified via flash column chromatography on silica gel eluting with 50% EtOAc/hexanes, affording 18a (3.30 g, 100% yield) as an off-white foam that typically entrains 10–15% ethyl acetate.
1H NMR (MeOH-d4, 300 MHz) δ 5.85 (s, 2H), 3.95 (t, J = 8.1 Hz, 1H), 3.67 (dd, J = 18.6 Hz, 7.5 Hz, 1H), 2.62–2.57 (m, 1H), 2.48–2.35 (m, 3H), 2.14 (t, J = 6.0 Hz, 1H), 1.35 (s, 3H), 0.94 (s, 3H). 13C NMR (MeOH-d4, 75 MHz) δ 220.2, 158.5, 157.3, 108.9, 95.9, 59.3, 48.8, 43.2, 38.8, 30.0, 26.6, 24.9, 22.4.
Synthesis of 23
To a solution of 18a (1.02 g, 3.89 mmol; the mass of pure ketone 18a was 1.20 g; ethyl acetate was present as an impurity) in 300 mL of MeNO2 at 0 °C was added TMSOTf (1.76 mL, 9.73 mmol) dropwise. The resulting mixture was stirred for 2.5 h at 0 °C and then quenched with solid K2CO3 and stirred for 45 min at rt. The solids were filtered off and the solvent removed under reduced pressure. Crude 23 was used without further purification in the next step.
1H NMR (MeOH-d4, 300 MHz) δ 5.85 (d, J = 2.4 Hz, 1H), 5.76 (d, J = 2.4 Hz, 1H), 3.81 (dd, J = 15.0 Hz, 3.0 Hz, 1H), 2.79–2.70 (m, 1H), 2.47–2.25 (m, 2H), 2.18–2.02 (m, 2H), 1.92 (td, J = 12.3 Hz, 2.4 Hz, 1H), 1.57–1.45 (m, 1H), 1.42 (s, 3H), 1.08 (s, 3H). 13C NMR (MeOH-d4, 75 MHz) δ 214.6, 158.3, 156.6, 111.2, 104.3, 96.7, 96.6, 77.8, 46.8, 41.5, 36.0, 28.1, 27.9, 19.0.
Synthesis of 24
To crude ketone 23 (1.02 g, 3.89 mmol) in 40 mL of DCM at 0 °C was added TEA (1.62 mL, 11.7 mmol) followed by dropwise addition of N-phenyltrifluoromethanesulfonimide (1.60 g, 4.47 mmol) in 40 mL of DCM via cannula. The reaction mixture was allowed to warm to room temperature over 12 h, was quenched with 1 N HCl, and washed with water. The aqueous layer was back extracted with DCM, washed with brine, and dried over MgSO4. The crude product was purified via flash column chromatography on silica gel eluting with 20, 30, and 40% EtOAc/hexanes affording 24 (874 mg, 57% yield over 2 steps) as a white semisolid.
1H NMR (CDCl3, 300 MHz): δ 6.39 (d, J = 2.4 Hz, 1H), 6.33 (d, J = 2.4 Hz, 1H), 4.13 (d, J = 15.0 Hz, 1H), 2.94–2.84 (m, 1H), 2.73–2.66 (m, 1H), 2.59–2.47 (m, 1H), 2.25–1.95 (m, 3H), 1.62–1.49 (m, 4H), 1.29–1.12 (m, 4H). 13C NMR (CDCl3, 75 MHz): δ 215.9, 156.6, 155.6, 148.8, 110.7, 102.3, 100.9, 77.8, 46.9, 44.2, 40.7, 34.7, 27.6, 26.8, 18.8. IR (neat, cm−1): 3415(br),1617, 1423, 1246, 1211, 1141, 1099. Mass spec calcd for C16H17F3O6S, 394.0698; found, 394.0681. EI +(amu): 394 (M+, 13), 279 (28), 270 (72), 243 (69), 225 (80), 207 (100), 195 (70); [α]23D −45.0° (c 0.006, CH2Cl2).
(6aS,9R,10aR)-6a,7,8,9,10,10a-Hexahydro-1,9-dihydroxy-6,6-dimethyl-6H-benzo[c]chromen-3-yl trifluoromethanesulfonate 27
To ketone 24 (583 mg, 1.48 mmol) in 15 mL of MeOH at 0 °C was added NaBH4 (280 mg, 7.40 mmol) in 3 portions over 5 min. The reaction mixture was then stirred for 1 h, quenched with dropwise addition of 1 N HCl, and diluted with EtOAc. The organic layer was washed with brine and dried over MgSO4. The crude product was then purified via flash column chromatography on silica gel eluting with 30% then 40% EtOAc/hexanes, affording alcohol 27 and minor alcohol 28 (570 mg, 97% combined yield; the minor diastereomer is easily removed during column chromatography after the subsequent protection) as a white foam.
1H NMR (CDCl3, 300 MHz): δ 8.19 (br s, 1H), 6.27 (s, 1H), 6.18 (s, 1H), 4.00–3.89 (m, 1H), 3.68–3.60 (m, 1H), 2.50–2.42 (m, 2H), 2.20–2.15 (m, 1H), 1.93–1.88 (m, 1H), 1.51–1.42 (m, 2H), 1.37 (s, 3H), 1.20–1.11 (m, 1H), 1.06–0.99 (m, 4H). 13C NMR (CDCl3, 75 MHz): δ 156.5, 155.9, 148.3, 119.9, 102.3, 100.6, 77.8, 71.5, 47.7, 37.1, 35.7, 33.3, 27.6, 25.8, 18.9. IR (neat, cm−1): 3245(br), 2937, 2873, 1597, 1420, 1245, 1213, 1141, 989, 857. Mass spec calcd for C16H19F3O6S, 396.0855; found, 396.0863. EI+(amu): 396 (M+, 79), 378 (45), 336 (57), 335 (100), 186 (29), 69 (71); [α]23D −63.9° (c 0.015, CH2Cl2).
(6aS,9S,10aR)-6a,7,8,9,10,10a-Hexahydro-1,9-dihydroxy-6,6-dimethyl-6H-benzo[c]chromen-3-yl trifluoromethanesulfonate 28
To ketone 24 (299 mg, 0.76 mmol) in 8 mL of THF at −78 °C was added a 1 M solution of L-Selectride in THF (3.00 mL, 3.00 mmol). The reaction was maintained at −78 °C for 2 h and then stirred at room temperature for 1 h. The flask was cooled to −78 °C and solid NaHCO3 (930 mg, 11.1 mmol) was added followed by dropwise addition of a 30% aqueous solution of H2O2 (1.60 mL). After the addition of 30% H2O2 was complete, the cooling bath was removed and the reaction mixture was stirred for 1 h at rt. A saturated solution of sodium thiosulfate (5 mL) was added and the reaction mixture was stirred for an additional 30 min. Ether was added and the organic layer was separated, then washed with brine and dried over MgSO4. The crude product was purified via flash column chromatography on silica gel eluting with 40% EtOAc/hexanes, affording alcohol 28 (269 mg, 90% yield) as a white solid. Alcohol 27 was not observed in the 1H NMR at 300 MHz.
1H NMR (CDCl3, 300 MHz): δ 7.66 (br s, 1H), 6.30 (d, J = 2.5 Hz, 1H), 6.26 (d, J = 2.5 Hz, 1H), 4.35 (s, 1H), 3.24 (d, J = 14.1 Hz, 1H), 2.93–2.86 (m, 1H), 2.54 (br s, 1H), 1.99–1.94 (m, 1H), 1.77–1.68 (m, 2H), 1.56–1.46 (m, 2H), 1.37–1.26 (m, 4H), 0.99 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 156.1, 148.3, 112.8, 102.9, 101.3, 77.7, 67.5, 48.8, 35.3, 33.5, 28.9, 27.2, 22.5, 18.8; mp: 188.5–192.5 °C. IR (neat, cm−1): 3210 (br), 2938, 1597, 1506, 1419, 1245, 1212, 1140, 1102, 987, 879, 839, 735. Mass spec calcd for C16H19F3O6S–H2O, 378.0749; found, 378.0743. EI +(amu): 396 (M+, 8), 378 (64), 335 (56), 309 (31), 202 (14), 151 (42), 101 (39), 92 (19), 69 (100); [α]23D −57.7° (c 0.014, CH2Cl2).
(6aS,9R,10aR)-6a,7,8,9,10,10a-Hexahydro-1,9-bis(methoxy-methoxy)-6,6-dimethyl-6H-benzo[c]chromen-3-yl trifluoromethanesulfonate 29
Alcohol 27 (430 mg, 1.08 mmol) was dissolved in 10 mL of DCM, was cooled to 0 °C, and was treated with DIPEA (1.13 mL, 6.48 mmol) and dropwise addition of MOMCl (492 μL, 6.48 mmol). After 45 min, the cooling bath was removed and the reaction mixture was stirred at room temperature for another 1 h 45 min. Saturated NaHCO3 was added to quench the reaction, and the resulting mixture was diluted with Et2O. The organic layers were washed with CuSO4 and brine and then dried over MgSO4. The crude product was purified via flash column chromatography on silica gel eluting with 20% EtOAc/hexanes, affording 29 (487 mg, 93% yield) as a clear, colorless oil.
1H NMR (CDCl3, 300 MHz): δ 6.56 (d, J = 2.5 Hz, 1H), 6.40 (d, J = 2.5 Hz, 1H), 5.19–5.12 (m, 2H), 4.74–4.69 (m, 2H), 3.75–3.67 (m, 1H), 3.47 (s, 3H), 3.69 (s, 3H), 3.35–3.30 (m, 1H), 2.44 (td, J = 11.3 Hz, 2.4 Hz, 1H), 2.21–2.16 (m, 1H), 1.92–1.86 (m, 1H), 1.55–1.37 (m, 5H), 1.19–1.03 (m, 5H). 13C NMR (CDCl3, 75 MHz): δ 157.1, 155.4, 148.5, 114.2, 104.3, 99.5, 94.8, 94.6, 77.8, 75.5, 56.3, 55.1, 48.1, 36.1, 33.7, 33.0, 27.4, 25.9, 18.8. IR (neat, cm−1): 3197, 3105, 2940, 2789, 1603. Mass spec calcd for C20H27F3O8S, 484.1379; found, 484.1403. EI +(amu): 484 (M+, 7), 379 (33), 378 (100), 335 (25), 245 (19); [α]23 +D−67.6° (c 0.017, CH2Cl2).
(6aS,9R,10aR)-6a,7,8,9,10,10a-Hexahydro-1,9-bis(methoxy-methoxy)-6,6-dimethyl-6H-benzo[c]chromene-3-carbonitrile 31
To triflate 29 (435 mg, 0.90 mmol) was added Zn(CN)2 (84 mg, 0.72 mmol), Pd2(dba)3 (82 mg, 0.090 mmol), and PPh3 (188 mg, 0.718 mmol) followed by PMHS (44 mg, 10 wt %) and 27 mL of DMF under an atmosphere of argon. The reaction mixture was further degassed by bubbling argon through the mixture for 15 min. The reaction mixture was heated to 60 °C and stirred for 8 h. The solvent was removed under reduced pressure, and the residue was adsorbed onto celite. The crude product was subjected to flash column chromatography on silica gel eluting with 10%, 20%, and 30% EtOAc/hexanes, affording 31 as a clear, colorless viscous oil (313 mg, 96% yield).
1H NMR (CDCl3, 300 MHz): δ 6.90 (d, J = 1.6 Hz, 1H), 6.77 (d, J = 1.6 Hz, 1H), 5.21 (d, J = 6.8 Hz, 1H), 5.16 (d, J = 6.8 Hz, 1H), 4.73 (dd, J = 8.0 Hz, 7.1 Hz, 2H), 3.79–3.68 (m, 1H), 3.49 (s, 3H), 3.40–3.30 (m, 4H), 2.49 (td, J = 11.4 Hz, 2.4 Hz, 1H), 2.23–2.18 (m, 1H), 1.94–1.89 (m, 1H), 1.571.89 (m, 1H1.39 (m, 5H), 1.201.89 (m, 1H1.03 (m, 5H). 13C NMR (CDCl3, 75 MHz): δ 156.7, 155.2, 119.8, 118.8, 115.6, 110.9, 108.7, 94.9, 94.5, 77.8, 75.5, 56.4, 55.2, 48.2, 36.1, 34.1, 33.0, 27.5, 25.9, 18.7. IR (neat, cm−1): 2939, 2880, 2228, 1565, 1423, 1369, 1336, 1207, 1155, 1103, 1058. Mass spec Calcd for C20H27NO5, 361.1889; found, 361.1880. EI +(amu): 361 (M+, 11), 285 (19), 255 (100), 240 (25), 212 (75), 69 + (10); [α]23D −77.9 ° (c 0.007, CH2Cl2).
(6aS,9R,10aR)-6a,7,8,9,10,10a-Hexahydro-1,9-bis(methoxy-methoxy)-6,6-dimethyl-6H-benzo[c]chromene-3-carboxylic Acid 33
To nitrile 31 (52 mg, 0.15 mmol) in a screw cap vial was added MeOH:H2O (4:1, 1 mL) and LiOH (61 mg, 1.45 mmol) and the mixture was heated to 70 °C in an oil bath for 3 days. Conc HCl was added to the reaction mixture, and the resultant milky solution was extracted with CHCl3, washed with saturated brine, and dried over Na2SO4. Acid 33 was obtained as a clear, colorless oil (50 mg, 91% yield). No purification was necessary.
1H NMR (CDCl3, 300 MHz): δ 7.29 (s, 1H), 7.24 (s, 1H), 5.26 (d, J = 6.6 Hz, 1H), 5.20 (d, J = 6.6 Hz, 1H), 4.74 (dd, J = 9.3 Hz, 6.9 Hz, 2H), 3.81 –3.70 (m, 1H), 3.51 (s, 3H), 3.47 –3.40 (m, 4H), 2.51 (td, J = 11.1 Hz, 1.8 Hz, 1H), 2.251.89 (m, 1H2.18 (m, 1H), 1.921.89 (m, 1H1.87 (m, 1H), 1.601.89 (m, 1H1.40 (m, 5H), 1.251.89 (m, 1H1.04 (m, 5H). 13C NMR (CDCl3, 75 MHz): δ 171.2, 156.3, 154.8, 128.7, 120.1, 113.7, 106.5, 94.8, 94.5, 77.4, 75.7, 56.4, 55.2, 48.4, 36.2, 34.3, 33.1, 27.6, 26.1, 18.7. IR (neat, cm−1): 2939, 1719, 1690, 1574, 1424, 1375, 1211, 1149, 1100, 1051. Mass spec calcd for C20H28O7, 380.1835; found, 380.1827. EI (amu): 380 (M, 3), 293 (11), 149 (100), 71 (26); [α]23 + D 86.0° (c 0.014, CH + 2Cl2).
β-C9 Oxaza Amide 35 (Procedure for Amidation Reaction)
To a solution of 33 (30 mg, 0.079 mmol) in 2 mL of DCM was added amine I (30 mg, 0.12 mmol) followed by DMAP (39 mg, 0.32 mmol) and EDCI (30 mg, 0.16 mmol). The flask was sealed with a Teflon cap and stirred overnight at rt. The mixture was diluted with EtOAc, washed with 1 N HCl and brine and dried over MgSO4. The crude product was directly adsorbed onto celite and purified via flash column chromatography eluting with 80% EtOAc/hexanes, affording amide 35 as a clear, colorless oil (36 mg, 91% yield).
1H NMR (CDCl3, 300 MHz): δ 6.66 (s, 1H), 6.49 (s, 1H), 5.21 (d, J = 6.6 Hz, 1H), 5.12 (d, J = 6.6 Hz, 1H), 5.05 (br s, 1H), 4.72 (dd, J = 9.6 Hz, 6.9 Hz, 2H), 4.24 –4.18 (m, 3H), 3.78 –3.68 (m, 1H), 3.47 (s, 3H), 3.431.89 (m, 1H3.34 (m, 4H), 2.47 (td, J = 11.2 Hz, 2.0 Hz, 1H), 2.211.89 (m, 1H1.75 (m, 10H), 1.571.89 (m, 1H1.37 (m, 5H), 1.211.89 (m, 1H1.02 (m, 5H). 13C NMR (CDCl3, 75 MHz): δ 169.1, 156.6, 154.8, 135.4,115.5, 109.4, 103.9, 94.7, 94.5, 77.2, 75.6, 66.7, 56.2, 55.1, 49.0, 48.4, 42.9, 36.3, 35.1, 34.3, 33.9, 33.1, 27.6, 26.0, 18.8. IR (neat, cm−1): 2937, 1626, 1566, 1424, 1370, 1148, 1101, 1048. Mass spec calcd for C28H39NO7, 501.2727; found, 501.2702. EI (amu): 501 (M+, 28), 457 (23), 395 (100), 380 (13), 352 (20), 257 + (30), 167 (17), 149 (44), 95 (21), 71 (24), 69 (30); [α]23D −33.3° (c 0.011, CH2Cl2).
Procedure for Deprotection of MOM Ethers (4–11). β-C9 Oxaza Amide 4
To a solution of amide 35 (36 mg, 0.072 mmol) and n-BuSH (180 μL, 1.68 mmol) in 2 mL of DCM was added ZnBr2 (81 mg, 0.36 mmol) all in one portion. The reaction flask was placed in an oil bath and heated at 45 °C for 8 h. The flask was then cooled to room temperature, diluted with EtOAc, and quenched with saturated NaHCO3. The organic layer was washed with brine and dried over Na2SO4. The crude product was adsorbed onto Celite and subjected to column chromatography on silica gel using a gradient elution of 2.5, 5, 10% MeOH/DCM. Amide 4 is a white glass obtained in 77% yield (23 mg). Chiral HPLC (0.46 cm ×25 cm Chiralcel AD-H, 50% 2-propanol in hexanes, 1 mL/min, UV detection at 280 nm) 7.50 min and 98.6% pure.
1H NMR (MeOH-d4, 500 MHz): δ 6.33 (d, J = 1.8 Hz, 1H), 6.29 (d, J = 1.8 Hz, 1H), 4.96 (br s, 1H), 4.20 (br s, 3H), 3.77–3.71 (m, 1H), 3.53–3.49 (m, 1H), 2.50 (td, J = 11.3 Hz, 2.5 Hz, 1H), 2.18–2.09 (m, 3H), 2.06–2.00 (m, 2H), 1.93–1.80 (m, 5H), 1.46 (td, J = 11.8 Hz, 2.3 Hz, 1H), 1.42–1.33 (m, 4H), 1.24–1.15 (m, 1H), 1.04 (s, 3H), 0.98–0.91 (m, 1H). 13C NMR (MeOH-d4, 126 MHz): δ 171.7, 158.4, 156.7, 136.0, 115.4, 107.7, 105.8, 78.4, 71.3, 68.2(2), 51.0, 50.1, 44.8, 39.6, 36.6, 36.0, 35.9, 35.2, 35.1, 28.1, 27.2, 19.1. IR (neat, cm−1): 3454 (br), 2932, 1738, 1727, 1604, 1572, 1441, 1381, 1240, 1057. Mass spec calcd for C24H31NO5, 413.2234; found, 413.2322. EI +(amu): 413 (M+, 100), 395 (26), 352 (20), 275 (30), 149 (56); [α]23D −92.6° (c 0.007, MeOH).
((6aS,9R,10aR)-6a,7,8,9,10,10a-Hexahydro-1,9-bis(methoxy-methoxy)-6,6-dimethyl-6H-benzo[c]chromen-3-yl)methanol 41
To carboxylic acid 33 (12 mg, 0.032 mmol) in 0.1 mL of THF was added excess BH3 · THF (100 μL, 1 mmol, 1 M) at 0 °C, and the mixture was allowed to warm to room temperature overnight. Six N HCl was added slowly and carefully at 0 °C, and the mixture was diluted with CHCl3. The organic layers were washed with brine and dried over Na2SO4. The crude product was purified via flash column chromatography on silica gel using 50% EtOAc/hexanes as the eluent which resulted in alcohol 41 (10 mg, 88% yield) as a clear, colorless oil.
1H NMR (CDCl3, 500 MHz): δ 6.63 (s, 1H), 6.50 (s, 1H), 5.21 (d, J = 6.3 Hz, 1H), 5.16 (d, J = 6.3 Hz, 1H), 4.72 (dd, J = 13.3 Hz, 6.8 Hz, 2H), 4.57 (br s, 2H), 3.77–3.71 (m, 1H), 3.50 (s, 3H), 3.44–3.39 (m, 4H), 2.47 (td, J = 11.3 Hz, 2.5 Hz, 1H), 2.23–2.17 (m, 1H), 1.93–1.88 (m, 1H), 1.56–1.40 (m, 2H), 1.39 (s, 3H), 1.18–1.06 (m, 2H), 1.04 (s, 3H). 13C NMR (CDCl3, 126 MHz): δ 156.7, 154.9, 140.8, 113.3, 109.8, 104.2, 94.8, 94.4, 76.9, 75.7, 65.2, 56.3, 55.2, 48.6, 36.5, 33.9, 33.2, 27.7, 26.1, 18.8. IR (neat, cm−1): 3409 (br), 2918, 2849, 1577, 1431, 1056, 1042. Mass spec calcd for C20H30O6, 366.2042; found, 366.2035. EI +(amu): 366 (M, 26), 260 (100), 245 (28), 217 (34), 177 (10);+ [α]23D −65.8° (+c 0.012, CH2Cl2).
β-C9 Oxaza Benzyl Amine 43
Alcohol 41 (55 mg, 0.15 mmol) was dissolved in 2 mL of THF under N2, was cooled to −40 °C, and was treated with NEt3 (125 μL, 0.90 mmol) and MsCl (50 μL, 0.65 mmol). The reaction mixture was allowed to stir at this temperature for 45 min and then was warmed to 0 °C and stirred for an additional 30 min. A solution of LiBr (130 mg, 1.50 mmol) in 2 mL of THF was added via cannula, and the reaction mixture was allowed to warm to room temperature and was stirred for 4 h. The reaction mixture was quenched with ice cold saturated NaHCO3, extracted with Et2O, washed with brine, and dried over Na2SO4. The crude bromide and amine I (24 mg, 0.17 mmol) were dissolved in 1 mL of DMF under N2. K2CO3 (124 mg, 0.90 mmol) was added, and the mixture was stirred overnight. The solvent was removed under vacuum and then diluted with EtOAc, washed with water and brine and dried over Na2SO4. The crude product was subjected to column chromatography on silica gel eluting with 50% then 80% EtOAc/hexanes, affording amine 43 (55 mg, 75% yield over 2 steps) as a clear, colorless oil.
1H NMR (MeOH-d4, 300 MHz): δ 6.70 (d, J = 1.5 Hz, 1H), 6.47 (d, J = 1.5 Hz, 1H), 5.21 (d, J = 6.5 Hz, 1H), 5.16 (d, J = 6.5 Hz, 1H), 4.71 (s, 2H), 4.22–4.06 (m, 3H), 3.80 (s, 2H), 3.78–3.67 (m, 1H), 3.51–3.43 (m, 4H), 3.37 (s, 3H), 3.06–3.00 (m, 2H), 2.49 (td, J = 11.3 Hz, 2.4 Hz, 1H), 2.24–2.03 (m, 5H), 1.96–1.85 (m, 5H), 1.52–1.36 (m, 5H), 1.20 (td, J = 12.6 Hz, 3.5 Hz, 1H), 1.02 (s, 3H). 13C NMR (MeOH-d4, 126 MHz): δ 158.0, 156.0, 114.5, 112.9, 107.7, 95.9, 95.8, 77.9, 77.4, 68.9, 57.7, 56.6, 55.5, 50.9, 50.3, 38.2, 35.2, 34.4, 33.5, 32.8, 28.1, 27.1, 19.0. IR (neat, cm−1): 2930, 1573, 1429, 1335, 1154, 1106, 1057. Mass spec calcd for C28H41NO6, 487.2934; found, 487.2950. EI+(amu): 487 (M+, 61), 364 (19), 258 (100), 215 (62), 152 (35), 95 (51), 69 (73); [α]23D −60.3° (c 0.029, MeOH).
(6aS,9R,10aR)-6a,7,8,9,10,10a-Hexahydro-1,9-dihydroxy-6,6-dimethyl-6H-benzo[c]chromene-3-carbonitrile 45
To nitrile 31 (132 mg, 0.365 mmol) in 3.5 mL of DCM at rt was added n-BuSH (390 μL, 3.65 mmol) followed by ZnBr2 (544 mg, 2.41 mmol) all at once. The reaction mixture was stirred for 15 min and then diluted with EtOAc, washed with saturated NaHCO3 and brine, and dried over Na2SO4. The crude product was subjected to flash column chromatography on silica gel eluting with 50% then 80% EtOAc/hexanes, resulting in nitrile 45 (90 mg, 90% yield) as a white solid.
1H NMR (MeOH-d4, 300 MHz): δ 6.55 (s, 2H), 4.62 (br s, 1H), 3.80–3.70 (m, 1H), 3.52–3.44 (m, 1H), 2.50 (td, J = 11.2 Hz, 2.4 Hz, 1H), 2.17–2.08 (m, 1H), 1.94–1.86 (m, 1H), 1.52–1.31 (m, 5H), 1.22–1.16 (m, 1H), 1.03 (s, 3H), 1.00–0.89 (m, 1H). 13C NMR (MeOH-d4, 75 MHz): δ 158.7, 157.0, 119.8, 119.6, 113.6, 111.3, 110.6, 78.8, 71.1, 49.7, 39.2, 36.4, 35.2, 28.0, 27.0, 19.1; mp: 212.1 °C (dec). IR (neat, cm−1): 3234 (br), 2982, 2972, 2864, 2224, 1711, 1568, 1424, 1344, 1270, 1057. HRMS calcd for C16H19NO3, 273.1365; found, 273.11360. EI +(amu): 273 (M+, 72), 240 (53), 212 (100), 186 (18), 69 (36); [α]23+D −152.6° (c 0.010, + MeOH).
(6aS,9R,10aR)-6a,7,8,9,10,10a-Hexahydro-1,9-dihydroxy-6,6-dimethyl-6H-benzo[c]chromene-3-carbaldehyde 47
To an ice cold solution of nitrile 45 (140 mg, 0.51 mmol) in 5 mL of DCM was added DIPEA (460 μL, 2.56 mmol) followed by dropwise addition of TESCl (300 μL, 1.31 mmol). The reaction mixture was stirred for 20 min and then quenched with ice cold saturated NaHCO3, diluted with Et2O, washed with brine, and dried over Na2SO4. The crude nitrile was dissolved in 5 mL of DCM, cooled to −78°C, and stirred for 10 min. DIBAL in PhMe (1.10 mL, 1.32 mmol, 1.2 M) was added dropwise, and the resulting mixture was stirred for 1 h. Excess DIBAL was quenched with acetone at −78°C, and the reaction mixture was stirred with saturated Rochelle’s salt at room temperature until the biphasic mixture was clear. EtOAc was added and the organic layer was separated, washed with brine, and dried over Na2SO4. The crude product was purified on a plug of silica gel eluting with 5% EtOAc/hexanes with 2% TEA present. The silylated aldehyde was dissolved in 5 mL of THF, treated with TBAF (550 mg, 1.74 mmol) at rt, and stirred until the reaction was shown to be complete by TLC analysis. Solid CaCO3 was added to the flask and stirred for 15 min. EtOAc was added and the organic layer was separated, washed with brine, and dried over Na2SO4. The crude product was purified via flash column chromatography on silica gel eluting with 50% then 80% EtOAc/hexanes, resulting in aldehyde 47 (44 mg, 61% yield over 3 steps) as a white foam.
1H NMR (CDCl3, 300 MHz): δ 9.76 (s, 1H), 6.85 (d, J = 1.4 Hz, 1H), 6.79 (d, J = 1.4 Hz, 1H), 4.01–3.90 (m, 1H), 3.67–3.63 (m, 1H), 2.55 (td, J = 11.1 Hz, 2.1 Hz, 1H), 2.25–2.16 (m, 1H), 1.97–1.86 (m, 1H), 1.57–1.41 (m, 5H), 1.25–1.15 (m, 2H), 1.07 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 193.9, 158.7, 157.0, 137.4, 120.8, 112.5, 107.2, 78.4, 71.2, 49.9, 39.3, 36.5, 35.5, 28.1, 27.1, 19.1. IR (neat, cm−1): 3338 (br), 2976, 2934, 2872, 1716,1684, 1577, 1558, 1338, 1144, 1057. Mass spec calcd for C16H20O4, 276.1362; found, 276.1375. EI +(amu): 276 (M+, 75), 258 (42), 243 (33), 215 (100), 189 (54), + 142 (66); [α]23+D −154.4° (c 0.007, MeOH).
Procedure for Reductive Amination (12–15). β-C9 1-Adamantyl Benzylamine 12
To a round-bottom flask equipped with a stir bar, reflux condenser, and Dean–Stark trap was added aldehyde 47 (10 mg, 0.036 mmol), 1-adamantanamine (6 mg, 0.040 mmol), and two 4Å molecular sieve beads in benzene. The reaction mixture was heated at reflux overnight. The progress of the reaction was followed by IR, monitoring the disappearance of the carbonyl absorption. The solvent was removed under vacuum, and the crude imine was dissolved in dry MeOH and was treated with a spatula tip of 10% Pd/C. The reaction flask was purged with H2 gas three times and the mixture was allowed to stir at room temperature overnight. The Pd/C was filtered off through a plug of celite. The crude product was purified on silica gel using 2.5, 5, 10% MeOH/DCM as the eluent resulting in amine 12 as a brown oil (12 mg, 81% yield over the 2 steps). HPLC (0.10 cm ×25 cm Luna C8(2) 5 μ, 20–60% MeCN in water (both containing 0.1% HCO2H) over 30 min, 3 mL/min, UV detection at 280 nm) 15.87 min and 96.1% pure.
1H NMR (MeOH-d4, 500 MHz): δ 6.26 (d, J = 1.0 Hz, 1H), 6.24 (d, J = 1.0 Hz, 1H), 3.77–3.70 (m, 1H), 3.52 (br s, 3H), 2.44 (td, J = 11.2 Hz, 2.3 Hz, 1H), 2.14–2.06 (m, 4H), 1.92–1.88 (m, 1H), 1.81–1.63 (m, 13H), 1.47–1.29 (s, 5H), 1.19 (td, J = 12.8 Hz, 3.3 Hz, 1H), 1.01 (s, 3H), 0.95–0.88 (m, 1H). 13C NMR (MeOH-d4, 126 MHz): δ 158.1, 156.3, 140.8, 112.2, 109.8, 108.6, 77.7, 71.3, 52.4, 50.3, 45.3, 42.8, 40.0, 37.7, 36.7, 35.0, 31.0, 28.2, 27.2, 19.1. IR (neat, cm−1): 3282 (br), 2976, 2906, 2844, 1576, 1363, 1232, 1134, 1054. Mass spec calcd for C26H37NO3, 411.2773; found, 411.2760. EI+(amu): 411 (M+, 10), 207 (23), 151 (82), 135 (28), 94 (100), 77 (30), 67 (23); [α]23D −109.5° (c 0.008, MeOH).
Procedure for Suzuki–Miyaura Coupling. β-C9 Styrenyl ketone 54
A 4:1 solution of DMF/EtOH(abs) over 4Å molecular sieves was degassed by bubbling Ar through the solution for 20 min. In a separate flask equipped with a stir bar was added triflate 29 (65 mg, 0.13 mmol), boronate 53 (45 mg, 0.17 mmol), K2CO3 (62 mg, 0.45 mmol), and PdCl2(dppf) 3 DCM (12 mg, 0.015 mmol). The reaction flask was evacuated and purged with Ar three times, then 2 mL of the DMF/EtOH mixture was added and the reaction mixture was heated at 70 °C for 6 h. The flask was cooled to room temperature; the mixture was filtered through cCelite and concentrated directly onto celite. The crude product was purified via flash column chromatography on silica gel eluting with 10, 20, 30, and 40% EtOAc/hexanes, resulting in ketone 54 as a clear, colorless oil as a mixture of diastereomers (55 mg, 87% yield).
1H NMR (CDCl3, 300 MHz): δ 6.63 (d, J = 6.0 Hz, 1H), 6.51–6.47 (m, 1H), 6.12 (d, J = 5.7 Hz, 1H), 5.22–5.12 (m, 2H), 4.73 (dd, J = 10.5 Hz, 6.9 Hz, 1H), 3.79–3.68 (m, 1H), 3.49 (s, 3H), 3.43–3.34 (m, 4H), 2.91–2.83 (br s, 1H), 2.80–1.84 (m, 13H), 1.55–1.25 (m, 5H), 1.20–1.03 (m, 5H). IR (neat, cm−1): 2924, 2853, 1712, 1608, 1564, 1422, 1367, 1209, 1141, 1105, 1056, 1041. Mass spec calcd for C28H38O6, 470.2668; found, 470.2667. EI+ (amu): 470 (M+, 26), 85 (100), 83 (51), 69 (26), 67 (21).
Procedure for NaBH4 Reduction of Styrenyl Ketones. β-C9 endo-Alcohol 56
Ketone 54 (55 mg, 0.117 mmol) in 2 mL of MeOH was cooled to 0 °C and NaBH4 (22 mg, 0.58 mmol) was added all at once and stirred for 30 min. The reaction was quenched with brine and the crude product was extracted with EtOAc and dried over Na2SO4. The crude product was quickly purified via flash column chromatography eluting with 50% EtOAc/hexanes, affording a diastereomeric mixture of endoal-cohol 56 as a clear, colorless oil (50 mg, 90% yield).
1H NMR (C6D6, 300 MHz): δ 6.99–6.95 (m, 2H), 6.43 (d, J = 6.0 Hz, 1H), 5.03–5.00 (m, 1H), 4.92–4.90 (d, J = 6.6 Hz, 1H), 4.77–4.70 (dd, J = 14.6 Hz, 6.8 Hz, 2H), 3.93 (br s, 1H), 3.74–3.67 (m, 2H), 3.28–3.23 (m, 7H), 2.60–2.48 (m, 4H), 2.20–1.81 (m, 5H), 1.68 (dt, J = 14.7 Hz, J = 5.1 Hz, 1H), 1.57–1.22 (m, 8H), 0.96 (s, 3H), 0.82–0.69 (m, 1H). IR (neat, cm−1): 3566 (br), 2974, 2923, 2825, 1610, 1561, 1153, 1106, 1055, 1042. Mass spec calcd for C28H40O6, 472.2825; found, 472.2823. EI+(amu): 472 (M+, 100), 366 (94), 360 (14), 279 (14), 149 (44), 91 (31), 79 (30), 69 (35).
Deprotection and Cyclization Reaction. β-C9 Oxaadamantane 16
To alcohol 56 (26 mg, 0.055 mmol) in 2 mL of DCM was added n-BuSH (135 μL, 1.27 mmol) at rt followed by ZnBr2 (62 mg, 0.28 mmol) all at once. The reaction mixture was stirred for 20 min and then diluted with EtOAc and saturated NaHCO3. The organic layer was washed with brine and dried over Na2SO4. The crude product was purified via flash column chromatography eluting with 40% and then 50% EtOAc/hexanes, affording oxaadamantane 16 (20 mg, 95% yield) as a white glass. Chiral HPLC (0.46 cm ×25 cm Chiralcel AD-H, 50% 2-propanol in hexanes, 1 mL/min, UV detection at 280 nm) 7.50 min and 98.1% pure.
1H NMR (C6D6, 500 MHz): δ 8.51 (br s, 1H, OH), 6.87 (s, 1H), 6.79 (s, 1H), 4.24 (br s, 1H), 4.01–3.99 (m, 1H), 3.85–3.80 (m, 1H), 2.56 (br t, J = 10.8 Hz, 1H), 2.15–1.87 (m, 8H), 1.63–1.15 (m, 12H), 0.97 (s, 3H), 0.81–0.74 (m, 1H). 13C NMR (C6D6, 126 MHz): δ 156.8, 155.6, 147.6, 111.0, 105.8, 104.3, 76.6, 73.0, 71.6, 69.2, 48.5, 42.3, 41.9, 38.6, 35.8, 35.7, 35.2, 34.2, 28.0, 26.4, 19.3. IR (neat, cm−1): 3336 (br), 2975, 2928, 2852, 1622, 1577, 1418, 1051. Mass spec calcd for C24H32O4 + H+, 385.2380; found, 385.2379. [α]23D −98.7° (c 0.008, MeOH)
Binding Assays
Rat brain CB1, mouse and human CB2 binding assays. Compounds were tested for their affinities for the CB1 and CB2 receptors using membrane preparations from rat brain or HEK293 cells expressing either mCB2 or hCB2, respectively, and [3H]CP-55,940, as previously described.22,26–9 Results from the competition assays were analyzed using nonlinear regression to determine the IC50 values for the ligand; Ki values were calculated from the IC5030 (Prizm by GraphPad Software, Inc.). Each experiment was performed in triplicate and Ki values determined from one experiment.
Supplementary Material
Acknowledgment
We thank The National Institute on Drug Abuse (DA07215, 2 P01 DA09158, 5 R01 DA03801) for generous support of this research. The contributions of Yan Peng, Shivangi Joshi, and Han Zhou in performing the binding assays are also acknowledged by the authors. We thank Dr. David Le Goanvic for supplying compounds 2 and 3.
Footnotes
Abbreviations: CB1, cannabinoid 1 receptor; CB2, cannabinoid 2 receptor; DCM, dichloromethane; DIBAL, diisobutylaluminum hydride; DIPEA, N,N-diisopropylethylamine; HEK, human embryonic kidney; L-Selectride, lithium tri-sec-butylborohydride; MOMCl, methyl chloromethyl ether; OPLS, optimized potentials for liquid simulations; PdCl2(dppf), 1,1′-bis(diphenylphosphino)ferrocene palladium(II) dichloride; Pd2(dba)3, tris(dibenzylideneacetone)dipalladium(0); PinB-BPin, bis-pinacolato diborane; rmsd, root-mean-square deviation; SAR, structure–activity relationship; TBAF, tetrabutylammonium fluoride; TEA, triethylamine; TESCl, triethylsilyl chloride; TMSBr, trimethylsilyl bromide; TMSCl, trimethylsilyl chloride; TMSOTf, trimethylsilyl trifluoromethanesulfonate.
Supporting Information Available: Optimized experimental procedures for the synthesis of I. Preparation of vinyl boronate 53. Spectroscopic data for 30, 32, 36–40, 5, 7–11, 42, 44, 46, 48, 13–15, 55, 57, 17, 49–53; reproductions of 1H and 13C NMR spectra of 4–17, 24, 25, 27–50, 52, 53. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Devane WA, Dysarz FA, III., Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- (2).Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structures of a cannabinoid receptor and functional expression of the cloned cDNA. Nature (London) 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- (3).Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- (4).For leading references see: Huffman JW, Miller JRA, Liddle J, Yu S, Thomas BF, Wiley JL, Martin BR. Structure–activity relationships for 1′,1′-dimethylalkyl-Δ8-tetra-hydrocannabinols. Bioorg. Med. Chem. 2003;11:1397–1410. doi: 10.1016/s0968-0896(02)00649-1. Papahatjis DP, Nikas SP, Kourouli T, Chari R, Xu W, Pertwee RG, Makriyannis A. Pharmacophoric requirements for the cannabinoid side chain. Probing the cannabinoid receptor subsite at C1′. J. Med. Chem. 2003;46:3221–3229. doi: 10.1021/jm020558c. Papahatjis DP, Nikas SP, Andreou T, Makriyannis A. Novel 1′,1′-chain substituted Δ8-tetrahydrocannabinols. Bioorg. Med. Chem. Lett. 2002;12:3583–3586. doi: 10.1016/s0960-894x(02)00785-0. Khanolkar AD, Lu D, Fan P, Tian X, Makriyannis A. Novel conformationally restricted tetracyclic analogs of Δ8-tetrahydrocannabinol. Bioorg. Med. Chem. Lett. 1999;9:2119–2124. doi: 10.1016/s0960-894x(99)00355-8. Huffman JW, Liddle J, Duncan SG, Jr., Yu S, Martin BR, Wiley JL. Synthesis and pharmacology of the isomeric methylheptyl-Δ8-tetrahydrocannabinols. Bioorg. Med. Chem. 1998;6:2383–2396. doi: 10.1016/s0968-0896(98)80014-x. Papahatjis DP, Kourouli T, Abadji V, Goutopoulos A, Makriyannis A. Pharmacophoric requirements for cannabinoid side chains: multiple bond and C1′-substituted Δ8-tetrahydrocannabinols. J. Med. Chem. 1998;41:1195–1200. doi: 10.1021/jm970277i. Busch-Petersen J, Hill WA, Fan P, Khanolkar A, Xie X-Q, Tius MA, Makriyannis A. Unsaturated side chain β-11-hydroxyhexahydrocannabinol analogs. J. Med. Chem. 1996;39:3790–3796. doi: 10.1021/jm950934b.
- (5).(a) Roesch S, Ramer R, Brune K, Hinz B. (R)-Methanandamide and other cannabinoids induce the expression + of cyclooxygenase-2 and matrix metalloproteinases in human nonpigmented ciliary epithelial cells. J. Pharm. Exp. Ther. 2006;316:1219–1228. doi: 10.1124/jpet.105.092858. [DOI] [PubMed] [Google Scholar]; (b) Jarvinen T, Pate DW, Laine K. Cannabinoids in the treatment of glaucoma. Pharmacol. Ther. 2002;95:203–220. doi: 10.1016/s0163-7258(02)00259-0. [DOI] [PubMed] [Google Scholar]
- (6).(a) Huang SM, Walker JM. In: Cannabinoids as Therapeutics. Raphael M, editor. Birkh€auser Verlag; Basel: 2005. p. 149. [Google Scholar]; (b) Mbvundula EC, Rainsford KD, Bunning RAD. Cannabinoids in pain and inflammation. Inflammopharmacology. 2004;12:99–114. doi: 10.1163/1568560041352275. [DOI] [PubMed] [Google Scholar]; (c) Smith PF. Cannabinoids in the treatment of pain and spasticity in multiple sclerosis. Curr. Opin. Invest. Drugs. 2002;3:859–864. [PubMed] [Google Scholar]
- (7).(a) Barth F, Rinaldi-Carmona M. In: Cannabinoids as Therapeutics. Raphael M, editor. Birkh€auser Verlag; Basel: 2005. p. 219. [Google Scholar]; (b) Koch JE. Δ9-THC stimulates food intake in Lewis rats: effects on chow, high-fat and sweet high-fat diets. Pharmacol., Biochem. Behav. 2001;68:539–543. doi: 10.1016/s0091-3057(01)00467-1. [DOI] [PubMed] [Google Scholar]
- (8).(a) Adams R. Marihuana. Harvey Lect. 1942;37:168–197. [Google Scholar]; (b) Adams R, Harfenist M, Lowe S. New analogs of tetrahydrocannabinol. XIX. J. Am. Chem. Soc. 1949;71:1624–1628. [Google Scholar]
- (9).Lu D, Meng Z, Thakur GA, Fan P, Steed J, Tartal CL, Hurst DP, Reggio PH, Deschamps JR, Parrish DA, George C, J€arbe TUC, Lamb RJ, Makriyannis A. Adamantyl cannabinoids: a novel class of cannabinergic ligands. J. Med. Chem. 2005;48:4576–4585. doi: 10.1021/jm058175c. See also: McLaughlin PJ, Lu D, Winston KM, Thakur G, Swezey A, Makriyannis A, Salamone JD. Behavioral effects of the novel cannabinoid full agonist AM 411. Pharmacol., Biochem. Behav. 2005;81:78–88. doi: 10.1016/j.pbb.2005.02.005. J€arbe TUC, DiPatrizio NV, Lu D, Makriyannis A. (–)-Adamantyl-Δ8-tetrahydrocannabinol (AM-411), a selective cannabinoid CB1 receptor agonist: effects on open-field behaviors and antagonism by SR-141716 in rats. Behav. Pharmacol. 2004;15:517–521. doi: 10.1097/00008877-200411000-00008.
- (10).Le Goanvic D, Tius MA. Oxaza adamantyl cannabinoids. A new class of cannabinoid receptor probes. J. Org. Chem. 2006;71:7800–7804. doi: 10.1021/jo061352c. [DOI] [PubMed] [Google Scholar]
- (11).(a) Stetter H, Heckel K. Compounds with the adamantane structure synthesized by cyclization reactions with N,N-dibromo-p-toluenesulfonamide. Tetrahedron Lett. 1972;13:801–804. [Google Scholar]; (b) Stetter H, Heckel K. Compounds with urotropine structure. LII. Cyclization reactions with cis,cis-1,5-cyclooctadiene as the precursor. Chem. Ber. 1973;106:339–348. [Google Scholar]
- (12).(a) Portmann RE, Ganter C. Heterotricyclodecane. XVI. 2-Oxa-6-azaadamantane and derivatives. Helv. Chim. Acta. 1973;56:1962–1985. [Google Scholar]; (b) Ganter C, Portmann RE. Synthesis of 2-thia-6-azaadaman-tanes and derivatives. Helv. Chim. Acta. 1971;54:2069–2077. [Google Scholar]
- (13).An improved synthesis of oxazaadamantane I is described in the Supporting Information.
- (14).Archer RA, Blanchard WB, Day WA, Johnson DW, Lavagnino ER, Ryan CW, Baldwin JE. Cannabinoids. 3. Synthetic approaches to 9-ketocannabinoids. Total synthesis of nabilone. J. Org. Chem. 1977;42:2277–2284. doi: 10.1021/jo00433a020. [DOI] [PubMed] [Google Scholar]
- (15).Modified procedure of: Chittiboyina AG, Reddy CR, Watkins EB, Avery MA. First synthesis of antimalarial Machaeriols A and B. Tetrahedron Lett. 2004;45:1689–1691.
- (16).(a) Kubota H, Rice KC. Palladium-catalyzed cyanation of hindered, electron-rich aryl triflates by zinc cyanide. Tetrahedron Lett. 1998;39:2907–2910. [Google Scholar]; (b) Martin MT, Liu B, Cooley BE, Jr., Eaddy JF. Open air palladium catalyzed cyanation-the use of PMHS to protect from oxygen. Tetrahedron Lett. 2007;48:2555–2557. [Google Scholar]
- (17).Sohn J-H, Waizumi N, Zhong HM, Rawal VH. Total synthesis of mycalamide A. J. Am. Chem. Soc. 2005;127:7290–7291. doi: 10.1021/ja050728l. [DOI] [PubMed] [Google Scholar]
- (18).Shibuya M, Tomizawa M, Suzuki I, Iwabuchi Y. 2-Azaadamantane N-oxyl (AZADO) and 1-Me-AZADO: highly efficient organocatalysts for oxidation of alcohols. J. Am. Chem. Soc. 2006;128:8412–8413. doi: 10.1021/ja0620336. [DOI] [PubMed] [Google Scholar]
- (19).Ishiyama T, Takagi J, Kamon A, Miyaura N. Palladium-catalyzed cross-coupling reaction of bis(pinacolato)diboron with vinyl triflates β-substituted by a carbonyl group: efficient synthesis of β-boryl-R,β-unsaturated carbonyl compounds and their synthetic utility. J. Organomet. Chem. 2003;687:284–290. [Google Scholar]
- (20).Ghosh S, Kinney WA, Gauthier DA, Lawson EC, Hudlicky T, Maryanoff BE. Convenient preparation of aryl-substituted nortropanes by Suzuki–Miyaura methodology. Can. J. Chem. 2006;84:555–560. [Google Scholar]
- (21).(a) Tius MA, Makriyannis A, Zou XL, Abadji V. Conformationally restricted hybrids of CP-55,940 and HHC: stereoselective synthesis and activity. Tetrahedron. 1994;50:2671–2680. [Google Scholar]; (b) Tius MA, Hill WAG, Zou XL, Busch-Petersen J, Kawakami JK, Fernandez-Garcia MC, Drake DJ, Abadji V, Makriyannis A. Classical/nonclassical cannabinoid hybrids; stereochemical requirements for the southern hydroxyalkyl chain. Life Sci. 1995;56:2007–2012. doi: 10.1016/0024-3205(95)00182-6. [DOI] [PubMed] [Google Scholar]
- (22).Dodd PR, Hardy JA, Oakley AE;, Edwardson JA, Perry EK;, Delaunoy JP. A rapid method for preparing synaptosomes: comparison, with alternative procedures. Brain Res. 1981;226:107–118. doi: 10.1016/0006-8993(81)91086-6. [DOI] [PubMed] [Google Scholar]
- (23).Jorgensen WL, Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- (24).Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with Accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- (25).Macromodel. Schrodinger, LLC; New York, NY: 2007. version 9.5. [Google Scholar]
- (26).Abadji V, Lucas-Lenard JM, Chin C, Kendall DA. Involvement of the carboxyl terminus of the third intracellular loop of the cannabinoid receptor in constitutive activation of Gs. J. Neurochem. 1999;72:2032–2038. doi: 10.1046/j.1471-4159.1999.0722032.x. [DOI] [PubMed] [Google Scholar]
- (27).Lan R, Lui Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A. Structure–activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J. Med. Chem. 1999;42:769–776. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- (28).Morse KL, Fournier DJ, Li X, Grzybowska J, Makriyannis A. A novel electrophilic high affinity irreversible probe for the cannabinoid receptor. Life Sci. 1995;56:1957–1962. doi: 10.1016/0024-3205(95)00176-7. [DOI] [PubMed] [Google Scholar]
- (29).Guo Y, Abadji V, Morse KL, Fournier DJ, Li X, Makriyannis A. (–)-11-Hydroxyl-7′-isothiocyanato-1′,1′-dimethylheptyl-D8-THC: a novel, high-affinity irreversible probe for the cannabinoid receptor in the brain. J. Med. Chem. 1994;37:3867–3870. doi: 10.1021/jm00049a002. [DOI] [PubMed] [Google Scholar]
- (30).Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.