

Abstract
The chimeric fusion protein, AML1-ETO, generated by translocation of t(8;21), abnormally recruits histone deacetylase (HDAC) to the promoters of AML1 target genes, resulting in transcriptional repression of the target genes and development of t(8;21) acute myeloid leukemia. Abnormal expression of cyclin-dependent kinase inhibitors, especially p21, is considered a possible mechanism of the arrested maturation and differentiation seen in leukemia cells. A new generation of HDAC inhibitors is becoming an increasing focus of attention for their ability to induce differentiation and apoptosis in tumor cells and to block the cell cycle. Our previous research had demonstrated that valproic acid induces G0/G1 arrest of Kasumi-1 cells in t(8;21) acute myeloid leukemia. In this study, we further confirmed that valproic acid inhibits the growth of Kasumi-1 cells in a murine xenograft tumor model, and that this occurs via upregulation of histone acetylation in the p21 promoter region, enhancement of p21 expression, suppression of phosphorylation of retinoblastoma protein, blocking of transcription activated by E2F, and induction of G0/G1 arrest.
Keywords: valproic acid, acute myeloid leukemia, AML1-ETO, p21, E2F
Introduction
Acute myeloid leukemia (AML) is the most common malignant myeloid disease in adults, and is a heterogeneous clonal disorder of hematopoietic progenitor cells. The t(8;21)(q22;q22) translocation is one of the most frequent chromosomal rearrangements in AML. AML1-ETO fusion protein, a protein product resulting from t(8;21) chromosomal translocation, abnormally recruits histone deacetylase (HDAC) and inhibits transcription of AML1 target genes via deacetylation of histone, which plays a central role in the development of t(8;21) AML.1 Arrest of the cell cycle is crucial for cell differentiation, and p21, an important regulator of the cell cycle and the first recognized broad-spectrum cyclin-dependent kinase (CDK) inhibitor gene, induces suppression of almost all cyclin-CDK complexes and blockage of phosphorylation of retinoblastoma protein (Rb), resulting in G0/G1 arrest.2–4 It has been found that the p21WAF1/CIP1 promoter contains an AML1 recognition sequence and that mSin3A interacts with AML1 to modulate transcription of p21WAF1/CIP1.5 However, p21 is not expressed in some tumor cells, although p21WAF1/CIP1 is considered a possible target gene in AML.6–8
As already reported, several HDAC inhibitors can induce cell cycle arrest in various tumor cell lines via activation of CDK p21WAF1/CIP expression.9–12 Recently, valproic acid, a short chain fatty acid, was found to have HDAC-inhibiting properties, which induced inhibition of proliferation, apoptosis, and cell cycle arrest when used in the treatment of malignancies.
Kasumi-1 cells, which carry the characteristic AML1-ETO fusion protein, are an ideal cell line for the study of t(8;21) AML. Our previous research has demonstrated that valproic acid significantly inhibits growth of Kasumi-1 cells via G0/G1 arrest.13 In the present study, we established a nude mouse xenograft model of t(8;21) AML to explore further the ability of valproic acid to inhibit tumor growth and arrest the cell cycle.
Materials and methods
Cell culture and reagents
Kasumi-1 cells grown from human AML with t(8;21) were cultured in RPMI 1640 medium containing 10% fetal bovine serum at 37°C in a 5% CO2 incubator under saturated humidity conditions. Valproic acid was sourced from Sigma (St Louis, MO, USA) and diluted with normal saline to 25 mg/mL. Female BALB/c null mice (weight 10–15 g, aged 4–6 weeks) from Beijing Vital River Laboratories (Beijing, People’s Republic of China) were housed in a specific pathogen-free environment.
In vivo antitumor activity test
One week after splenectomy, when the incision had healed the mice were irradiated with 400 cGy 137Cs rays for 48–72 hours. After irradiation, Kasumi-1 cells in the logarithmic growth phase were subcutaneously injected into the mice at the right axilla. The amount of inoculation was approximately 2 × 107 cells per mouse (0.15–0.2 mL). When the tumor volume had reached about 200 mm3 (10 days after inoculation), the mice were randomly divided into two groups (n = 6 per group) to receive daily intraperitoneal treatment with normal saline 0.2 mL (control group) or valproic acid 500 mg/kg in an injection volume of 0.2 mL (valproic acid group). A caliper was used to measure the maximum diameter (a) and minimum diameter (b) of the mouse xenograft tumors every three days. Tumor volume (V) was calculated according to the formula V = 4π/3 × (b/2)2 × (a/2); the relative tumor volume (RTV) was calculated as the measured volume each time/initial volume. Antitumor activity was estimated using the inhibition rate (IR%); IR% = (1 − RTV of valproic acid arm/RTV of control arm) ×100%. After two weeks of daily injections, the mice were euthanized for analysis.
Detection of cell cycle by flow cytometry
Freshly harvested xenograft tumor tissues from the control and treatment groups were washed with phosphate-buffered solution and then soaked further in phosphate-buffered solution. The tissues were then divided into small pieces using eye scissors and filtered through a 200-mesh sieve to prepare a unicellular suspension for examination of the cell cycle.
Reverse transcriptase polymerase chain reaction
RNA extraction from the xenograft tissues was done using the one-step method recommended in the manual for TRIzol® (Invitrogen, Carlsbad CA, USA). The concentration of RNA was adjusted to 1 μg/μL. Polymerase chain reaction (PCR) amplification was performed using Taq polymerase (Takara Biotechnology, Dalian, People’s Republic of China) with cDNA synthesized by Moloney murine leukemia virus (M-MuLV) reverse transcriptase (MBI Company, New York, NY, USA) as a template and β-actin serving as the internal control. The primers used for PCR were: cyclin D1, 5′-CTGTCCTCCCTCACACGTCA-3′, 5′-GAAAG-TAGGGACCTCAGAGG-3′; cyclin E1, 5′-GTCACAT-ACGCAAACTGG-3′, 5′-TTTCTTGAGCAACACCCT-3′; CDK4, 5′-CTGAGAATGGCTACCTCTCGATATG-3′, 5′-AGAGTGTAACAACCACGGGTGTAAG-3′; CDK6, 5′-CCGAGTAGTGCATCGCGATCTAA-3′, 5′-CTTTGCCTAGTTCATCGATATC-3′; and p21WAF1/CIP, 5′-GCCCGCTCTACATCTTCTG-3′, 5′-GTGCCATCTGTTTACTTCTCAA-3′.
Western blotting
First, 1 mL of precooled phosphate-buffered solution was added to 100 mg of tumor tissue to make a homogenate, which was then washed three times until the liquid appeared clear. RIPA lysis buffer was added to the cell pellet and incubated on ice for 40 minutes. A 12,000× g centrifugation was then performed at 4°C, the supernatant was collected and the protein concentration was detected using the Bradford method. Next, 50 μg of protein was analyzed by polyacrylamide gel electrophoresis and electrotransferred to a polyvinylidene difluoride membrane. After a two-hour incubation with 5% skim milk powder at room temperature, primary antibodies (mouse antihuman cyclin D1 monoclonal antibody, 1:400; mouse antihuman E2F monoclonal antibody, 1:500; mouse antihuman β-actin monoclonal antibody, 1:1000) were added to the polyvinylidene difluoride membrane for overnight shake incubation at 4°C. Antirabbit secondary antibody (1:5000) labeled with horseradish peroxidase was used for detection of protein expression.
Chromatin immunoprecipitation
This assay was operated according to the instructions of a chromatin immunoprecipitation assay kit (Upstate Biotechnology, Lake Placid, NY, USA). Briefly, after the cells were prepared with 1% formaldehyde and kept at room temperature for 10 minutes, small pieces of tumor tissue from each treatment group were resuspended and precipitated using cell lysis solution. Ultrasonic processing was performed five times (80 W, for 10 seconds each time; interval 60 seconds). 20 μL of preparation served as input control, 3 μg of rabbit antihuman Ac-H3 monoclonal antibody were added to the preparation and incubated at 4°C overnight. Protein A agarose slurry was added to the preparation, followed by incubation at 4°C for one hour to collect the antibody-protein-DNA complex. After incubation, centrifugation was performed and the supernatant was discarded. The precipitate was washed five times with wash buffer provided by kit (low salt, high salt, LiCl immune complex wash buffer and 1× TE buffer, in that order). The protein-DNA complex was isolated using elution buffer. The complex was then incubated at 65°C with 8 μL of 0.9% NaCl over four hours for decrosslinking. Next, the dissociated DNA fragment was extracted for further purification. PCR amplification was performed using p21 promoter primer under conditions of 94°C for three minutes, 94°C for 20 seconds, 57°C for 30 seconds, 72°C for 30 seconds, and 72°C for two minutes, for a total of 32 cycles. Anti-RNA polymerase served as the positive control and immunoglobulin G as the negative control.
Protein immunoprecipitation
First, 1 mL of precooled phosphate-buffered solution was added to 100 mg of tumor tissue to make a homogenate, which was washed three times until the liquid appeared clear. After incubation with 1 mL of RIPA cell lysis solution for 30 minutes, the supernatant protein extract was obtained by 20 minutes of centrifugation at 12,000 rpm. The protein extract was then incubated with 60 μL of protein G agarose and 4 μg of immunoglobulin G at 4°C over two hours of mild shaking to prevent nonspecific binding. Next, a one-minute centrifugation at 4000 rpm was performed at 4°C, and the supernatant was transferred to another centrifuge tube. The supernatant was divided into two portions (one with 5 μg of E2F antibody, the other with 4 μg of immunoglobulin G as the negative control), and followed by mild shaking overnight at 4°C. Meanwhile, two solutions (containing 60 μL of protein G agarose [according to the antibody] and 1 mL of RIPA cell lysis solution) were incubated overnight at 4°C with mild shaking. The next day, the solutions containing protein G agarose were pretreated with a one-minute centrifugation at 4000 rpm and 4°C, and the supernatant was removed. The pretreated protein G agarose was then added to the protein extract with E2F or immunoglobulin G, respectively, for three hours of mild shaking. A further one-minute centrifugation (4000 rpm) was performed at 4°C and the supernatant was discarded. The precipitate was washed (at 4°C with mild shaking) with precooled RIPA cell lysis solution for 10 minutes five times. Once again, a one-minute centrifugation was performed at 4000 rpm. Next, 15 μL of 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer was added to each sample for a 10-minute boiling. The supernatant was used for loading the DNA samples. Electrophoresis was then performed, and Rb monoclonal antibody was used for the final hybridization.
Statistical analysis
Each experiment was repeated at least three times, and the results were analyzed with Statistical Package for the Social Sciences version 16.0 software (SPSS Inc, Chicago, IL, USA) using the Student’s t-test. A statistically significant difference was considered at P < 0.05 or P < 0.01.
Results
Establishment of a xenograft tumor model using Kasumi-1 cells
Xenograft tumors appeared three days after inoculation of Kasumi-1 cells. The tumor formation rate was 100% and tumor volume increased gradually (Figure 1A). Pathological examination showed no tumor involvement in the peripheral blood, lung, and liver. Expression of the typical AML1/ETO fusion gene in the tumor tissue was confirmed by reverse transcriptase PCR, indicating successful establishment of a xenograft model of Kasumi-1 cells (Figure 1B).
Figure 1.
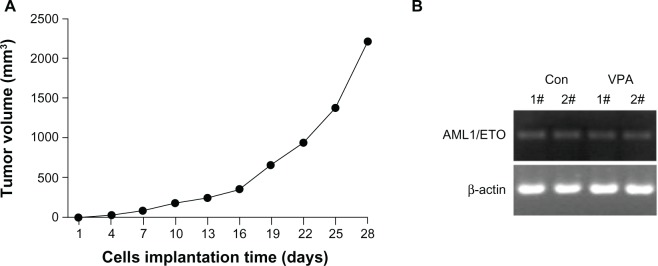
Establishment of Kasumi-1 cell xenograft tumor. (A) growth of xenograft tumor after inoculation of Kasumi-1 cells. Data are represented as the mean. (B) Expression of the typical AML1/ETO fusion gene detected by reverse transcriptase polymerase chain reaction in the control group and the valproic acid group after 28 days of treatment.
Abbreviations: Con, control group; VPA, valproic acid group.
Inhibition of xenograft by valproic acid
Treatment with valproic acid was started when the tumor volume reached a certain size. Growth of the xenograft was compared between the treatment and control groups. As shown in Figure 2A, compared with the control group, the group treated with valproic acid showed a marked reduction in tumor size. Tumor size in the xenografted valproic acid group after 13 days of treatment was also much smaller than in the control group (Figure 2B and C). The mice were then euthanized and the tumors were isolated. A significant decrease in tumor volume was observed after treatment with valproic acid (Figure 2D and E). These results indicate that treatment with valproic acid significantly inhibited tumor growth in a xenograft animal model of Kasumi-1 cells.
Figure 2.

Inhibition of Kasumi-1 xenograft tumor growth by valproic acid. (A) Curve chart demonstrated a significant difference in tumor growth between control arm and valproic acid group after two weeks of treatment. Data are shown as the mean ± standard error of the mean. (B and C) Comparison of the nude mice with xenografts between the control group and the valproic acid group. (D and E) Comparison of tumor size after sacrifice of the mice.
Abbreviations: Con, control group; VPA, valproic acid group.
Valproic acid induces G0/G1 arrest
Using flow cytometry to investigate how valproic acid decreases tumor burden, we found that valproic acid significantly increased the number of cells in G0/G1 phase (74.05% ± 3.06% in the valproic acid group versus 60.93% ± 1.18% in the control group, P < 0.01) and markedly decreased the number of cells in S phase (20.37% ± 1.78% in the valproic acid group versus 30.03% ± 1.02% in the control group, P < 0.01, Figure 3). These results suggest that valproic acid can cause cell cycle arrest in G0/G1 phase to reduce tumor burden.
Figure 3.
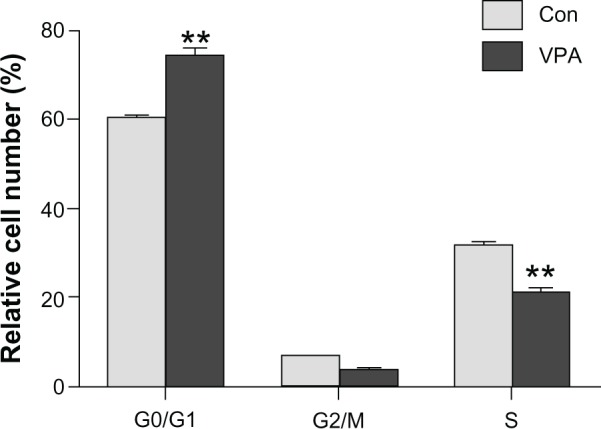
Detection of the cell cycle in Kasumi-1 cell xenograft tumor model by flow cytometry. Analysis of cell cycle after two weeks of treatment with valproic acid. **P < 0.01.
Abbreviations: Con, control group; VPA, valproic acid group.
Valproic acid downregulates transcription of cyclin D1, cyclin E1, CDK4, and CDK6, and upregulates p21 transcription and protein expression
It is well known that the cell cycle in mammals is controlled via regulation of the cyclin-CDK complex in G1 phase. G1 cyclins include cyclin D1, cyclin D2, cyclin D3, cyclin C, and cyclin E. Cyclin D1 is probably a mediator that couples external signals with the cell cycle. The cyclin D1-CDK4/CDK6 complex helps cells to pass through the G1 checkpoint via activation of CDK, phosphorylation of Rb, release of transcription factor E2F, and inducing expression of genes which drive cells to S phase. Cyclin E, synthesized in mid G1 phase and activated by costimulation of E2F and Myc, is a necessary regulator of G1/S transition. Formation of the cyclin D-CDK4/CDK6 complex induces phosphorylation of Rb and release of E2F, resulting in expression of cyclin E and CDK2 and positive feedback on phosphorylation of Rb, giving rise to further phosphorylation of Rb, thereby enabling the cell to pass through the G1/S checkpoint.
Semiquantitative reverse transcriptase PCR was performed to determine whether or not valproic acid influences cyclin expression, and showed that valproic acid induced a significant reduction in mRNA expression of cyclin D1, cyclin E1, CDK4, and CDK6 in the xenografts (Figure 4A). Western blotting also revealed that expression of cyclin D1 decreased markedly after treatment with valproic acid (Figure 4B). These results indicate that valproic acid induced G0/G1 arrest by inhibition of expression of cyclin D1-CDK4/6 and cyclin E1-CDK2.
Figure 4.
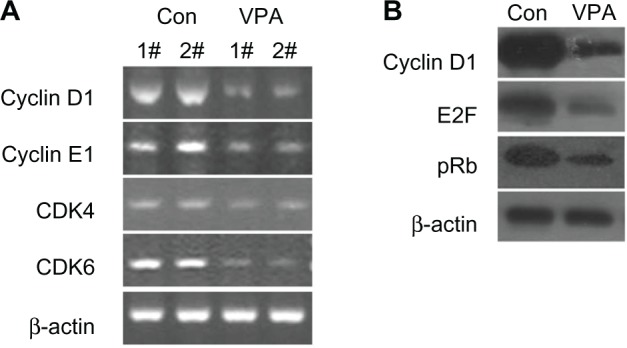
Valproic acid downregulates transcription of cyclin D1, cyclin E1, CDK4, and CDK6, and upregulates transcription and protein expression of p21. Comparison between control group and valproic acid after two weeks of treatment. (A) Expression of cyclin D1, cyclin E1, CDK4, and CDK6 detected by reverse transcriptase polymerase chain reaction. (B) Expression of cyclin D1, E2F, and retinoblastoma protein by Western blotting.
Abbreviations: Con, control group; VPA, valproic acid group; CDK, cyclin-dependent kinase.
Valproic acid influences Rb, E2F, and Rb-E2F complexes
In G1 phase, E2F is not released until formation of Rb resulting from phosphorylation of cyclin-CDK. E2F release promotes the necessary protein transcription of cyclin and CDK, facilitating transition to G1/S. To test whether suppression of cyclin D1-CDK4/6 and cyclin E1-CDK2 expression by valproic acid leads to G0/G1 arrest via reduction of free E2F1, we initially detected expression of Rb and E2F (Figure 4B), which was downregulated after treatment with valproic acid. A chromatin immunoprecipitation assay then showed that treatment with valproic acid induced a significant increase in Rb/E2F complexes compared with the control group (Figure 5). These findings indicate that valproic acid induces G0/G1 arrest via reduction of free E2F.
Figure 5.
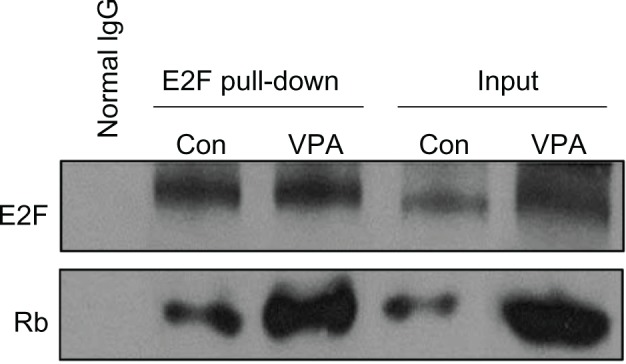
Valproic acid influences retinoblastoma protein, E2F-1, and retinoblastoma protein-E2F complex. Detection of E2F and retinoblastoma protein binding by the protein complex immunoprecipitation method in mice after treatment with valproic acid for two weeks.
Abbreviations: Con, control group; VPA, valproic acid group.
Valproic acid upregulates p21 expression by enhancing acetylation of histone H3 in the p21 promoter region
p21, a CDK inhibitor, is an important cell cycle regulator, and participates in negative control of the cell checkpoint by blocking CDK activity. It had been shown that AML1-ETO upregulates expression of p21 and that AML1-ETO (+) leukemia can be inhibited by activation of the p21 pathway (Liu S, Klisovic RB, et al, 2007; and Peterson LF, Yan M, et al, 2007).27,28 Using semiquantitative reverse transcriptase PCR to test whether valproic acid upregulates expression of p21, we found that valproic acid induced a significant increase in expression of both p21 mRNA (Figure 6A) and p21 protein (Figure 6B) in a mouse xenograft model.
Figure 6.
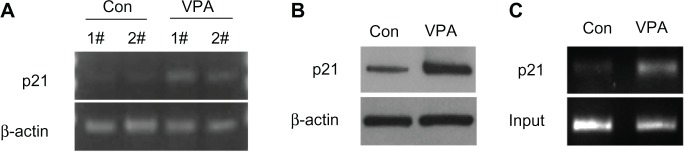
Valproic acid upregulates p21 expression by enhancing acetylation of histone H3 in p21 promoter region. Comparison between control group and valproic acid group after two weeks of treatment. (A) Detection of p21 expression by reverse transcriptase polymerase chain reaction. (B) Detection of p21 expression by Western blotting. (C) Detection of histone H3 acetylation level in p21 promoter by chromatin immunoprecipitation.
Abbreviations: Con, control group; VPA, valproic acid group.
To investigate how valproic acid inhibits p21 gene transcription, we assumed that valproic acid downregulates p21 transcription via upregulation of histone acetylation in the p21 promoter region. Using chromatin immunoprecipitation analysis, we found that acetylation of histone H3 in the p21 promoter region was significantly enhanced after treatment with valproic acid (Figure 6C).
Discussion
Histone is a component of chromatin and plays a vital role in the regulation of gene expression. This regulation occurs via the combination and dissociation of histone and DNA, resulting from local acetylation of histone.14–16 The acetylation state of histone is dynamically regulated by histone acetyltransferases which promote histone acetylation, resulting in a loose chromatin structure in favor of gene transcription, whereas HDAC induces deacetylation of histone, leading to a tight chromatin structure which prevents gene transcription.17 Abnormal recruitment of HDAC and the relevant gene change associated with chromatin modification may lead to alteration in gene expression.18,19 It has already been found that HDAC is associated with development and progression of leukemia,20,21 and that an abnormally recruited HDAC complex plays a critical role in AML1-ETO fusion protein-related oncogenesis in t(8;21) AML.22 Valproic acid, a widely used anticonvulsant, was recently found to have HDAC-inhibiting properties, and induces inhibition of proliferation, apoptosis, and arrest of the cell cycle when used in the treatment of solid tumors and hematological malignancies.
We established a murine model of a Kasumi-1 xenograft tumor for leukemia. An in vivo study demonstrated that valproic acid has an antileukemic effect, in that two weeks of treatment with this agent retarded xenograft tumor growth significantly in comparison with growth in a control group. Flow cytometry further demonstrated significant G0/G1 arrest in tumor cells. Thus, our study confirmed that valproic acid can induce cell cycle arrest, thereby inhibiting proliferation of leukemic cells.
CDK inhibitors are important regulators of the cell cycle, and include p21, p27, p15, and p16.23 Numerous studies have shown that cell cycle arrest is partly due to attenuation of expression of CKI p21 by HDAC inhibitors; promoter of P21 contains AML1 recognition sequence, and a combination of AML1 and mSin3A modulates transcription of p21WAF1/CIP1. Therefore, p21WAF1/CIP1 is considered to be one of the target genes of AML1. The alteration in p21 expression caused by valproic acid and the associated mechanism were explored in this study.
Using reverse transcriptase PCR and Western blotting, we found that valproic acid induced increased expression of p2 protein in a Kasumi-1 xenograft tumor model. The p21WAF1/ CIP promoter locus had been identified as the target of HDAC inhibitors, including butyrate and (R)-trichostatin A.24 In our study, a chromatin immunoprecipitation assay further demonstrated enhancement of histone acetylation in the p21 promoter region. This confirms that valproic acid upregulates p21 expression via suppression of HDAC, histone acetylation enhancement in the promoter region, and promotion of transcription.
In fact, numerous CDK inhibitors, including p21, exert their effects by blocking phosphorylation of Rb.25 p21 inhibits the activity of CPK, resulting in blockage of Rb phosphorylation; with binding of hypophosphorylated Rb to E2F, downstream target genes regulated by E2F are suppressed, thereby preventing replication of DNA, leading to arrest of the cell cycle at G0/G1. In this study, downregulation of pRb and E2F expression was detected by Western blotting, while the rise in Rb/E2F complex was confirmed by protein complex immunoprecipitation.
In addition, we also found that valproic acid induced direct downregulation of protein/gene expression of cyclin D1, cyclin E1, CDK4, and CDK6 in a xenograft tumor model. There is also a possibility, as we have already speculated, that valproic acid regulates the cell cycle by directly inhibiting the expression of cyclin D1, cyclin E1, CDK4, and CDK6. Jin et al26 also reported that the HDAC inhibitor, vorinostat, inhibits HDAC activity by downregulating cyclin D1 protein expression in colonic carcinoma cells in xenografted mice.
In conclusion, our study confirms that valproic acid significantly suppresses growth of Kasumi-1 cell xenograft tumors in nude mice via cell cycle arrest. The antitumor activity of valproic acid is possibly attributable to its direct downregulation of cyclin D1, CDK4/6, and cyclin E1 expression, blockage of Rb phosphorylation, reduction of free E2F, inhibition of HDAC activity, attenuation of histone acetylation in the p21 promoter region, upregulation of p21 expression, and induction of cell cycle arrest. The working mechanism of valproic acid in treating Kasumi-1 xenograft tumors in mice is summarized in Figure 7.
Figure 7.
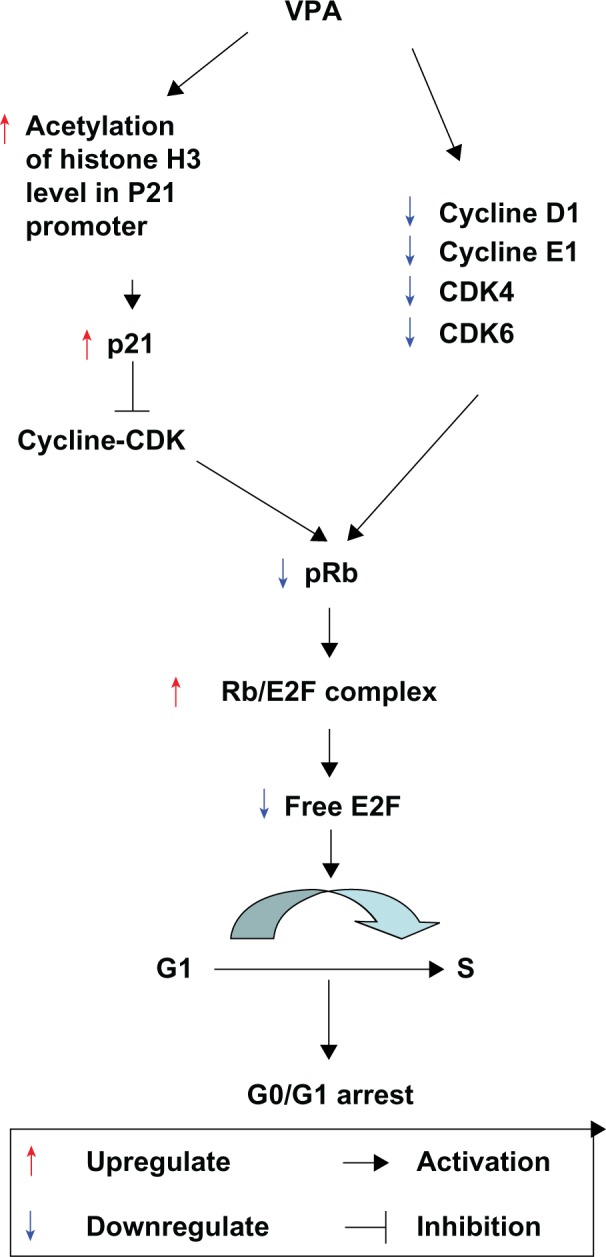
Working mechanism of valproic acid in treating Kasumi-1 cell xenograft tumors in mice. Valproic acid directly downregulates cyclin D1, CDK4/6, and cyclin E1 expression, blocks retinoblastoma protein phosphorylation, reduces free E2F, inhibits HDAC activity, attenuates histone acetylation in the p21 promoter region, upregulates p21 expression, and induces cell cycle arrest.
Abbreviations: Con, control group; VPA, valproic acid group; CDK, cyclin-dependent kinase.
Acknowledgment
This study was supported by the Natural Science Foundation of Hebei Province (C2008000660).
Footnotes
Disclosure
The authors declare that they have no competing interests in this work.
References
- 1.Hug BA, Lazar MA. ETO interacting proteins. Oncogene. 2004;23:4270–4274. doi: 10.1038/sj.onc.1207674. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 3.Lee YM, Sicinski P. Targeting cyclins and cyclin-dependent kinases in cancer: lessons from mice, hopes for therapeutic applications in human. Cell Cycle. 2006;5:2110–2114. doi: 10.4161/cc.5.18.3218. [DOI] [PubMed] [Google Scholar]
- 4.Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell. 2003;4:13–18. doi: 10.1016/s1535-6108(03)00165-x. [DOI] [PubMed] [Google Scholar]
- 5.Lutterbach B, Westendorf JJ, Linggi B, Isaac S, Seto E, Hiebert SW. A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. J Biol Chem. 2000;275:651–656. doi: 10.1074/jbc.275.1.651. [DOI] [PubMed] [Google Scholar]
- 6.Ogino S, Nosho K, Shima K, et al. p21 expression in colon cancer and modifying effects of patient age and body mass index on prognosis. Cancer Epidemiol Biomarkers Prev. 2009;18:2513–2521. doi: 10.1158/1055-9965.EPI-09-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allan LA, Duhig T, Read M, Fried M. The p21(WAF1/CIP1) promoter is methylated in Rat-1 cells: stable restoration of p53-dependent p21(WAF1/CIP1) expression after transfection of a genomic clone containing the p21(WAF1/CIP1) gene. Mol Cell Biol. 2000;20:1291–1298. doi: 10.1128/mcb.20.4.1291-1298.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roman-Gomez J, Castillejo JA, Jimenez A, et al. 5′ CpG island hypermethylation is associated with transcriptional silencing of the p21(CIP1/WAF1/SDI1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood. 2002;99:2291–2296. doi: 10.1182/blood.v99.7.2291. [DOI] [PubMed] [Google Scholar]
- 9.Sakajiri S, Kumagai T, Kawamata N, Saitoh T, Said JW, Koeffler HP. Histone deacetylase inhibitors profoundly decrease proliferation of human lymphoid cancer cell lines. Exp Hematol. 2005;33:53–61. doi: 10.1016/j.exphem.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Rosato RR, Almenara JA, Cartee L, Betts V, Chellappan SP, Grant S. The cyclin-dependent kinase inhibitor flavopiridol disrupts sodium butyrate-induced p21WAF1/CIP1 expression and maturation while reciprocally potentiating apoptosis in human leukemia cells. Mol Cancer Ther. 2002;1:253–266. [PubMed] [Google Scholar]
- 11.Sasakawa Y, Naoe Y, Inoue T, et al. Effects of FK228, a novel histone deacetylase inhibitor, on human lymphoma U-937 cells in vitro and in vivo. Biochem Pharmacol. 2002;64:1079–1090. doi: 10.1016/s0006-2952(02)01261-3. [DOI] [PubMed] [Google Scholar]
- 12.De Schepper S, Bruwiere H, Verhulst T, et al. Inhibition of histone deacetylases by chlamydocin induces apoptosis and proteasome-mediated degradation of survivin. J Pharmacol Exp Ther. 2003;304:881–888. doi: 10.1124/jpet.102.042903. [DOI] [PubMed] [Google Scholar]
- 13.Zhao L, Zhang ZH, Zhu CM, et al. Inhibitory effect of valproic acid on cell cycle of Kasumi-1 cell line and its mechanism. Zhonghua Xue Ye Xue Za Zhi. 2008 Dec;29(12):802–805. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19176032. [PubMed] [Google Scholar]
- 14.Davie JR. Covalent modifications of histones: expression from chromatin templates. Curr Opin Genet Dev. 1988;8:173–178. doi: 10.1016/s0959-437x(98)80138-x. [DOI] [PubMed] [Google Scholar]
- 15.Kouzarides T. Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev. 1999;9:40–48. doi: 10.1016/s0959-437x(99)80006-9. [DOI] [PubMed] [Google Scholar]
- 16.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 18.Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25:1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 20.Batty N, Malouf GG, Issa JP. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett. 2009;280:192–200. doi: 10.1016/j.canlet.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Green SR, Choudhary AK, Fleming IN. Combination of sapacitabine and HDAC inhibitors stimulates cell death in AML and other tumour types. Br J Cancer. 2010;103:1391–1399. doi: 10.1038/sj.bjc.6605922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slack JL. The biology and treatment of acute progranulocytic leukemia. Curr Opin Oncol. 1999;11:9–13. doi: 10.1097/00001622-199901000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19. doi: 10.1017/S1462399408000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milutinovic S, Knox JD, Szyf M. DNA methyltransferase inhibition induces the transcription of the tumor suppressor p21(WAF1/CIP1/sdi1) J Biol Chem. 2000;275:6353–6359. doi: 10.1074/jbc.275.9.6353. [DOI] [PubMed] [Google Scholar]
- 25.Lohr K, Moritz C, Contente A, Dobbelstein M. p21/CDKN1A mediates negative regulation of transcription by p53. J Biol Chem. 2003;278:32507–32516. doi: 10.1074/jbc.M212517200. [DOI] [PubMed] [Google Scholar]
- 26.Jin JS, Tsao TY, Sun PC, Yu CP, Tzao C. SAHA inhibits the growth of colon tumors by decreasing histone deacetylase and the expression of cyclin D1 and survivin. Pathol Oncol Res. 2012;18:713–720. doi: 10.1007/s12253-012-9499-7. [DOI] [PubMed] [Google Scholar]
- 27.Liu S, Klisovic RB, Vukosavljevic T, et al. Targeting AML1/ETO-histone deacetylase repressor complex: a novel mechanism for valproic acid-mediated gene expression and cellular differentiation in AML1/ETO-positive acute myeloid leukemia cells. J Pharmacol Exp Ther. 2007;321(3):953–960. doi: 10.1124/jpet.106.118406. [DOI] [PubMed] [Google Scholar]
- 28.Peterson LF, Yan M, Zhang DE. The p21Waf1 pathway is involved in blocking leukemogenesis by the t(8;21) fusion protein AML1-ETO. Blood. 2007 May 15;109(10):4392–4398. doi: 10.1182/blood-2006-03-012575. [DOI] [PMC free article] [PubMed] [Google Scholar]