

Abstract
The transient receptor potential (melastatin) 2 (TRPM2), is an oxidant-activated nonselective cation channel, that is widely expressed in mammalian tissues including the vascular endothelium. Oxidative stress, through the generation of oxygen metabolites including H2O2, stimulates intracellular ADP-ribose formation which, in turn, opens TRPM2 channels. These channels act as an endogenous redox sensor for mediating oxidative stress/ROS-induced Ca2+ entry and the subsequent specific Ca2+-dependent cellular reactions such as endothelial hyper-permeability and apoptosis. This review summarizes recent findings on the mechanism by which oxidants induce TRPM2 activation, the role of these channels in the signaling vascular endothelial dysfunctions, and the modulation of oxidant-induced TRPM2 activation by PKCα and phospho-tyrosine phosphates L1.
Keywords: TRPM2, Oxidative stress, Endothelial cells
Introduction
Tissue damage caused by oxidative stress has a role in a number of pathophysiological conditions including neurodegenerative disorders (Alzheimer’s and Parkinson’s diseases), diabetes mellitus, atherosclerosis, autoimmune disease, ischemia/reperfusion injury (1–3). The vascular endothelium regulates the passage of macromolecules and circulating cells from blood to tissues and is a major target of oxidant stress. Reactive oxygen species (ROS) generated at sites of inflammation and injury are important mediators of vascular barrier dysfunction, increase endothelial barrier permeability and edema formation (4–7), and, therefore, play a critical role in the pathophysiology of several vascular diseases and disorders.
Oxidants increase Ca2+ permeability of cell membrane, which is recognized to promote interendothelial gap formation (8–10). Among the members of the TRP superfamily potentially responsible for oxidative damage and cell death in the endothelium (11–13), we identified the critical role of transient receptor potential melastatin (TRPM) channel 2 (3, 14). TRPM2, first named TRPC7 and later LTRPC-2 (15), is a voltage-independent, calcium-permeable non-selective cation channel that confers susceptibility to cell death through the activation of caspases and poly-ADP-ribose polymerase (14). The channel is ubiquitously expressed in mammalian tissues including the brain (16, 17), pancreatic b-cells (18), peripheral blood cells such as neutrophils (19), bone marrow, heart (13, 20), lungs (3, 13, 20, 21) and vascular endothelium (3, 13). Recently, we have established the role of TRPM2 in mediating H2O2-induced Ca2+ entry and endothelial hyper-permeability in cultured pulmonary artery endothelial cells (3). In this review, we will emphasize on the oxidative pathways that mediate TRPM2 activation, and their biological relevance. TRPM2 is likely to be a key player in signaling endothelial cell injury or cell death in response to oxidative stress.
TRPM2 channel, structure and function
The basic channel structure
Functional TRPM2 molecules are tetramers. The channel protein consists of six putative transmembrane spans with the pore formed by loops between the fifth and sixth segments (20). The N-terminal cytosolic tail of about 700 amino acids contains calmodulin-binding domains that mediate Ca2+ regulation of TRPM2 activity (22–24), a high affinity binding site for PKCα and two PxxP motifs implicated in protein-protein interactions (24). The N-terminal part of TRPM2 is thus likely to be crucial for the proper interaction with essential regulatory cytosolic components, assembly of the channel units and membrane trafficking (24). A region of high coiled coil character in the C-terminus may also play a role in ion channel subunit multimerization or in recruitment of regulatory proteins (25). After being exposed to oxidants, TRPM2 gating is induced by the binding of the intracellular second messenger adenosine diphosphoribose (ADP-ribose) or related molecules to a nudix-box sequence motif (NUDT9-H) (26–28) in its C-terminus domain (ADP-ribose) (27, 30, 31). Since the nudix-box has significant homology with a pyrophosphatase, NUDT9 ADP-ribose hydrolase (17, 27, 29–34), TRPM2 was dubbed a “chanzyme”. TRPM2 NUDT9-H displays a high binding affinity for ADP-ribose but a weak enzymatic activity (30, 31). However, enhancing the enzymatic activity of the nudix box abolishes the ADP-ribose gating of TRPM2, pointing to the requirement of prolonged substrate binding rather than degradation (29–31).
Whole-cell current measurements show a linear current-voltage relationship for TRPM2 cationic currents with a reversal potential at 0 mV indicating that TRPM2 functions as a non-specific cation permeable ion channel. While highly permeant to Na+ and K+, TRPM2 also shows considerable permeability to Ca2+ with a permeability ratio PCa:PNa of about 0.6–0.7 (17, 29). Ca2+ permeability is functionally important as it causes substantial increase in cytosolic Ca2+ concentration and ensuing Ca2+-dependent cellular response upon TRPM2 stimulation. The single channel properties of TRPM2 are unique as the channel exhibits extremely long opening times with a single channel conductance of 58–76 pS (17, 19, 27, 29).
TRPM2 channel variants
Subunit composition of TRPM2 tetrameric complexes is a factor in regulation of the channel opening. Several physiological splice variants of TRPM2 have been described: In addition to full-length functional TRPM2 (TRPM2-L), TRPM2-ΔN lacking the amino acids 538–557 in the N-terminus (31) and TRPM2-ΔC missing the amino acids 1292–1325 (nudix-box sequence motif) in the C-terminal (32) isoforms have been identified in human hematopoietic cells. Another variant of approximately, lacking the N-terminal 214 amino acid residues but still maintaining oxidant-induced Ca2+ influx activity, was shown to be particularly expressed in human brain (SSF-TRPM2) (35). The short splice variant TRPM2-S (22) lacks the entire carboxyl terminus of the long variant including the putative Ca2+-permeable pore, and functions in a dominant-negative fashion to inhibit TRPM2-L activity (22, 36). Thus, TRPM2-S is an important isoform of TRPM2 that may influence channel activity, vascular endothelial injury (3) and cell death induced by oxidative stress that activates TRPM2-L (24).
Structural elements and amino acid residues engaged in TRPM2 activation
The structural elements determining the TRPM2 channel activation are not fully understood. There is indeed increasing interest in identifying the amino acids residues of the pore loop involved in oxidant-induced TRPM2 activation and Ca2+ permeability. Mei (37) demonstrated the requirement of two conserved cysteine residues (at positions 996 and 1008) in the putative pore region of the human TRPM2; a substitution of either cysteine residue by alanine or serine abolished TRPM2 activation by ADP-ribose while not affecting the protein expression, trafficking or membrane localization, and the ability of TRPM2 to interact with neighboring subunits that is required for channel assembly. The same group of investigators recently depicted the crucial role of three additional pore residues in the channel function, the glutamate 960, the glutamine 981 and the aspartate 987, and established their contribution in defining the Ca2+ permeation properties of the TRPM2 channel. Although a substitution of either residues by alanine led to a loss of the channel properties, substitution of the aspartate residue 987 by a glutamate showed a much greater permeability to Ca2+ (38). In agreement with studies previously published by Mederos Y Schnitzler (39), incorporation of a glutamate residue in place of glutamine at position 981 also substantially increases Ca2+ permeability of the TRPM2 (38, 39).
Oxidative pathways leading to generation of ADP-ribose and subsequent activation of TRPM2
TRPM2 channels participate in signaling oxidative stress-induced Ca2+ entry and thereby eliciting Ca2+ -dependent cellular processes. Although most investigators can demonstrate an indirect action of oxidants on TRPM2 through ADP-ribose generation, direct action of oxidants on TRPM2 has been also proposed for neutrophil granulocytes, as the TRPM2-ΔC splice variant lacking the C-terminal NUDT9-H domain was stimulated by H2O2 but did not respond to ADP-ribose (31, 32, 40, 41).
ADP-ribose generation during oxidative stress
Oxidants, applied externally or produced in the cytosol during oxidative stress (14, 32), stimulate ADP-ribose formation in the nucleus and mitochondria (28, 42). In vascular endothelial cells, ROS may be generated in significant amount as a consequence of mitochondrial dysfunction. Pathologies in which endothelial mitochondrial oxidative stress plays a key role include atherosclerosis (43), ischemia-reperfusion injury (44, 45) and acute hypoxia. The free radical intermediates produced in the cytosol during oxidative stress include superoxide anion (O2), H2O2, nitric oxide (NO), and a more damaging compound, hydroxyl radical (OH.). These radicals contribute to DNA oxidation and damage, which initiate poly-ADP ribose polymerase (PARP)-mediated ADP-ribose generation. PARP binds to single-stranded and double-stranded DNA breaks and catalyses the breakdown of NAD into nicotinamide and poly(ADP-ribose) to initiate DNA repair mechanisms (22, 28, 44). Free ADP-ribose is then generated from poly(ADP-ribose) degradation by a poly(ADP-ribose) glycohydrolase (PARG) (28). Although H2O2 and O2− activate PARP, they may act through conversion to OH.
Alternative supply of ADP-ribose may results from the direct hydrolysis of NAD+ into nicotinamide and ADP-ribose or indirectly through cyclic ADP-ribose (cADP-ribose) formation. This reaction is catalyzed by NAD+ glycohydrolases (NADases) that are not only located at cell surface, but also in mitochondria and the nucleus (46–48). Nevertheless, as confirmed by our findings, oxidative stress-mediated activation of the PARP/PARG pathway remains however the major source of free ADP-ribose production in endothelial cells (3). H2O2-mediated Ca2+ entry through TRPM2 in pulmonary artery endothelial cells was reduced by at least 65 % in cells treated with a PARP inhibitor to prevent ADP-ribose agonist formation, either the 3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone (DPQ) or the 3-aminobenzamide (3-AB) (3).
TRPM2 gating by ADP-ribose or related molecules
The channel opening is induced by the binding of ADP-ribose to the nudix-box sequence motif in its intracellular C-terminus domain. Besides ADP-ribose, second messengers 2′-O-acetyl-ADP-ribose (49), cyclic ADP-ribose (cADP-ribose) and nicotinic acid adenine dinucleotide phosphate (NAADP) also directly gate the TRPM2 channel (37, 50–52), but requires higher concentrations than ADP-ribose to activate the channel. Their gating mechanisms are unknown, even though they are likely related to those of ADP-ribose (31, 50). Although patch-clamp studies suggest that nicotinamide adenine dinucleotide (NAD) also can directly induce opening of TRPM2 (14, 29), other evidence suggests this is secondary to conversion to or contamination by ADP-ribose (27, 28, 32, 53).
Synergistic gating behavior of TRPM2
Intracellular Ca2+ acts as a mandatory and dose-dependent modulator and cofactor of TRPM2 gating by ADP-ribose (54), as demonstrated for several other members of the TRP family. Ca2+ does not lead to channel activation by itself. However, it dramatically shifts the concentration-response curve to ADP-ribose to the left (27). Tong (23) identified calmodulin as the Ca2+ sensor responsible for Ca2+-dependent activation of TRPM2 (23). Through co-immunoprecipitation and mutagenesis studies, he indeed showed a direct interaction of calmodulin with the calmodulin binding motifs (also called calmodulin IQ-like motifs) in the N terminus of TRPM2 (23). Thus, oxidant-induced Ca2+ entry through TRPM2 enhances interaction of calmodulin with TRPM2, providing a positive feedback enhancement of TRPM2 activation.
Other TRPM2 co-factors include cADP-ribose, NAADP, NAD and H2O2 (41, 55). While cADP-ribose and NAADP can gate the TRPM2 channel by themselves at high micromolar range, they rather work at lower concentrations to synergize with ADP-ribose and potentiate TRPM2 activation (50, 55). Thus, NAADP and cADP-ribose generated in cells during oxidative stress regulate the efficacy and the sensitivity of TRPM2 channels in conjunction with internal Ca2+ (55). Interestingly, the channel is also temperature sensitive with the body temperature acting as ‘an endogenous co-activator’ for TRPM2 (56, 57).
Regulation of oxidant-induced Ca2+ entry and ensuing cellular processes by PKCα and PTPL1 modulation of TRPM2 activation
PKCα modulation of TRPM2 regulates H2O2-induced Ca2+ entry in endothelial cells
Because TRPM2 is implicated in endothelial dysfunction and many pathological states, elucidating the mechanisms of TRPM2 activation and regulation has gained significant interest. In pulmonary endothelial cells, both forms of TRPM2 (TRPM2-L and TRPM2-S) are expressed (figure 2); therefore, the control of their physical interaction is an enticing potential regulatory mechanism of TRPM2 activity in these cells. Our current studies are establishing the role of PKCα in the regulation of TRPM2 activation (unpublished data). PKCα regulates major endothelial cell functions important to maintenance of microvascular homeostasis including angiogenesis, cell migration and microvascular permeability (7, 58). Whether the generated ROS oxidize and activate directly PKCα (59) in the endothelium during oxidative stress is still to be defined. There is evidence that TRPM2 in the endothelial plasma membrane is associated with its short isoform (TRPM2-S), which serves as a negative regulator of TRPM2 channel activity (3, 15). We observed that PKCα modulates Ca2+ entry through TRPM2 channel, and interestingly PKCα rapidly co-localizes with TRPM2-S in endothelial cells exposed to H2O2. The intracellular N-terminal domain of the TRPM2-S protein sequence possesses a high affinity binding motif for PKCα; it is therefore likely that PKCα regulates TRPM2-induced Ca2+ entry and endothelial injury by PKCα phosphorylation of TRPM2-S. Studies are needed to investigate the contribution of PKCα in the control of TRPM2 activity by using a mutagenesis approach and PKCα-deficient mice. PKCα through the modulation of TRPM2 activation may regulate ROS-induced lung vascular hyper-permeability and injury.
Figure 2. TRPM2-L and –S expression in Human pulmonary artery endothelial cells.
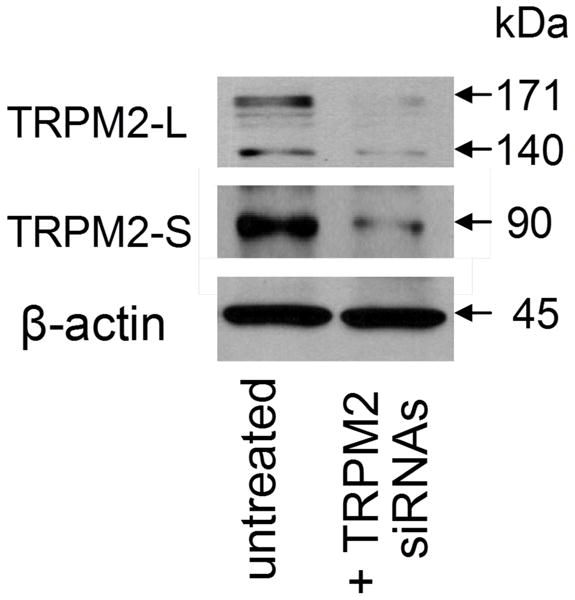
Western Blot showing endothelial expression of TRPM2-S and two TRPM2-L variants (of 140 and 170 kDa) and their decrease by TRPM2 silencing. The 140 kDa long variant is still unknown. Interestingly, the short striatum form (SSF-TRPM2) in which the N-terminal 214 amino acid residues of the TRPM2-L are removed has a molecular weight of 140 kDa.
Regulation of TRPM2-evoked Ca2+ entry by the tyrosine phosphatase PTPL1
In addition to PKCα, the phospho-tyrosine phosphatase L1 (PTPL1) also regulates oxidant-induced TRPM2 activation. A study by Zhang (60) showed a rapid tyrosine phosphorylation of TRPM2-L after stimulation with H2O2, which is important for its activation and function. Conversely, TRPM2-L dephosphorylation by the widely expressed PTPL1 resulted in channel inactivation. Thus, modulation of TRPM2 function by tyrosine phosphorylation may be another mechanism through which PTPL1 protects the cells against oxidative damage, and mediates resistance to cell death (60).
Relevance of oxidative stress-induced TRPM2 activation in the endothelium
TRPM2 as an oxidative stress sensor in the endothelium
There is increasing awareness of the contribution of TRPM2 in signaling oxidant-induced vascular endothelial injury; for instance, TRPM2 may be a factor in the development of atherosclerosis, which is initiated by mitochondrial dysfunction-induced ROS generation and subsequent endothelial injury (7, 42). TRPM2 has been identified in the heart vessel and pulmonary artery endothelium where it acts as an oxidant sensor and may play a key role in the activation of leukocytes, vascular endothelial permeability and injury (3, 13).
Poteser (61) described an interesting mechanism by which oxidants induced association of the TRPC3 and TRPC4 subunits to form a redox-sensitive cation channel and induce Na+ and Ca2+ entry into porcine aortic endothelial cells through mechanisms that are dependent on phospholipase C. We later identified in human pulmonary artery endothelial cells another TRP channel, the TRPM2, as being responsible for mediating the effects of oxidants on the endothelium by mechanisms that involve production of the second messenger ADP-ribose (3). As illustrated in figure 3, exposure of endothelial cell monolayers to sub-lytic concentrations of H2O2 (100 μM) induced increase in intracellular Ca2+ by stimulating Ca2+ entry without provoking Ca2+-store depletion. Ca2+ entry was entirely blocked by TRPM2 silencing, overexpression of TRPM2-S isoform or a treatment of the cells with specific TRPM2 blocking antibodies, while TRPC4 silencing did not modify it (3).
Figure 3. H2O2-induced Ca2+ entry occurs entirely via TRPM2 channels in endothelial cells.

Ca2+ mobilization assays using a Ca2+ add-back protocol. Human lung endothelial cells in culture were loaded with Fura-2, washed, and transferred to Ca2+-free medium. Left panel: H2O2 (100 μM) or control buffer (for baseline) was added at the arrow and CaCl2 (2.0 mM) was repleted at the fifth minute. Ionomycin (ion) addition is shown at the end of tracings. The abscissa indicates time in seconds; the ordinate, relative intracellular Ca2+ level. Right panel: Mean ratiometric values (± S.E.M) for steady-state intracellular [Ca2+] (n=4). In untreated cells, addition of H2O2 elicited a marked Ca2+ transient upon Ca2+ repletion that were abolished by TRPM2-silencing, a treatment of the cells with a specific TRPM2 blocking antibody, or overexpression of the short dominant negative TRPM2 isoform (TRPM2-S); therefore, Ca2+-repletion transient reflected Ca2+ entry through TRPM2.
Role of TRPM2 in inflammation and oxidant-induced vascular hyper-permeability
Oxidants generated at sites of inflammation and injury, by activation of neutrophils adherent to endothelial cells during sepsis, induced increase in vascular permeability (4–7). Although it was long believed that the resulting reactive oxygen species would directly damage the vascular endothelium, we provided evidence of the TRPM2 channel contribution in H2O2-induced increase in cytosolic Ca2+ and subsequent reduction of transendothelial resistance (3). The critical role of TRPM2 in the mechanism of endothelial barrier disruption following oxidative stress was demonstrated by suppressing endogenous TRPM2 expression and function by various means; that is, by small interfering RNAs, a specific TRPM2 blocking antibody, overexpression of TRPM2-S isoform (figure 4), and poly-ADP-ribose polymerase inhibitors that prevent generation of ADP-ribose (3, 26, 36, 42). All of these interventions abolished H2O2-induced Ca2+ influx via TRPM2 channels and the resulting increase in endothelial permeability.
Figure 4. Role of TRPM2 channels in H2O2-induced increase in endothelial barrier permeability.
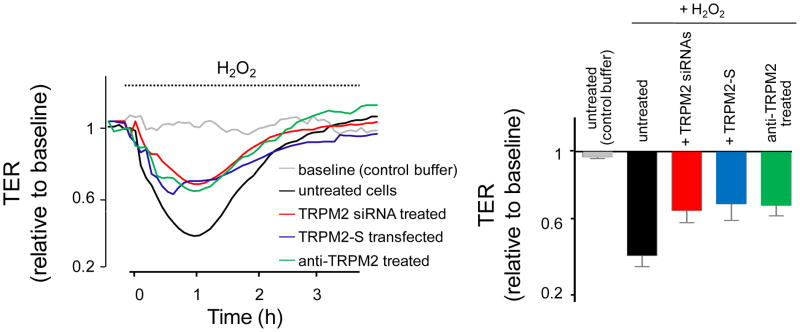
Confluent endothelial cell monolayers grown on gold microelectrodes were treated with H2O2 (300 μM), or control buffer (for baseline). Changes in transendothelial electrical resistance (TER), reflecting the paracellular permeability of endothelial monolayers were followed for 4 hr. Left panel: Original TER recordings (each trace is the average of 4 responses). Right, Mean value (± S.E.M) of peak TER responses to H2O2. H2O2 (300 μM) decreased trans-monolayer TER, indicating opening of interendothelial junctions. TRPM2-silencing, treatment with a specific TRPM2 blocking antibody, and TRPM2-S overexpression attenuated the peak TER response to H2O2. Thus TRPM2 channels contribute to H2O2-induced endothelial barrier hyper-permeability.
Oxidative stress is initiated by the products of activated lung macrophages and infiltrated neutrophils (4, 27). Specifically, oxidant stress through the production of oxygen metabolites such as H2O2, and chemotactic cytokines, also called chemokines, increases endothelial adhesivity of neutrophils and vascular endothelial permeability, both critical factors governing tissue edema formation and neutrophil extravasation (7, 62–64). Chemokine expression is inducible and is responsible for the recruitment of inflammatory cells to sites of infection or injury (65, 66). The recent work by Yamamoto (67) illustrates the functional role of monocyte TRPM2 channels in mediating chemokine production and neutrophil-induced lung injury. Specifically, H2O2 evokes Ca2+ influx through TRPM2 to activate Ca2+-dependent tyrosine kinase Pyk2 and amplify Erk signaling via Ras GTPase. This elicits nuclear translocation of nuclear factor-B (Nf-kB) essential to increase the production of the chemokines. Thus, TRPM2-elicited Ca2+ influx controls the oxidant-induced signaling cascade responsible for chemokine production, which in turn increases endothelial adhesivity of neutrophils and generation of ROS, and thus aggravates endothelial inflammation and injury (67). In chronic inflammation, the continued production of ROS by neutrophils causes extensive tissue damage.
Role of TRPM2 in oxidant-induced endothelial apoptosis
Oxidative stress-induced apoptosis can arise from TRPM2 activation (14, 42). Whereas low concentration of ROS are necessary in signaling regulation of fundamental cell activities such as cell growth, excessive ROS production and persistent Ca2+ entry may tilt the balance toward severe tissue damage and cell death. Cell death pathway is initiated by TRPM2-evoked Ca2+ entry, followed by caspase activation and PARP cleavage (42). Inhibition of endogenous TRPM2 function by downregulation with a specific siRNA or expression of TRPM2-S inhibited the rise in intracellular Ca2+ and enhanced cell viability after exposure to oxidants (42). Oxidant-induced cell death however involves additional mechanisms, as blockade of channel opening (evidenced by suppression of Ca2+ influx) in TRPM2-L overexpressing HEK293 does not entirely correlate with protection from cell death (56).
To investigate the role of TRPM2 in oxidant-induced endothelial hyperpermeability (3), we have transduced TRPM2-L in human pulmonary endothelial cells to cause overexpression of the functional channel (3). Cells transfected by TRPM2-L were susceptible to apoptosis; apoptosis was however suppressed by addition of a caspase 9 inhibitor (Ac-LEHD-CHO) to the medium (3). Overexpression of the full-length TRPM2 enhanced H2O2-mediated Ca2+ entry, cationic current, endothelial permeability increase, and cell death compared to the untransfected cells. While addition of the caspase inhibitor protected cell viability, it did not modify TRPM2-evoked Ca2+ entry or hyper-permeability in response to H2O2. Thus apoptosis does not cause oxidant-mediated endothelial hyper-permeability. Nevertheless, excessive and sustained exposure to oxidants, as we observed with H2O2 concentration over 500 μM, led to cell death and irreversible endothelial barrier disruption.
Conclusions and perspectives
Vascular endothelial injury, for instance in the setting of sepsis, is the result of oxidant generation by endothelial cells themselves and neutrophils and other inflammatory cells adherent to vessels. The mechanism of oxidant-mediated disruption of endothelial barrier function, in part, is attributable to a rise in intracellular Ca2+ mediated by Ca2+ entry through TRPM2 channels. There is no known specific inhibitor of the TRPM2 channel. However, because both forms of TRPM2 (TRPM2-L and TRPM2-S) are expressed in endothelial cells, the mechanisms that regulate their expression or control of their physical interaction could be targeted to reduce vascular endothelial injury due to oxidant production. Therefore, manipulating TRPM2 function in the endothelium may provide a useful therapeutic strategy for the treatment of endothelial barrier dysfunction and vascular inflammation.
Fig. 1. Schematic representation of cellular pathways leading to ADP-ribose formation and subsequent TRPM2 activation during oxidative stress.

The major sources of ADP-ribose include mitochondria and the nucleus. Dotted lines indicate minor pathways or relationships not generally accepted. Oxidants, generated extracellularly in setting such as sepsis or intracellularly as a result of mitochondrial oxidative stress, result in DNA damage and activation of the poly(ADP-ribose) polymerase. In the nucleus, formation of AD-ribose polymers is evoked after stimulation of PARP which are then hydrolyzed to ADP-ribose by poly(ADP-ribose) glycohydrolases. NAD conversion to ADP-ribose or cADP-ribose catalyzed by NADase and ADP-riboses cyclases inside the cell, including in mitochondria and the nucleus, provides additional sources of ADP-ribose. Upon gating by ADP-ribose, Ca2+ entering the cell through TRPM2 provides a positive feedback enhancement of TRPM2 activation. cADP-ribose, cyclic ADP-ribose; NAD, nicotinamide adenine dinucleotide; SOD, superoxide dismutase; AMP, adenosine mono phosphate.
References
- 1.Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 2000;29:323–33. doi: 10.1016/s0891-5849(00)00302-6. Review. [DOI] [PubMed] [Google Scholar]
- 2.Langley B, Ratan RR. Oxidative stress-induced death in the nervous system: cell cycle dependent or independent? J Neurosci Res. 2004;77:621–9. doi: 10.1002/jnr.20210. Review. [DOI] [PubMed] [Google Scholar]
- 3.Hecquet CM, Ahmmed GU, Vogel SM, et al. Role of TRPM2 Channel in Mediating H2O2-Induced Ca2+ Entry and Endothelial Hyperpermeability. Circ Res. 2008;102:347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- 4.Johnson A, Phillips P, Hocking D, et al. Protein kinase C inhibitor prevents pulmonary edema in response to H2O2. Am J Physiol Heart Circ Physiol. 1989;256:H1012–H1022. doi: 10.1152/ajpheart.1989.256.4.H1012. [DOI] [PubMed] [Google Scholar]
- 5.Stevens T, Garcia JG, Shasby DM, et al. Mechanisms regulating endothelial cell barrier function. Am J Physiol Lung Cell Mol Physiol. 2000;279:L419–L422. doi: 10.1152/ajplung.2000.279.3.L419. [DOI] [PubMed] [Google Scholar]
- 6.Barnard ML, Matalon S. Mechanisms of extracellular reactive oxygen species injury to the pulmonary microvasculature. J Appl Physiol. 1992;72:1724–1729. doi: 10.1152/jappl.1992.72.5.1724. [DOI] [PubMed] [Google Scholar]
- 7.Lum H, Roebuck K. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 8.Siflinger-Birnboim A, Goligorsky MS, Delvecchio PJ, et al. Activation of protein kinase C pathway contributes to hydrogen peroxide-induced increase in endothelial permeability. Lab Invest. 1992;67:24–30. [PubMed] [Google Scholar]
- 9.Dreher D, Junod AF. Differential effects of superoxide, hydrogen peroxide, and hydroxyl radical on intracellular calcium in human endothelial cells. J Cell Physiol. 1995;162:147–153. doi: 10.1002/jcp.1041620118. [DOI] [PubMed] [Google Scholar]
- 10.Volk T, Hensel M, Kox WJ. Transient Ca2+ changes in endothelial cells induced by low doses of reactive oxygen species: role of hydrogen peroxide. Mol Cell Biochem. 1997;171:11–21. doi: 10.1023/a:1006886215193. [DOI] [PubMed] [Google Scholar]
- 11.Groschner K, Rosker C, Lukas M. Role of TRP channels in oxidative stress. Novartis Found Symp. 2004;258:222–230. [PubMed] [Google Scholar]
- 12.Kwan HY, Huang Y, Yao X. TRP channels in endothelial function and dysfunction. Biochimica et Biophysica Acta (BBA) – Molec Basis of Disease. 2007;1772:907–914. doi: 10.1016/j.bbadis.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Dietrich A, Gudermann T. Another TRP to Endothelial Dysfunction: TRPM2 and Endothelial Permeability. Circ Res. 2008;102:275–277. doi: 10.1161/CIRCRESAHA.107.170548. [DOI] [PubMed] [Google Scholar]
- 14.Hara Y, Wakamori M, Ishii M, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 15.Zhang W, Chu X, Tong Q, et al. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem. 2003;278:16222–16229. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- 16.Nagamine K, Kudoh J, Minoshima S, et al. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics. 1998;54:124–131. doi: 10.1006/geno.1998.5551. [DOI] [PubMed] [Google Scholar]
- 17.Kraft R, Grimm C, Grosse K, et al. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol. 2004;286:C129–C137. doi: 10.1152/ajpcell.00331.2003. [DOI] [PubMed] [Google Scholar]
- 18.Inamura K, Sano Y, Mochizuki S, et al. Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J Membr Biol. 2003;191:201–207. doi: 10.1007/s00232-002-1057-x. [DOI] [PubMed] [Google Scholar]
- 19.Heiner I, Eisfeld J, Luckhoff A. Role and regulation of TRP channels in neutrophil granulocytes. Cell Calcium. 2003;33:533–540. doi: 10.1016/s0143-4160(03)00058-7. [DOI] [PubMed] [Google Scholar]
- 20.Eisfeld J, Lückhoff A. TRPM2: Handb Exp Pharmacol. 2007;179:237–252. doi: 10.1007/978-3-540-34891-7_14. [DOI] [PubMed] [Google Scholar]
- 21.Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97:853–863. doi: 10.1161/01.RES.0000187473.85419.3e. [DOI] [PubMed] [Google Scholar]
- 22.Zhang W, Chu X, Tong Q, et al. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem. 2003;278:16222–16229. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- 23.Tong Q, Zhang W, Conrad K, et al. Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J Biol Chem. 2006;281:9076–9085. doi: 10.1074/jbc.M510422200. [DOI] [PubMed] [Google Scholar]
- 24.Kühn FJ, Kühn C, Naziroglu M, et al. Role of an N-Terminal Splice Segment in the Activation of the Cation Channel TRPM2 by ADP-Ribose and Hydrogen Peroxide. Neurochem Res. 2008 doi: 10.1007/s11064-008-9755-0. accepted manuscript. [DOI] [PubMed] [Google Scholar]
- 25.Schmitz C, Perraud AL. The TRPM cation channels in the immune context. Curr Pharm Des. 2005;11:2765–2778. doi: 10.2174/1381612054546851. Review. [DOI] [PubMed] [Google Scholar]
- 26.Fonfria E, Marshall IC, Benham CD, et al. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol. 2004;143:186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perraud AL, Fleig A, Dunn CA, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 28.Perraud AL, Takanishi CL, Shen B, et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 29.Sano Y, Inamura K, Miyake A, et al. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- 30.Perraud AL, Shen B, Dunn CA, et al. NUDT9, a member of the Nudix hydrolase family, is an evolutionarily conserved mitochondrial ADP-ribose pyrophosphatase. J Biol Chem. 2003;278:1794–1801. doi: 10.1074/jbc.M205601200. [DOI] [PubMed] [Google Scholar]
- 31.Kuhn FJ, Luckhoff A. Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J Biol Chem. 2004;279:46431–46437. doi: 10.1074/jbc.M407263200. [DOI] [PubMed] [Google Scholar]
- 32.Wehage E, Eisfeld J, Heiner I, et al. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem. 2002;277:23150–23156. doi: 10.1074/jbc.M112096200. [DOI] [PubMed] [Google Scholar]
- 33.Perraud AL, Schmitz C, Scharenberg AM. TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium. 2003;33:519–553. doi: 10.1016/s0143-4160(03)00057-5. [DOI] [PubMed] [Google Scholar]
- 34.Heiner I, Eisfeld J, Halaszovich CR, et al. Expression profile of the transient receptor potential (TRP) family in neutrophil granulocytes: evidence for currents through long TRP channel 2 induced by ADP-ribose and NAD. Biochem J. 2003;371:1045–1053. doi: 10.1042/BJ20021975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uemura T, Kudoh J, Noda S, et al. Characterization of human and mouse TRPM2 genes: identification of a novel N-terminal truncated protein specifically expressed in human striatum. Biochem Biophys Res Commun. 2005;328:1232–1243. doi: 10.1016/j.bbrc.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 36.Zhang W, Hirschler-Laszkiewicz I, Tong Q, et al. TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am J Physiol Cell Physiol. 2006;290:C1146–C1159. doi: 10.1152/ajpcell.00205.2005. [DOI] [PubMed] [Google Scholar]
- 37.Mei ZZ, Mao HJ, Jiang LH. Conserved cysteine residues in the pore region are obligatory for human TRPM2 channel function. Am J Physiol Cell Physiol. 2006;291:C1022–C1028. doi: 10.1152/ajpcell.00606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia R, Mei ZZ, Mao HJ, et al. Identification of pore residues engaged in determining divalent cationic permeation in transient receptor potential melastatin subtype channel 2. J Biol Chem. 2008 doi: 10.1074/jbc.M801049200. Accepted manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mederos y Schnitzler M, Wäring J, Gudermann T, et al. Evolutionary determinants of divergent calcium selectivity of TRPM channels. FASEB J. 2008;22:1540–1551. doi: 10.1096/fj.07-9694com. [DOI] [PubMed] [Google Scholar]
- 40.Kühn FJ, Heiner I, Luckhoff A. TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflugers Arch. 2005;451:212–219. doi: 10.1007/s00424-005-1446-y. Review. [DOI] [PubMed] [Google Scholar]
- 41.Kolisek M, Beck A, Fleig A, et al. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 42.Miller BA. The role of TRP channels in oxidative stress-induced cell death. J Membrane Biol. 2006;209:1432–1424. doi: 10.1007/s00232-005-0839-3. [DOI] [PubMed] [Google Scholar]
- 43.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 44.Martin E, Rosenthal RE, Fiskum G. Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J Neurosci Res. 2005;79:240–247. doi: 10.1002/jnr.20293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrari R, Guardigli G, Mele D, et al. Oxidative stress during myocardial ischaemia and heart failure. Curr Pharm Des. 2004;10:1699–1711. doi: 10.2174/1381612043384718. [DOI] [PubMed] [Google Scholar]
- 46.Schuber F, Lund FE. Structure and enzymology of ADP-ribosyl cyclases: conserved enzymes that produce multiple calcium mobilizing metabolites. Curr Mol Med. 2004;4:249–61. doi: 10.2174/1566524043360708. Review. [DOI] [PubMed] [Google Scholar]
- 47.Partidá-Sánchez S, Rivero-Nava L, Shi G, et al. CD38: an ecto-enzyme at the crossroads of innate and adaptive immune responses. Adv Exp Med Biol. 2007;590:171–183. doi: 10.1007/978-0-387-34814-8_12. Review. [DOI] [PubMed] [Google Scholar]
- 48.Han MK, Cho YS, Kim YS, et al. Interaction of two classes of ADP-ribose transfer reactions in immune signaling. J Biol Chem. 2000;275:20799–20805. doi: 10.1074/jbc.M001189200. [DOI] [PubMed] [Google Scholar]
- 49.Grubisha O, Rafty LA, Takanishi CL, et al. Metabolite of SIR2 reaction modulates TRPM2 ion channel. J Biol Chem. 2006;281:14057–14065. doi: 10.1074/jbc.M513741200. [DOI] [PubMed] [Google Scholar]
- 50.Beck A, Kolisek M, Bagley LA, et al. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. 2006;20:962–964. doi: 10.1096/fj.05-5538fje. [DOI] [PubMed] [Google Scholar]
- 51.Heiner I, Eisfeld J, Warnstedt M, et al. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem J. 2006;398:225–232. doi: 10.1042/BJ20060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fliegert R, Gasser A, Guse AH. Regulation of calcium signalling by adenine-based second messengers. Biochem Soc Trans. 2007;35:109–114. doi: 10.1042/BST0350109. Review. [DOI] [PubMed] [Google Scholar]
- 53.Naziroğlu M. 2007 New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res. 2007;32:1990–2001. doi: 10.1007/s11064-007-9386-x. Review. [DOI] [PubMed] [Google Scholar]
- 54.McHugh D, Flemming R, Xu SZ, et al. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem. 2003;278:11002–11006. doi: 10.1074/jbc.M210810200. [DOI] [PubMed] [Google Scholar]
- 55.Lange I, Penner R, Fleig A, et al. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium. 2008 doi: 10.1016/j.ceca.2008.05.001. accepted manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Togashi K, Hara Y, Tominaga T, et al. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006;25:1804–1815. doi: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilkinson JA, Scragg JL, Boyle JP, et al. H2O2-stimulated Ca2+ influx via TRPM2 is not the sole determinant of subsequent cell death. Pflugers Arch. 2008;455:1141–1151. doi: 10.1007/s00424-007-0384-2. [DOI] [PubMed] [Google Scholar]
- 58.Siflinger-Birnboim A, Goligorsky MS, Delvecchio PJ, et al. Activation of protein kinase C pathway contributes to hydrogen peroxide-induced increase in endothelial permeability. Lab Invest. 1992;67:24–30. [PubMed] [Google Scholar]
- 59.Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 60.Zhang W, Tong Q, Conrad K, et al. Regulation of TRP channel TRPM2 by the tyrosine phosphatase PTPL1. Am J Physiol Cell Physiol. 2007;292:C1746–C1758. doi: 10.1152/ajpcell.00569.2006. [DOI] [PubMed] [Google Scholar]
- 61.Poteser M, Graziani A, Rosker C, et al. TRPC3 and TRPC4 Associate to Form a Redox-sensitive Cation Channel. Evidence for expression of native TRPC3-TRPC4 heterometric channels in endothelial cells. J Biol Chem. 2006;281:13588–13595. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- 62.Kaslovsky RA, Parker K, Siflinger-Birnboim A, et al. Increased endothelial permeability after neutrophil activation occurs by a diffusion-dependent mechanism. Microvasc Res. 1995;49:227. doi: 10.1006/mvre.1995.1018. [DOI] [PubMed] [Google Scholar]
- 63.Wang Q, Doerschuk CM. Neutrophil-induced changes in the biomechanical properties of endothelial cells: roles of ICAM-1 and reactive oxygen species. J Immunol. 2000;164:6487. doi: 10.4049/jimmunol.164.12.6487. [DOI] [PubMed] [Google Scholar]
- 64.Siflinger-Birnboim A, Malik AB. Regulation of endothelial permeability by second messengers. New Horiz. 1996;4:87. [PubMed] [Google Scholar]
- 65.Han XB, Liu X, Hsueh W, et al. I.G. Macrophage inflammatory protein-2 mediates the bowel injury induced by platelet-activating factor. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1220–G1226. doi: 10.1152/ajpgi.00231.2004. [DOI] [PubMed] [Google Scholar]
- 66.Buanne P, Di Carlo E, Caputi L, et al. Crucial pathophysiological role of CXCR2 in experimental ulcerative colitis in mice. J Leukoc Biol. 2007;82:1239–1246. doi: 10.1189/jlb.0207118. [DOI] [PubMed] [Google Scholar]
- 67.Yamamoto S, Shimizu S, Kiyonaka S, et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nature Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]