

Abstract
Low pH depolarizes the voltage-dependence of cardiac voltage-gated sodium (NaV1.5) channel activation and fast inactivation and destabilizes the fast-inactivated state. The molecular basis for these changes in protein behavior has not been reported. We hypothesized that changes in the kinetics of voltage sensor movement may destabilize the fast-inactivated state in NaV1.5. To test this idea, we recorded NaV1.5 gating currents in Xenopus oocytes using a cut-open voltage-clamp with extracellular solution titrated to either pH 7.4 or pH 6.0. Reducing extracellular pH significantly depolarized the voltage-dependence of both the QON/V and QOFF/V curves, and reduced the total charge immobilized during depolarization. We conclude that destabilized fast-inactivation and reduced charge immobilization in NaV1.5 at low pH are functionally related effects.
Introduction
Voltage-gated sodium (NaV) channels are responsible for action potential generation and propagation in most excitable cells. Each channel is composed of a pore-forming α-subunit and one or more modulating β-subunits (1–4). The α-subunit forms the functional pore of the channel and is composed of four homologous domains (DI–DIV), each consisting of six transmembrane helical segments (S1–S6) (1,5). Helices S5 and S6, and the extracellular loops linking them (P-loops), combine to form the channel pore and selectivity filter (6). The S1–S4 helices form the voltage-sensing domain (VSD). Voltage sensitivity is mediated primarily by a high concentration of positively charged lysines and arginines positioned every third residue within each S4 helix (7). Upon membrane depolarization the S4 helices move through the electric field, activating the channel, which leads to opening of the sodium conduction pathway (8,9).
After activation, the intracellular DIII-DIV linker, acting as a hinged lid, occludes the cytoplasmic side of the channel pore and blocks further ion permeation (10). This process is called “fast inactivation” because it occurs in the millisecond timeframe. Fluorescence recordings revealed that the movement of the S4 helices of DI–DIII occurs on a timescale that corresponds with the activation time constant (11). The S4DIV helix displays a slower component of fluorescence that more closely resembles the time constants of NaV channel fast inactivation (11). Extracellular immobilization of S4DIV attenuates fast inactivation (12) and mutations of the four most extracellular arginine residues within S4DIV modulate fast inactivation, whereas mutations in S4 helices of other domains typically modulate activation (13–15). Therefore the S4DIV helix is often termed the voltage sensor for fast inactivation.
During depolarization, the voltage sensors in DIII and DIV become immobilized (15–17). Gating currents therefore display biexponential decay upon membrane repolarization. The integral of the fast and slow components of gating current decay represent nonimmobilized and immobilized charge, respectively; up to 60% of the gating charge is immobilized during a 44-ms pulse in NaV1.5 (17). Charge immobilization also occurs with a time course that is similar to fast inactivation (11,15,16). Additionally, disrupting the fast-inactivated state, either through mutations within the S4DIV or by chemically interfering with the inactivation lid, attenuates charge immobilization (13,17). For example, exposing the intracellular membrane to pronase removes both fast inactivation and charge immobilization (8,9).
Extracellular protons destabilize the fast-inactivated state of NaV1.5. Protons slow open-state onset of and accelerate recovery from fast inactivation leading to increased persistent and window currents (19). The majority of effects on fast inactivation by protons occur from the open state (19). Interestingly, charge immobilization occurs faster and with greater magnitude when NaV channels inactivate from the open state (20,21). We therefore hypothesized that destabilized fast-inactivation during acidosis is the result of protons modulating the kinetics and magnitude of charge immobilization. To test this idea, we expressed NaV1.5 in Xenopus oocytes and recorded gating currents using a cut-open voltage-clamp with extracellular solution titrated to either pH 7.4 (control) or pH 6.0. Reducing extracellular pH triggered depolarizing shifts in the QON/V and QOFF/V curves and led to a reduction in the total charge immobilized during depolarization. These data are, to our knowledge, the first reported NaV1.5 gating currents recorded at reduced pH and suggest a molecular basis for the destabilized fast-inactivated state observed in ionic currents during acidosis. These data were previously presented in abstract form (22).
Materials and Methods
Molecular biology
The human variant of the pore-forming α-subunit, hNaV1.5, in SP6-4T was graciously donated by Dr. Chris Ahern (University of British Columbia, Canada). hNaV1.5 DNA was linearized using XbaI (Invitrogen, Carlsbad, CA). Transcription was completed using an Sp6 mMESSAGE mMACHINE High Yield Capped RNA Transcription Kit (Applied Biosystems, Carlsbad, CA).
Oocyte preparation
Female Xenopus laevis (Boreal Northwest, St. Catharines, Canada) were terminally anesthetized in 2 g/L tricaine solution. Oocytes were surgically removed and theca and follicular layers were enzymatically removed by ∼1 h agitation of semiintact lobes in a calcium-free solution containing 96 mM NaCl, 2 mM KCl, 20 mM MgCl2, 5 mM HEPES, and supplemented with 1 mg/mL Type 1A collagenase (Sigma-Aldrich, Oakville, Canada). Oocytes were then washed and sorted in calcium-free solution and finally incubated overnight at 19°C with SOS+ media containing 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, 2.5 mM sodium pyruvate, supplemented with 100 mg/L gentamicin sulfate and 5% horse serum. Stage V–VI oocytes were injected with 50 nL of cRNA encoding hNaV1.5. Injected oocytes were incubated at 19°C in SOS+ media for 5–10 days before recording. All surgical and animal care procedures were completed in accordance with the policies and procedures of the Simon Fraser University Animal Care Committee and the Canadian Council of Animal Care (23).
Data acquisition
Macroscopic current recordings were made using a CA-1B amplifier (Dagan, Minneapolis, MN) in the cut-open mode. Data were low-pass-filtered at 10 kHz, digitized at 50 kHz using an ITC-16 interface (HEKA Elektronics, Mahone Bay, Canada), and recorded using Patchmaster version 2x65 (HEKA Elektronics) running on an iMac (Apple Canada, Markham, Canada). Oocytes were placed in a cut-open voltage-clamp triple bath setup, as previously described in Cha et al. (11). Cells were then permeabilized via bottom bath perfusion with intracellular solution containing 120 mM MES, 120 mM NMG, 10 mM HEPES, 2 mM EGTA, titrated to pH 7.4 and supplemented with 0.1% saponin. After 1–2 min exposure, saponin-free intracellular solution was washed in. Extracellular solution of the top and middle chambers contained 120 mM MES, 120 mM NMG, 10 mM HEPES, 2 mM Ca(MES)2. Bath chambers were temperature-controlled at 21°C using a Peltier device run by a TC-10 temperature controller (Dagan, Minneapolis, MN).
Pulse protocols
Cells were maintained at a holding potential of −100 mV between all protocols. Leak subtraction was completed online, using a p/4 protocol from a holding potential of 40 mV. At 40 mV, >99% of voltage sensors are depolarized and therefore small changes in membrane potential do not affect total gating charge. This allowed subtraction of leak current without subtracting the gating current. Because protons reduce tetrodotoxin-NaV1.5 binding affinity (24), ionic currents were blocked using 500 μM tetrodotoxin (TTX) to ensure a saturating concentration of TTX in each recording solution.
Charge-voltage (Q/V) relationships were determined by recording gating currents during 20-ms depolarizations ranging between −130 mV and +60 mV, in 10-mV increments, from a 500-ms, −130 mV prepulse. Measured currents were integrated to determine charge movement, and the charge was plotted as a function of test potential. Charge recovery was measured using a double pulse protocol. The membrane was first depolarized to 0 mV for 500 ms to fully immobilize the gating charge. The membrane was then stepped to a conditioning potential of −120 mV for durations ranging from 0 to 100 ms. The extent of IgON recovery during each conditioning pulse was assessed during a final test pulse to 0 mV. To measure the onset rate of charge immobilization, the membrane potential was held at −130 mV for 100 ms and stepped to a 0-mV conditioning pulse, which varied in duration from 0 to 100 ms. IgOFF was then recorded upon repolarization to −130 mV. All recordings were paired between pH 7.4 and pH 6.0.
Data analysis
Analysis was completed using Fitmaster Ver.. 2x65 (HEKA Elektronics) and Igor Pro v. 5.01 (WaveMetrics, Lake Oswego, OR) run on an iMac (Apple Canada). For Q/V relationships, integrals of IgON and IgOFF were plotted as a function of test potential and fitted with a modified Boltzmann function,
| (1) |
where I1 and I2 are the maximum and minimum values in the fit, z is the apparent valence, e0 is the elementary charge, Vm is the membrane potential, V1/2 is the midpoint of the curve, T is the recording temperature in °K, and k is the Boltzmann constant. The decay of IgOFF and the integrals of IgON (taken from the recovery protocol and plotted as a function of time) were fitted with a double exponential equation,
| (2) |
where Y0 is the asymptote of the fit, A1 is the relative component of the first exponent, τ1 is the slow time constant, A2 is the relative component of the second exponent, τ2 is the fast time constant, and x is time. The decay of IgON and the integral of the slow time constant of decay of IgOFF (taken from the onset protocol and plotted as a function of time) were fitted with a single exponential function,
| (3) |
where Y0 is the asymptote of the fit, A is the relative component of the exponent, τ is the time constant, and x is time.
Statistical analysis was completed using Student’s t-test and analysis of variance (ANOVA), where appropriate (INSTAT, GraphPad, La Jolla, CA). All data are reported as mean ± SE and statistical significance was accepted at p < 0.05.
Results
Protons depolarize Q/V curves
The β-subunit has limited effects on NaV1.5 activation and fast inactivation in oocytes (2). Moreover, we previously showed that proton modulation of fast inactivation in NaV1.5 is not altered by the β-subunit (19). We therefore assessed proton modulation of NaV1.5 gating currents in the absence of the β-subunit. We recorded NaV1.5 gating currents at different test potentials and calculated the charge-voltage (Q/V) relationship with extracellular solution titrated to either pH 7.4 or pH 6.0. Gating currents recorded during 20-ms test potentials, from −130 through +60 mV in 10-mV intervals, were integrated, plotted as a function of test potential, and fitted with a Boltzmann function (Eq. 1). Off-gating currents (IgOFF) recorded after a depolarizing pulse were measured before and after the application of 500 μM TTX. Fig. 1, A–C, displays sample traces recorded at pH 7.4 as well as pH 7.4 and pH 6.0 in the presence of 500 μM TTX. TTX significantly hyperpolarized the QOFF/V curve from −37.6 ± 3.01 mV in the absence of TTX to −52.8 ± 4.3 mV in its presence (n = 6 and 4, respectively, p < 0.01). TTX also significantly reduced the total QOFF by 16.7 ± 3.1% (p < 0.01). QOFF contamination by ionic tail currents is expected to be negligible in the absence of TTX because fast inactivation typically inhibits >99% of ionic currents, even at reduced extracellular pH (19). These findings are consistent with previous reports that TTX causes hyperpolarizing shifts in NaV gating (26) and reduces total gating charge (26,27).
Figure 1.
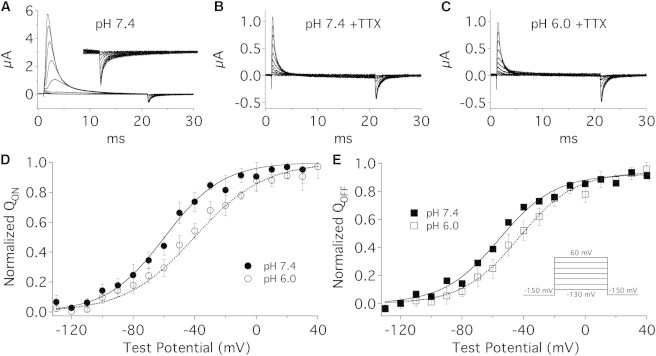
Reducing extracellular pH depolarizes the V1/2 of NaV1.5 Q/V curves. (A–C) Sample NaV1.5 gating currents recorded at pH 7.4 (A), pH 7.4+TTX (B), and pH 6.0+TTX (C). (Inset in A) IgOFF recorded in the absence of TTX but expanded to the same scale as panels B and C. (D and E) Normalized charge (Q) was plotted as a function of test potential and fitted with a Boltzmann function (Eq. 1). The V1/2 of NaV1.5 QON (D) was significantly depolarized from −46.2 ± 4.8 mV at pH 7.4 (solid circles) to −36.0 ± 4.4 mV at pH 6.0 (open circles) (n = 6, p < 0.05). The V1/2 of NaV1.5 QOFF (E) was significantly depolarized from −52.8 ± 4.3 mV at pH 7.4 (solid squares) to −40.3 ± 6.5 mV at pH 6.0 (open squares) (n = 6, p < 0.01). No significant change was observed in the apparent valence of QON or QOFF. Pulse protocol is displayed in the inset of panel E. All data points are mean ± SE.
In the presence of 500 mM TTX, the V1/2 of both the QON/V and the QOFF/V curve were significantly depolarized by extracellular solution titrated to pH 6.0, from −46.2 ± 4.8 mV at pH 7.4 to −36.0 ± 4.4 at pH 6.0 (n = 6, p < 0.05, Fig. 1 D) and −52.8 ± 4.3 mV to −40.3 ± 6.5 mV (n = 6, p < 0.01, Fig. 1 E), respectively. This was expected based on previous work studying the effects of surface charge on channel gating (28). Extracellular pH 6.0 had no effect on the total gating charge or the apparent valence of either the QON/V or QOFF/V curves.
Protons modulate Ig kinetics
Reducing extracellular pH slows open state inactivation, observed as a slowing of the decay of macroscopic ionic current after membrane depolarization (19). A similar effect on the kinetics of NaV1.5 IgON was also observed (Fig. 2, A and C). We fitted the decay of IgON with a single exponential function (Eq. 3). Previous studies have reported two exponents of decay (τ1 ≈ 1 ms, τ2 ≈ 0.1 ms) (29). The slower τ1 was dominant at all potentials and τ2 diminished substantially at more depolarized potentials (>−50 mV) (29). Our signal/noise ratio was insufficient to accurately resolve such a small component of decay; however, our data are comparable to the previously reported dominant component of IgON decay (Fig. 2 C) (29). Extracellular pH 6.0 significantly increased the time constant (τ) of decay of IgON at −50 mV and −30 mV through +10 mV, compared to pH 7.4 (n = 6, p < 0.05, Fig. 2 C).
Figure 2.
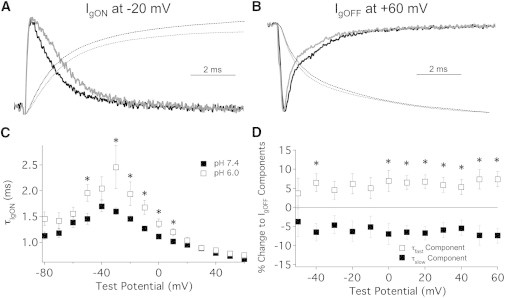
Extracellular pH modulates gating current kinetics. (A and B) Normalized sample IgON traces recorded at −20 mV (A) and IgOFF traces recorded at −130 mV after depolarization to +60 mV (B) with extracellular solution titrated to pH 7.4 (solid traces) and pH 6.0 (shaded traces). (Dotted lines) Integral of each trace therefore indicates the corresponding QON (A) and QOFF (B). pH 6.0 significantly accelerated the time course of τfast of IgOFF at −40 mV, −30 mV, and 10 mV. (C) Time constants (τ) of decay from IgON plotted as a function of test potential recorded at pH 7.4 (solid squares) and pH 6.0 (open squares). pH 6.0 significantly accelerated the τ of IgON at −50 mV and −30 mV through +10 mV. Our data recorded at pH 7.4 are comparable to previous reports of IgON decay recorded under similar conditions (29). (D) Change in the relative fast (open squares) and slow (solid squares) components of IgOFF decay plotted as a function of test potential. pH 6.0 significantly reduced the component of slow decay in IgOFF at −40 mV and 0 mV through +60 mV. The asterisk (∗) denotes statistical significance at p < 0.05. Pulse protocol from the inset of Fig 1E. n = 3–7.
In contrast to the effect on IgON, we observed a trend toward an acceleration of the decay of IgOFF at acidic pH. IgOFF was fitted with a double exponential function and pH 6.0 significantly reduced the τfast of IgOFF at −40 mV, −30 mV, and +10 mV (n = 6, p < 0.05, Fig. 2 B). Surprisingly, there was no significant change in the time course of the slow component of IgOFF decay (data not shown). pH 6.0 also significantly increased the relative component of the fast decay (and subsequently reduced the relative component of slow decay) in IgOFF at −40 mV and 0 mV through +60 mV (n = 6, p < 0.05, Fig. 2 D). These data demonstrate that reducing extracellular pH slows IgON and accelerates the time course of IgOFF. The slowed IgON and accelerated IgOFF decays, along with the positive shift in the V1/2 of each Q/V, are consistent with a proton-dependent charge screening mechanism. The uniform and voltage-independent shift in the balance of the fast and slow components of IgOFF decay, in conjunction with an unchanged time course of slow IgOFF decay, suggests that there is a reduction in the total charge immobilized after depolarization not attributable to surface charge effects on channel gating (28).
Protons inhibit charge immobilization
Protons destabilize the fast-inactivated state (19). Given the evidence correlating fast inactivation with charge immobilization (8,9,20,21), we hypothesized that charge immobilization is inhibited at acidic extracellular pH. Impaired charge immobilization could underlie the destabilization of the fast-inactivated state at reduced extracellular pH. To test this hypothesis, we directly measured the rates of onset and recovery of charge immobilization at pH 7.4 and pH 6.0.
To assess the rate of charge immobilization onset, QOFF was recorded 0–110 ms after a 0-mV conditioning pulse. The immobilized charge, calculated from the integral of the slow decay of IgOFF, was normalized to the total QOFF for that pulse, plotted as a function of time, and fitted with a single exponential function (Eq. 3). The data show that protons significantly increased the time constant for the onset of charge immobilization from 1.1 ± 0.3 ms at pH 7.4 to 1.5 ± 0.4 ms at pH 6.0 (n = 6, p = 0.05, Fig. 3 A). Additionally, we measured the total charge immobilized after a 110-ms pulse to 0 mV. Extracellular protons significantly reduced the maximum probability of charge immobilization from 70.5 ± 5.7% at pH 7.4 to 62.4 ± 6.1% at pH 6.0 (n = 6, p < 0.05, Fig. 3 A).
Figure 3.
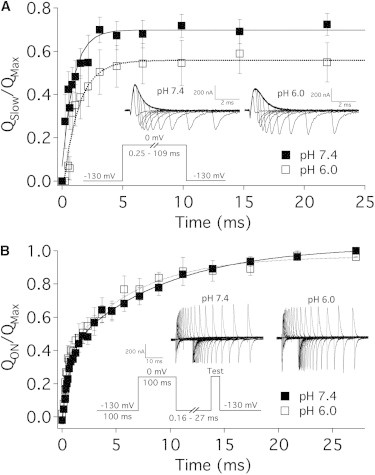
Protons slow the onset of gating charge immobilization in NaV1.5. (A) To assess the onset of charge immobilization, immobilized charge was normalized to the total QOFF of each sweep, plotted as a function of time, and fitted with a single exponential function (Eq. 3) for pH 7.4 (solid squares) and pH 6.0 (open squares). pH 6.0 significantly increased the time constant (τ) of charge immobilization onset from 1.1 ± 0.3 ms to 1.5 ± 0.4 ms and reduced the total charge immobilized from 70.5 ± 5.7 to 62.4 ± 6.1% (n = 6, p < 0.05). (Insets) Pulse protocols used. (B) Normalized QON is plotted as a function of time and fitted with a double exponential function (Eq. 2). We observed no statistically significant effect on the recovery of gating charge at pH 6.0 (open squares) compared to pH 7.4 (solid squares) (n = 5).
To assess the recovery of immobilized charge, IgON was integrated 0–100 ms into a −130 mV recovery pulse. QON was normalized to the total QON and plotted as a function of time. The data were then fitted with a double exponential (Eq. 2, Fig. 3 B). Extracellular protons had no effect on the recovery time course of immobilized charge.
Discussion
Previous studies on proton modulation of NaV channel gating currents, in a variety of tissues, displayed a depolarizing effect on the midpoints of the Q/V curves but mixed results on the kinetics of Ig decay, total charge, and charge immobilization (30–34). These previous data suggest tissue-specific modulation of gating currents, but did not directly assess proton modulation of a single NaV channel isoform. Describing NaV1.5 gating current proton modulation contributes to a comprehensive understanding about the effects of protons on NaV1.5 channels and cardiac excitability.
The data presented here, to our knowledge, represent the first NaV1.5 gating current recordings at reduced extracellular pH. We report that lowering the extracellular pH from 7.4 to 6.0 depolarized the V1/2 of QON/V and QOFF/V relationships, slowed IgON kinetics, and accelerated fast IgOFF kinetics. These effects are consistent with a charge screening effect. More notably, we demonstrate that protons induce a voltage-independent reduction of slow QOFF and suggest the resulting acceleration of IgOFF decay kinetics is attributable to reduced charge immobilization and not merely a shift in NaV1.5 voltage-dependence, as would be expected from a charge screening effect (28). The destabilized charge immobilization reported here implies a direct proton-protein interaction, most likely at the extracellular side of the VSD. Our data do not definitively determine whether disruption of charge immobilization by protons leads to destabilized fast inactivation, or whether destabilized fast inactivation by protons affects charge immobilization.
The voltage-sensing domain: a putative proton-binding site
In NaV1.5, the DIVS4 becomes immobilized after activation but before inactivation; the inactivation lid subsequently binds, blocking Na+ current, and, finally, the DIIIS4 is immobilized (17). Similar results have been observed in NaV1.4 channels (21). If immobilization precedes open-state fast inactivation in NaV1.5, then it seems reasonable to speculate that disruption of charge immobilization would alter fast inactivation. Horn et al. (12) showed that extracellular immobilization of the NaV1.4 DIVS4 with a photoactivated cross linker drastically slowed fast inactivation and increased peak currents (12). Additionally, the site-3 toxin, anthopleurin-A, reduced total charge displacement during depolarization (15). This presumably occurs by inhibiting movement of the S4DIV helix. Anthopleurin-A displays state-dependent binding, concomitantly accelerating fast inactivation and charge immobilization onset at membrane potentials below the threshold of ionic current (<−30 mV in NaV1.4) but slowing fast inactivation and immobilization onset at potentials above threshold (20). These effects underscore the ability of extracellular factors to concurrently modulate charge immobilization and fast inactivation through the VSD of DIV.
In the crystal structure of NaVAb, the VSD displays a region of negatively charged and polar residues that are predicted to stabilize positively charged arginines of the S4 voltage sensor in its activated state (35). This cluster lines an aqueous cleft that penetrates from the extracellular surface ∼10 Å into the VSD, making said arginines readily accessible to protonation (35). The same region in NaV1.5 correlates with a group of Long-QT-syndrome type III-causing mutations, E1225K, E1231K, and E1295K (36,37). Abriel et al. (36) explored the biophysical consequences of these mutations. They reported depolarized voltage dependencies of activation, fast inactivation, and window currents, slowed open-state fast inactivation onset, and accelerated fast inactivation recovery in E1295K compared to wild-type channels (36). These characteristics are remarkably similar to those observed in wild-type channels at reduced extracellular pH (19). These findings underscore the ability of extracellular positive charge at the NaV channel DIII and DIV VSD to destabilize the fast-inactivated state.
A similar region is present in KV channels with similar accessibility to positively charged ions (38–40). Most recently, Hoshi and Armstrong (41) demonstrated in Shaker channels that application of external La3+ displayed a more dramatic effect on the kinetics of gating currents compared to the application of Ca2+ (41). These effects were only observed when the cation was applied at hyperpolarized potentials. At depolarized potentials the negatively charged residues form a salt bridge with the positive charged residues of S4, thereby blocking any potential modification by extracellular cations (42–44). and Hoshi and Armstrong (41) postulated that cations such as Ca2+ or La3+ modulate KV channel gating by binding extracellular negatively charged residues E183 and E226 found in S1 and S2.
Another possible explanation for our results is that protons exert charge screening, which could explain the slowing of IgON kinetics and the depolarizing shifts of the Q/V curves. We think this is an unlikely explanation, however, because immobilized charge was reduced even at extreme voltages, i.e., +60 mV (Fig. 2). Strong depolarization would have been expected to rescue total charge immobilization if screening were the sole trigger of the observed effects. Additionally, mutational analysis of proton binding sites within the pore had no effect on proton modulation of channel fast inactivation, so these sites are unlikely to be responsible for proton-dependent changes in charge immobilization (45–47) Therefore, the effects of protons on charge immobilization most likely occur through protonation of extracellular amino-acid residues of the DIII or DIV voltage sensors, thereby disrupting charge immobilization directly.
Conclusion
Cations at the extracellular side of the NaV channel VSD modulate fast inactivation as well as gating charge (11–13,15,17,21,36,37). The data presented herein demonstrate that the immobilization of gating charge is hindered during acidosis. These low pH-induced effects correlate well with the effects previously described on NaV1.5 channel ionic currents and may explain the observed effects of protons on fast inactivation (19). We postulate that protons disrupt charge immobilization leading to the destabilization of the fast-inactivated state, and speculate that this effect occurs through a direct interaction with extracellular carboxylates of the VSDs of DIII or DIV.
Acknowledgments
The authors thank Stanislav Sokolov for his helpful comments.
This work was funded by Natural Sciences and Engineering Research Council Discovery Grants to P.C.R. and to T.W.C., and a Canadian Foundation for Innovation Leaders Opportunity Fund to T.W.C. and P.C.R. We note that T.W.C. was also supported by a Heart and Stroke Foundation of Canada New Investigator Award and a Michael Smith Foundation for Health Research Career Scholar Award. D.K.J. was supported by a Vanier Graduate Scholarship from the Canadian Institutes for Health Research.
References
- 1.Gellens M.E., George A.L., Jr., Kallen R.G. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. USA. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Makita N., Bennett P.B., George A.L., Jr. Molecular determinants of β1 subunit-induced gating modulation in voltage-dependent Na+ channels. J. Neurosci. 1996;16:7117–7127. doi: 10.1523/JNEUROSCI.16-22-07117.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meadows L., Malhotra J.D., Ragsdale D.S. The intracellular segment of the sodium channel β1 subunit is required for its efficient association with the channel α-subunit. J. Neurochem. 2001;76:1871–1878. doi: 10.1046/j.1471-4159.2001.00192.x. [DOI] [PubMed] [Google Scholar]
- 4.Noda M., Ikeda T., Numa S. Expression of functional sodium channels from cloned cDNA. Nature. 1986;322:826–828. doi: 10.1038/322826a0. [DOI] [PubMed] [Google Scholar]
- 5.Noda M., Shimizu S., Numa S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- 6.Terlau H., Heinemann S.H., Numa S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991;293:93–96. doi: 10.1016/0014-5793(91)81159-6. [DOI] [PubMed] [Google Scholar]
- 7.Stühmer W., Conti F., Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong C.M., Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bezanilla F., Armstrong C.M. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patton D.E., West J.W., Goldin A.L. Amino acid residues required for fast Na+-channel inactivation: charge neutralizations and deletions in the III-IV linker. Proc. Natl. Acad. Sci. USA. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cha A., Ruben P.C., Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- 12.Horn R., Ding S., Gruber H.J. Immobilizing the moving parts of voltage-gated ion channels. J. Gen. Physiol. 2000;116:461–476. doi: 10.1085/jgp.116.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kühn F.J., Greeff N.G. Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. J. Gen. Physiol. 1999;114:167–183. doi: 10.1085/jgp.114.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones D.K., Ruben P.C. Biophysical defects in voltage-gated sodium channels associated with long QT and Brugada syndromes. Channels (Austin) 2008;2:70–80. doi: 10.4161/chan.2.2.6000. [DOI] [PubMed] [Google Scholar]
- 15.Sheets M.F., Kyle J.W., Hanck D.A. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys. J. 1999;77:747–757. doi: 10.1016/S0006-3495(99)76929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheets M.F., Hanck D.A. Charge immobilization of the voltage sensor in domain IV is independent of sodium current inactivation. J. Physiol. 2005;563:83–93. doi: 10.1113/jphysiol.2004.077644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheets M.F., Kyle J.W., Hanck D.A. The role of the putative inactivation lid in sodium channel gating current immobilization. J. Gen. Physiol. 2000;115:609–620. doi: 10.1085/jgp.115.5.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reference deleted in proof.
- 19.Jones D.K., Peters C.H., Ruben P.C. Extracellular proton modulation of the cardiac voltage-gated sodium channel, NaV1.5. Biophys. J. 2011;101:2147–2156. doi: 10.1016/j.bpj.2011.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groome J., Lehmann-Horn F., Holzherr B. Open- and closed-state fast inactivation in sodium channels: differential effects of a site-3 anemone toxin. Channels (Austin) 2011;5:65–78. doi: 10.4161/chan.5.1.14031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groome J.R., Dice M.C., Ruben P.C. Charge immobilization of skeletal muscle Na+ channels: role of residues in the inactivation linker. Biophys. J. 2007;93:1519–1533. doi: 10.1529/biophysj.106.102079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones D.K., Claydon T.W., Ruben P.C. Proton modulation of cardiac voltage-gated sodium channel gating currents. Biophys. J. 2012;102:326a. doi: 10.1016/j.bpj.2011.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drummond G.B. Reporting ethical matters in the Journal of Physiology: standards and advice. J. Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henderson R., Ritchie J.M., Strichartz G.R. Evidence that tetrodotoxin and saxitoxin act at a metal cation binding site in the sodium channels of nerve membrane. Proc. Natl. Acad. Sci. USA. 1974;71:3936–3940. doi: 10.1073/pnas.71.10.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reference deleted in proof.
- 26.Heggeness S.T., Starkus J.G. Saxitoxin and tetrodotoxin. Electrostatic effects on sodium channel gating current in crayfish axons. Biophys. J. 1986;49:629–643. doi: 10.1016/S0006-3495(86)83690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capes D.L., Arcisio-Miranda M., Chanda B. Gating transitions in the selectivity filter region of a sodium channel are coupled to the domain IV voltage sensor. Proc. Natl. Acad. Sci. USA. 2012;109:2648–2653. doi: 10.1073/pnas.1115575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cukierman S., Zinkand W.C., Krueger B.K. Effects of membrane surface charge and calcium on the gating of rat brain sodium channels in planar bilayers. J. Gen. Physiol. 1988;92:431–447. doi: 10.1085/jgp.92.4.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheets M.F., Hanck D.A. Voltage-dependent open-state inactivation of cardiac sodium channels: gating current studies with Anthopleurin-A toxin. J. Gen. Physiol. 1995;106:617–640. doi: 10.1085/jgp.106.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keynes R.D., Rojas E. Kinetics and steady-state properties of the charged system controlling sodium conductance in the squid giant axon. J. Physiol. 1974;239:393–434. doi: 10.1113/jphysiol.1974.sp010575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wanke E., Testa P.L., Carbone E. High intracellular pH reversibly prevents gating-charge immobilization in squid axons. Biophys. J. 1983;44:281–284. doi: 10.1016/S0006-3495(83)84300-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schauf C.L. Evidence for negative gating charges in Myxicola axons. Biophys. J. 1983;42:225–231. doi: 10.1016/S0006-3495(83)84390-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumcke B., Schwarz W., Stämpfli R. Increased charge displacement in the membrane of myelinated nerve at reduced extracellular pH. Biophys. J. 1980;31:325–331. doi: 10.1016/S0006-3495(80)85062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell D.T. Sodium channel gating currents in frog skeletal muscle. J. Gen. Physiol. 1983;82:679–701. doi: 10.1085/jgp.82.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Payandeh J., Scheuer T., Catterall W.A. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abriel H., Cabo C., Kass R.S. Novel arrhythmogenic mechanism revealed by a long-QT syndrome mutation in the cardiac Na+ channel. Circ. Res. 2001;88:740–745. doi: 10.1161/hh0701.089668. [DOI] [PubMed] [Google Scholar]
- 37.Tester D.J., Will M.L., Ackerman M.J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 38.Starace D.M., Bezanilla F. Histidine scanning mutagenesis of basic residues of the S4 segment of the Shaker K+ channel. J. Gen. Physiol. 2001;117:469–490. doi: 10.1085/jgp.117.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Starace D.M., Stefani E., Bezanilla F. Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron. 1997;19:1319–1327. doi: 10.1016/s0896-6273(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 40.Starace D.M., Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 2004;427:548–553. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- 41.Hoshi T., Armstrong C.M. Initial steps in the opening of a Shaker potassium channel. Proc. Natl. Acad. Sci. USA. 2012;109:12800–12804. doi: 10.1073/pnas.1209665109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long S.B., Campbell E.B., MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 43.Fernandez D., Ghanta A., Sanguinetti M.C. Molecular mapping of a site for Cd2+-induced modification of human ether-à-go-go-related gene (hERG) channel activation. J. Physiol. 2005;567:737–755. doi: 10.1113/jphysiol.2005.089094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abbruzzese J., Sachse F.B., Sanguinetti M.C. Modification of hERG1 channel gating by Cd2+ J. Gen. Physiol. 2010;136:203–224. doi: 10.1085/jgp.201010450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones D.K., Peters C.H., Ruben P.C. Proton sensors in the pore domain of the cardiac voltage-gated sodium channel. J. Biol. Chem. 2013;288:4782–4791. doi: 10.1074/jbc.M112.434266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khan A., Kyle J.W., Fozzard H.A. Isoform-dependent interaction of voltage-gated sodium channels with protons. J. Physiol. 2006;576:493–501. doi: 10.1113/jphysiol.2006.115659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khan A., Romantseva L., Fozzard H.A. Role of outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block. J. Physiol. 2002;543:71–84. doi: 10.1113/jphysiol.2002.021014. [DOI] [PMC free article] [PubMed] [Google Scholar]