

Abstract
The presence of a regulatory site for monovalent cations that affects the conformation of the MgATP-binding pocket leading to enzyme activation has been demonstrated for ribokinases. This site is selective toward the ionic radius of the monovalent cation, accepting those larger than Na+. Phosphofructokinase-2 (Pfk-2) from Escherichia coli is homologous to ribokinase, but unlike other ribokinase family members, presents an additional site for the nucleotide that negatively regulates its enzymatic activity. In this work, we show the effect of monovalent cations on the kinetic parameters of Pfk-2 together with its three-dimensional structure determined by x-ray diffraction in the presence of K+ or Cs+. Kinetic characterization of the enzyme shows that K+ and Na+ alter neither the kcat nor the KM values for fructose-6-P or MgATP. However, the presence of K+ (but not Na+) enhances the allosteric inhibition induced by MgATP. Moreover, binding experiments show that K+ (but not Na+) increases the affinity of MgATP in a saturable fashion. In agreement with the biochemical data, the crystal structure of Pfk-2 obtained in the presence of MgATP shows a cation-binding site at the conserved position predicted for the ribokinase family of proteins. This site is adjacent to the MgATP allosteric binding site and is only observed in the presence of Cs+ or K+. These results indicate that binding of the monovalent metal ions indirectly influences the allosteric site of Pfk-2 by increasing its affinity for MgATP with no alteration in the conformation of residues present at the catalytic site.
Introduction
The characterization of ions as allosteric effectors of an enzyme’s activity addresses two fundamental aspects of enzymology: mechanism and specificity (1,2). As the structural databases grow and evolutionary relationships are clearly established, it should be possible to extend our understanding of ion sites from specific cases to the level of trends in homologous enzymes.
For members of the ribokinase family, the divalent cation Mg2+ is required for catalysis (3,4) and several x-ray structures of these kinases in the presence of ATP or its analogs suggest its direct involvement in phosphoryl transfer (5,6). However, monovalent cations such as K+ and Cs+ have also been shown to play important roles in the activation of ribokinases from different sources (7,8). In this case, the mechanism of activation is allosteric because the monovalent cation-binding site is in the vicinity of the active site (as demonstrated for Escherichia coli ribokinase) but plays no direct role in catalysis. For this enzyme, the structural effects of monovalent cation binding have been inferred from comparison with adenosine kinases from human and Toxoplasma gondii (7). It was proposed that the monovalent cation-binding site helps to organize the formation of the nucleotide-binding pocket of the active site. It seems that this site is able to bind K+ and Cs+ (apparent Kd of 5 and 17 mM, respectively) but not Li+ or Na+ (at concentrations up to 140 mM). More recently, the Staphylococcus aureus ribokinase (8) has been crystallized in the absence of monovalent ions and shows a disorganized monovalent cation site resulting in an occlusion of the nucleotide-binding pocket when compared to E. coli ribokinase bound to Cs+.
Among the ribokinase family members, allosteric regulation was first described for phosphofructokinase-2 (Pfk-2) from E. coli (9,10). For this enzyme, substrate inhibition by MgATP occurs at low concentrations of the cosubstrate fructose-6-P. The saturation kinetics for the sugar-P changes from hyperbolic to sigmoid when the concentration of MgATP is increased over the milimolar inhibitory range (11). Indeed, Pfk-2 was first crystallized in the presence of MgATP (12) and showed two ATP molecules bound per monomer: one at the nucleotide-binding pocket of the active site, and a second (the allosteric ATP) whose binding requires the presence of the first. Thus, the ribokinase monovalent cation-binding site and the allosteric ATP of Pfk-2 are both closely associated with the nucleotide-binding pocket of their respective structures. At this point, it is reasonable to hypothesize that if Pfk-2 possesses a monovalent cation site it could potentially affect either catalysis, substrate inhibition, or both. Evaluating this question will help to establish a new, to our knowledge, family feature, shedding light on the conservation of mechanism and selectivity of the monovalent cation-binding site.
In this work, we first observed the effect of different monovalent cations on Pfk-2 kinetics. We show that Pfk-2 is sensitive to the presence of cations with ionic radii larger than Na+. This effect does not occur at the level of the kinetic parameters for substrates per se but rather affects binding of the allosteric MgATP, leading to a strengthening of substrate inhibition. To obtain structural evidence for the location and properties of the monovalent cation-binding site in Pfk-2, we crystallized the enzyme under conditions compatible with the presence of the allosteric ATP (12) but in the presence of Cs+ or K+ instead of Na+. The resulting structures show that the monovalent cation-binding site is homologous to that found in ribokinase, but differs with respect to some features of the metal ion coordination. The K+-bound structure was used for molecular simulation under a free energy perturbation (FEP) protocol, where the energetic difference was observed to favor K+ over Na+. We conclude that although the monovalent cation-binding site of ribokinase is conserved in Pfk-2, the binding of metals to this site has the opposite effect with respect to enzyme activity. Thus, we suggest that this site could be a widespread feature of the ribokinase family with a common pattern of ion specificity but different regulatory outcomes depending on the specific features of each active site.
Materials and Methods
Enzymatic measurements
Phosphofructokinase activity was determined spectrophotometrically coupling the fructose-1,6-bisP production to the oxidation of NADH, according to Babul (13). In assays performed in the presence of salts, the concentration of auxiliary enzymes was increased three times to avoid the occurrence of lag phases in the time courses. Activity measurements were performed in 25 mM Tris, pH 8.2, using a constant excess of 5 mM MgCl2 with respect to the total concentration of ATP. This procedure maintains free magnesium close to 5 mM for each concentration of MgATP used (4,14). All of the measurements were carried out in 96-well plates and read with a microplate reader (Biotek, Synergy 25) at room temperature (25°C). Additionally, a discontinuous assay was implemented specifically to rule out the effect of salts on the auxiliary enzymes. Pfk-2 was incubated in the presence of substrates and aliquots were withdrawn and the reaction stopped by adding 50 mM EDTA. Kinetic assays were performed with ATP and fructose-6-P sodium salts purchased from Sigma. Fluorescence titration binding curves were performed with ATP di(tris) salt purchased from Sigma. The fructose-1,6-bisP concentration was determined by spectrophotometric titration using auxiliary enzymes (13).
Fluorescence measurements
Measurements were performed using a Shimadzu PC-5031 spectrofluorometer. The intrinsic fluorescence of Pfk-2 was obtained at several MgATP or fructose-6-P concentrations as described by Guixé et al. (15).
Crystallization and data collection, structure solution, and refinement
The enzyme was purified according to the method described by Parducci et al. (4). Crystals were grown by the vapor diffusion method in a hanging drop consisting of 2 μL of the protein stock solution (4 mg/ml of protein in 25 mM Tris-HCl pH 7.6 buffer, 10 mM MgCl2, 30 mM dithiotreitol, and 6 mM ATP) in the presence of 50 mM KCl or CsCl plus 2 μL of a reservoir solution comprising 13% PEG 6000, 0.1 M sodium acetate pH 5.3, 5 mM dithiotreitol. Crystals appeared after 3 days at 18°C. X-ray diffraction data were collected at 100 K. Measurements were made on the ID29 beamline of the European Synchrotron Radiation Facility (ESRF) (Å = 0.97627 Å) using a Pilatus 6M detector in fine slicing mode and were collected up to 1.7 Å for the K+ complex (Protein Data Bank (PDB)id 3UMO) and 1.85 Å for the Cs+ complex (PDBid 3UMP). The data were indexed, integrated, and scaled using the program XDS (16).
The structures of E. coli Pfk-2 in complex with MgATP and one of the two monovalent cations were both solved by molecular replacement using the program Phaser (17) employing a monomer of the previously solved Pfk-2 structure in complex with ATP as a search model (PDBid 3CQD). Model refinement was carried out with Phenix (18) employing TLS parameters in the final stages of refinement. Model building was performed with COOT (19), using σ-weighted 2Fo - Fc and Fo - Fc electron density maps. The cesium ion was readily located using an anomalous electron density map calculated with Phenix. In all cases, the behavior of Rfree was used as the principal criterion for validating the refinement protocol, and the stereochemical quality of the model was evaluated with Procheck (20) and Molprobity (21). Full statistics for data collection and refinement are summarized in Table S1 in the Supporting Material.
Alchemical FEP calculations
The selectivity of the monovalent cation-binding site of Pfk-2 was studied using FEP-molecular dynamics (MD) simulations. Simulations were carried out on the protein-cation complex described in this work by superimposing a Na+ ion onto each K+ ion bound per monomer in the dimer. K+ and Na+ ions were fixed during the simulations. This system was solvated in a 83 × 125 × 93 Å water box. The unbound state is modeled with both pairs of cations only, solvated in a 48 × 67 × 41 Å water box. Counter-ions were then added to obtain neutralized systems and were fixed in space through all the simulation. Both systems were prepared with Amber Tools 1.5 (22), using TIP3P waters and the AMBER99SB-ildn force field (23). The parameters for ATP in AMBER format were obtained from (24). Minimizations were carried out for 20,000 steps using the conjugated gradient algorithm, followed by a subsequent equilibration of 70 ps for the uncomplexed system and 3 ns for the K+-bound system. In the latter, a harmonic potential was applied over the backbone of Pfk-2 but gradually relaxed from 5 to 0 kcal/mol/Å2 except for those atoms related to the monovalent cation-binding site itself (residues 242 to 254 and 288 to 297). For each FEP-MD system, the forward and backward free energy change (K+⇔Na+) was calculated by changing the coupling parameter λ from 0 (initial state) to 1 (final state) by increments of 0.025, resulting in 40 windows with a total time of 0.64 ns (uncomplexed system) and 3.2 ns (protein-cation system). In these schemes, the change in free energy for each step was lower than 2 kBT (data not shown) and reasonable convergence was ensured. During all the FEP calculations, the electrostatic and van der Waals interactions were taken into account and the data analysis was performed with the ParseFEP plugin of VMD 1.9 (25). To validate our methodology, the FEP-MDs were computed for three independent runs. All MD calculations were performed using NAMD 2.8 (26), employing an integration step of 2 fs, a 10 Å cutoff for computations of nonbonded interactions, and the isothermal-isobaric (NPT) ensemble at 1 atm of pressure and a temperature of 300 K.
Results
Monovalent cations with ionic radii larger than Na+ promote Pfk-2 inhibition
Fig. 1 shows the effect of several chloride salts on the initial velocity of Pfk-2. The salts of K+, Cs+, and Rb+, but not Na+ or Li+, decrease the activity of Pfk-2 in a saturable fashion. The K0.5 values calculated from the salt-induced inhibition curves ranged from 1.6 to 8.9 mM for CsCl and RbCl, respectively. To discard a possible effect of the salts on the auxiliary enzymes of the assay mixture, the fructose-1,6-bisP produced by the reaction, was assayed discontinuously in the presence or absence of KCl. Under these conditions, potassium decreases the initial velocity of Pfk-2 (Fig. 1 A, inset). These results indicate that Pfk-2 and ribokinases have a common pattern of selectivity for the effect of monovalent cations on activity, but the actual consequence differs greatly, from activation in the case of ribokinase (7,8) to inhibition for Pfk-2.
Figure 1.
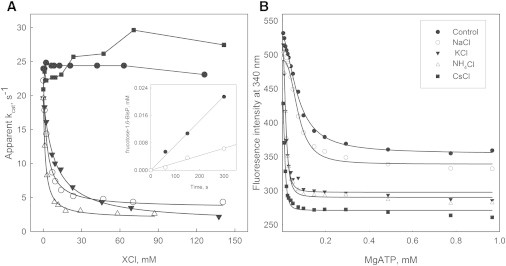
Effect of chloride salts on the activity of Pfk-2 and the binding isotherms of MgATP followed by intrinsic fluorescence. (A) Effect of chloride salts of monovalent metals on the initial velocity of Pfk-2 assayed with an inhibitory concentration of MgATP (5 mM) and 0.2 mM fructose-6-P. Chloride salts: LiCl (•), NaCl (■), KCl (o), CsCl (▵), RbCl (▾).The inset shows the progress curves of product formation (fructose-1,6-BisP) obtained with a discontinuous assay in the presence (o) or absence (•) of KCl (50 mM). (B) Changes of the intrinsic fluorescence intensity at 340 nm measured as a function of the MgATP concentration obtained using several chloride salts of monovalent cations at 50 mM; control (•), NaCl (o), KCl (▾), CsCl (■), and NH4Cl (▵). The continuous lines correspond to fit the MgATP saturation curves to the Hill equation.
Monovalent cations increase the affinity of the allosteric site for MgATP
In addition to the site where it acts as the phosphoryl donor, MgATP has a second site in Pfk-2 that negatively regulates the enzymatic activity and has important consequences for its in vivo behavior (27). Interestingly, it has been shown for Pfk-2 that the variation of its intrinsic fluorescence is a measure of the binding of MgATP to this allosteric site (15). Thus, to address the mechanism behind the observed inhibition by monovalent cations, we characterized the effect of the presence of 50 mM of several salts on the isothermal binding of MgATP (Fig. 1 B). It was observed that KCl and CsCl, but not NaCl, decrease the K0.5 for MgATP from 70 μM to values between 10 and 20 μM. Furthermore, the apparent Kd of Pfk-2 for potassium was determined to be 2.5 mM calculated from the saturable increment of the apparent affinity of MgATP induced by KCl (see Fig. S1). Therefore, the inhibition of Pfk-2 induced by monovalent cations seems to occur indirectly, through a mechanism involving the allosteric binding of MgATP. Further kinetic characterizations supported this hypothesis (see below).
Fig. 2 A shows the enzymatic activity of Pfk-2 as a function of MgATP concentration obtained in the presence or absence of 50 mM KCl. It was clearly observed that the inhibitory effect of high MgATP concentrations was strengthened when KCl was present, because the apparent constant for MgATP inhibition (Ki) decreased from 1 to 0.1 mM (Table 1). This effect was quite specific, occurring without affecting other kinetic parameters such as the kcat or KM for MgATP (Table 1). The allosteric inhibition of Pfk-2 is also evident from the behavior of the fructose-6-P saturation curves when performed at several and fixed concentrations of MgATP (11). In this case, MgATP induces a linear increase of the apparent K0.5 for the cosubstrate and decreases the apparent kcat, simultaneously promoting a transition from hyperbolic to sigmoidal saturation of the initial velocity curves. Fig. 2 B shows the saturation curves for fructose-6-P obtained at several fixed concentrations of KCl. Interestingly, the KCl concentration increases K0.5 and induced a sigmoidal behavior, along with a reduction in the apparent kcat. According to our hypothesis, these changes mimic the pattern of allosteric inhibition by MgATP.
Figure 2.
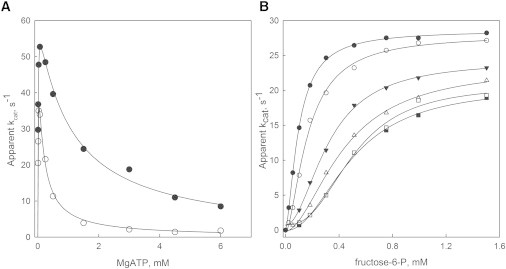
Effect of KCl on the allosteric inhibition of Pfk-2 induced by MgATP. (A) Initial velocity of Pfk-2 measured as a function of MgATP at 50 μM fructose-6-P in the presence (o) or absence (•) of 50 mM KCl. Continuous lines represent the fit to a model of uncompetitive substrate inhibition. Kinetic constants are shown in Table 1. (B) Initial velocities of Pfk-2 obtained with fructose-6-P as a variable substrate under inhibitory MgATP conditions (5 mM) at different fixed KCl concentrations of 0 (•), 7.5 (o), 17 (▾), 50 (▵), 75 (□), and 150 mM (■). Continuous lines represent the individual fit of each curve to the Hill equation.
Table 1.
Effect of KCl on the kinetic parameters of Pfk-2
| Control | 50 mM KCl | |
|---|---|---|
| akcat, s−1 | 61 ± 8 | 60 ± 9 |
| aKM ATP, μM | 8 ± 3 | 14 ± 4 |
| aKi, μM | 1026 ± 100 | 129 ± 55 |
| bKM fructose-6-P, μM | 6–10 | 10 |
| cKd fructose-6-P, μM | 6–8 | 8–10 |
Values obtained by fitting to an uncompetitive model of substrate inhibition. The initial velocity obtained as a function of MgATP using a fixed concentration of 0.2 mM fructose-6-P.
KM values for fructose-6-P extrapolated to zero concentration MgATP.
Values of Kd obtained from intrinsic fluorescence measurements (data not shown). Measurements obtained from b-c2 or a3 independent experiments.
In the case of the kinetic assays, variable concentrations of Na+ come from the stock solutions of ATP and fructose-6-P but binding isotherms obtained with fluorescence measurements were carried out in the absence of Na+ by titration with ATP (di(tris) salt form). As can be observed, the kinetics and binding experiments follow the same pattern of specificity irrespective of the presence of sodium.
The increase in K0.5 for fructose-6-P induced by K+ was not due to a decrease in the affinity for the sugar because the Kd for fructose-6-P (Table 1), determined in the absence of MgATP by fluorescence titrations, was unmodified by KCl. Furthermore, the extrapolated value of K0.5 at zero MgATP was quite similar regardless of the absence or presence of KCl (Table 1 and Fig. S2). Taken together, our biochemical results support the hypothesis that monovalent cations promote the allosteric binding of MgATP without causing alterations in the enzyme’s catalytic properties toward its substrates.
Atomic detail of the monovalent cation-binding site of Pfk-2
The presence and structural features of a monovalent cation-binding site have been shown for ribokinases from the x-ray structure of the E. coli enzyme in the presence of Cs+ (7). In both ribokinase (28) and Pfk-2 (12), the active site is located in a shallow groove formed between two domains. In ribokinase, the major domain contains the monovalent cation-binding site surrounded by two loops spanning residues from 244 to 254 and residues from 288 to 296, adjacent to the MgATP binding pocket (Fig. 3 A). Having determined the effect of K+ and Cs+ on Pfk-2 activity, we decided to obtain the x-ray structures of the ion-bound forms of the enzyme, to enable a direct comparison with the corresponding structure of E. coli ribokinase. Crystals were obtained essentially under the same conditions as those previously used to crystallize the inhibited, MgATP-bound, form of the enzyme, except for the addition of K+ or Cs+. Despite the presence of Na+ in the crystallization conditions (28 mM), the Pfk-2 structure bound to Cs+ shows one monovalent cation-binding site per monomer (PDBid 3UMP), unambiguously occupied by the metal and corresponding to a peak in the anomalous electron density map of ∼60 σ (Fig. 3 F). In the presence of potassium, electron density is observed occupying an equivalent position (PDBid 3UMO). Overall, the Cs+-bound and K+-bound structures were almost identical (0.10 Å root mean-square deviation (r.m.s.d.) for 617 Cα atoms in the dimer) and also very similar to that bound to MgATP alone (3CQD, 0.55 Å r.m.s.d.). All of these structures exhibit two MgATP molecules per subunit, one at the substrate site and the second (allosteric) juxtaposed to the first.
Figure 3.
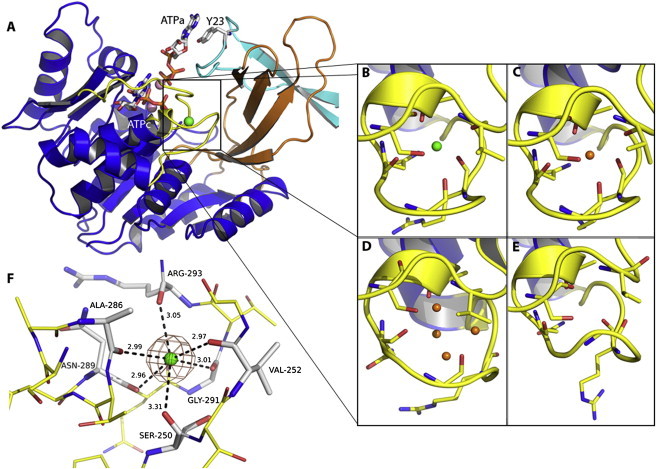
The monovalent cation-binding site of Pfk-2 from Escherichia coli. (A) Location of the cation binding site in the major domains (blue ribbons). The minor domain (orange ribbons) is shown packed against the partner subunit (cyan ribbons). The region around the Cs+ ion (green sphere) is formed by two loops (yellow), involving residues 288 to 296 and 244 to 254. Adjacent to this site the nucleotide substrate (ATPc) and the allosteric ATP (ATPa) sites are shown, highlighting the Y23 residue contributed from the partner subunit. The Mg2+ cations coordinated by the phosphoryl groups of ATPc and ATPa are represented as pink spheres. In panels (B to E), the side-chain residues around the cation-binding site are shown in licorice. (B) (PDBid 3UMP). The site is occupied by Cs+ (green sphere). (C) (PDBid 3N1C; chain A). In the presence of Na+ the site is organized around a single atom previously assigned as a water molecule (orange spheres). (D) (PDBid 3CQD, chain B). In the absence of cations the site is filled with four water molecules. (E) (PDBid 3N1C; chain D). The site could also be desolvated. (F) The coordination geometry of Cs+ (green sphere) and the carbonyl oxygen of the main chain are indicated by dashed lines.
In Pfk-2 the cation (either Cs+ or K+) is octahedrally coordinated (Fig. 3). For both ions, the first shell contains solely oxygen atoms coming from the backbone carbonyls of Ser-250, Val-252, Ala-286, Asn-289, Gly-291, and Arg-293 (Fig. 3 F). The mean bond distances are compatible with those expected for the given metal and coordination number (Table S3). For example, a typical K+-oxygen distance in octahedral coordination is 2.79 Å (according to the MESPEUS database (29)) compared with a measured mean distance of 2.93 Å, observed in our structure. In the case of Cs+, whose ionic radius is 1.81 Å (compared with 1.52 Å for K+), slightly longer coordinating bond distances are observed (mean values of 3.04 Å). In E. coli ribokinase the coordination geometry observed for Cs+ is best described as a pentagonal bipyramid where the ion interacts mainly with carbonyl oxygen atoms from the main chain, but the side chains of Asp-249 and Ser-294 are also within coordination distance (7).
Free energy perturbation proves the selectivity of the metal binding site of Pfk-2
Although our enzyme kinetic assays show that K+, but not Na+, enhances the allosteric inhibition of MgATP on Pfk-2 activity, this evidence is indirect and direct binding of K+ or Na+ to Pfk-2 has not been probed. Alchemical FEP calculations have been used to determine differences in the free energy of binding of ligands to proteins on the basis of potentials of mean force (30). Specifically, FEP calculations have been successfully applied to calculate the cation preferences of the selectivity filter in several ion-channels (31–35). Following this approach, we prepared two FEP systems, according to the thermodynamic cycle shown in Fig. 4 A (based on the K+-bound Pfk-2 x-ray structure). The difference in free energy between K+ and Na+ can be calculated as in (32,33), where ΔΔG(K+→Na+) = ΔG4(K→Na) - ΔG3(K→Na) = G(Na+)site – G(K+)site – (G(Na+)bulk – G(K+)bulk) and is larger than zero when K+ is preferred over Na+. FEP calculations yielded variations in free energies of <2 kBT for each step of change of the coupling parameter λ suggesting that our sampling scheme was sufficiently converged (data not shown). Furthermore, the difference between forward (K+→Na+) and backward (Na+→K+) simulations for the relative free energy change over all sampled windows did not exceed 1 kcal/mol (Fig. 4 B). The final values of free energy for ΔG3(K→Na) and ΔG4(K→Na) were obtained by combining the forward and backward calculations with the Bennett acceptance-ratio estimator (36). For the cation exchange reaction in solution, ΔG3(K→Na) was −34.3 ± 0.1 kcal/mol. This value is in agreement with the experimental solvation free energy difference reported for K+ versus Na+ (37), corrected for the fact that two ions were present in our system. For the K+-bound protein complex, the calculated free energy difference for the alchemical transformation ΔG4(K→Na) was −27.7 ± 0.1 kcal/mol. Upon the application of the thermodynamic cycle (ΔΔG = ΔG4(K→Na) - ΔG3(K→Na)) a free energy difference of 6.6 ± 0.1 kcal/mol is obtained for the alchemical transformation of two K+ ions into Na+. Therefore, our thermodynamic calculations indicate that each monovalent cation-binding site in Pfk-2 has a preference for K+ relative to Na+ of ∼3 kcal/mol in the presence of MgATP. These results are in agreement with the observed selectivity for the effect of cations on the enzymatic activity assays of Pfk-2.
Figure 4.
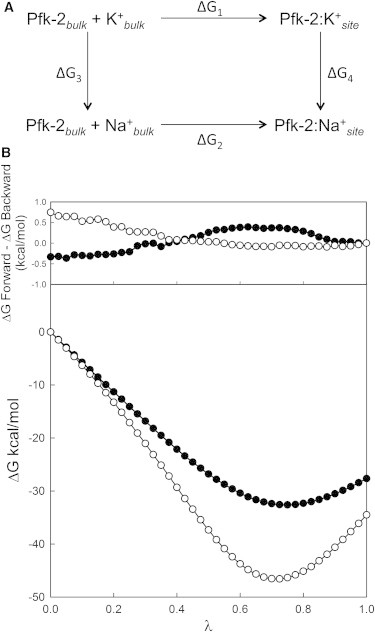
Alchemical free energy perturbation calculations. (A) Thermodynamic cycle for K+ to Na+ transformation. The two ensembles calculated by FEP-MD are labeled as ΔG3 and ΔG4. (B) The upper panel shows the free energy differences between the forward and backward FEP-MD simulations for each step of the K+⇔Na+ transformations in the absence (white circles) or bound to Pfk-2 (black circles). The bottom figure shows the values of ΔG3(K→Na) (white circles) and ΔG4(K→Na) (black circles) as a function of the coupling parameter λ. At λ = 1 the free energy difference between ΔG3(K→Na) and ΔG4(K→Na) gives a net gain of 3.3 kcal/mol for K+ over Na+ binding to each site.
Discussion
In this work, we demonstrate the presence of a monovalent cation-binding site in Pfk-2 by means of biochemical and structural methodologies together with molecular simulations. The x-ray structure of Pfk-2 in the presence of Cs+ unambiguously shows the presence of a monovalent cation-binding site, with structural features conserved in regard to that previously described for ribokinases. The binding of monovalent cations promotes enzymatic activation in ribokinases (60-fold and fivefold, respectively, in the case of the E. coli and S. aureus enzymes). This effect has been associated with a conformational change in the ribokinase active site that helps to shape the anion hole at the nucleotide-binding pocket, favoring phosphoryl transfer (7,28). The cation free x-ray structure of S. aureus ribokinase shows an unstructured cation-binding site that causes the occlusion of the nucleotide-binding pocket. The anion hole is also present in Pfk-2 at the N-terminal end of α-helix 8 and contains the crucial catalytic residue, Asp-256 (38). In this case the appropriate conformation of the nucleotide-binding pocket seems to be preformed in the absence of monovalent cations (PDBid 3CQD), which consistently had no effect on the kinetic parameters for substrates.
Selectivity of the monovalent cation-binding site of Pfk-2
Although the catalytic parameters of Pfk-2 are not dependent on the binding of monovalent cations, the allosteric inhibition was indeed enhanced. This effect was selective for monovalent cation of ionic radii larger than Na+, following the trend observed for ribokinases (7,8). Prior x-ray structures of Pfk-2 have been obtained in the presence of Na+ with either MgATP (PDBid 3CQD) or fructose-6-P (PDBid 3N1C). Together with those described previously, the structures reported here enable comparisons to be made with a view to discerning the pattern of change in the binding pocket, upon Cs+, K+ or Na+ binding. In the A subunit of Pfk-2 bound to fructose-6-P (3N1C) electron density is observed at the location where now we observe K+ and Cs+. In this case the density was modeled as a water molecule, but it could have been equally well described by a Na+ ion (Fig. 3 C). In particular, the protein structure around the suspected water molecule is effectively identical to that seen with K+ or Cs+ (Fig. 3, B and F), and the octahedral coordination is in fact more compatible with a Na+ ion than a water molecule, for which a tetrahedral arrangement of hydrogen bonds would be expected. Therefore, it seems possible that in some cases Na+ could have been observed bound to the monovalent cation site. However, it would be expected to be of much lower affinity due to the observed bond distances, which are barely compatible with those expected for octahedral Na+ coordination by oxygen. Typical coordination distances for Na+ are 2.49 Å (Mespeus server (29)) compared with the observed mean experimental distances of 3.02 Å. It would seem that the structure is constrained and unable to collapse around the Na+ to produce a more compact (and desirable) coordination sphere.
In the remaining subunits of the structures bound to either MgATP or fructose-6-P, clearly no metal ion is present. For example, in the case of the MgATP-Pfk-2 (3CQD) complex, despite being obtained in the presence of Na+, the binding site is either disordered (electron density missing for residues 289 to 293 in the A subunit) or structured in a way that is incompatible with monovalent cation binding (subunit B). In the latter case, the monovalent cation-binding site shows an altered conformation and is filled with four water molecules (Fig. 3 D). A similar phenomenon is repeated when the monovalent cation site is completely empty as in the D subunit of the fructose-6-P complex (Fig. 3 E). Overall, the evidence suggests that Na+ is generally absent from the cation-binding site (which tends to be somewhat disordered) or at best only weakly bound. Taken together, these observations give structural support to the selectivity of the cation-binding site of Pfk-2, according to our biochemical studies and the results of molecular simulation that suggest a significantly greater affinity for K+ than Na+.
Mechanism of monovalent cation-induced inhibition of Pfk-2
A unique characteristic of Pfk-2, as compared with the rest of the ribokinase family members, is its inhibition by the substrate MgATP. This is a regulatory feature also shown by the evolutionary unrelated phosphofructokinase-1 from E. coli (Pfk-1 (11,13)) that is physiologically relevant under gluconeogenic conditions: loss of this regulation causes substrate cycling of fructose-6-P and futile consumption of ATP, delaying growth in E. coli (27). As pointed out by Di Cera (2), the concentration of Na+ and K+ is tightly controlled in vivo, thus monovalent cations are not expected to function as regulators of enzyme activity. In E. coli the intracellular concentration of potassium is over 100 mM (39), which is enough to exert the maximum effect on Pfk-2. Kinetics (Fig. 1 A) and binding assay measurements (Fig. S1 B) show that the apparent Kd for K+ obtained under inhibitory concentrations of MgATP was between 2 and 5 mM suggesting that the monovalent cation-binding site exerts its function in vivo favoring the inhibited state of Pfk-2.
In Pfk-2 and ribokinase, the mechanism of action of the monovalent cation-binding site is indirect or allosteric. Furthermore, binding (Fig. 1 B and Fig. S1 A) and kinetic (Fig. 2) characterizations in Pfk-2 show that potassium exerts its effect by increasing the affinity of the allosteric binding site for MgATP. The comparison between the Pfk-2 MgATP-complex obtained in the presence K+ or Na+ gives some clues about the molecular mechanism affecting the MgATP allosteric site. Overall, the presence of K+ does not induce a large conformational change, but there are subtle but noticeable differences. The original complex of MgATP (obtained with Na+) shows some evidence for two alternative conformations for the adenosine moiety of the allosteric ATP in which interaction with the adenine of the catalytic ATP is clearly favored over a second conformation in which it lies over the side chain of Tyr-23 from the neighboring subunit (Fig. 3 A (12)). However, in the presence of either K+ or Cs+ this second conformation becomes favored (refined occupancies of 0.60 and 0.57 for the K+ and Cs+ complexes, respectively), prompting the idea that the presence of monovalent cations alters the equilibrium between conformational states of the allosteric ATP.
The most interesting (although still subtle) structural difference on metal binding is the position of the side chain of Thr-251, which is on the inside of the cation-binding region. As this loop becomes ordered in the presence of cations, the threonine side chain suffers a minor alteration in conformation, which is related to a change in the local water structure (Fig. 5). There are two waters bound in the case of the metal-free structure and three when metal is bound (Fig. 5). Furthermore, as the threonine shifts by slightly over 1 Å, it draws with it Glu-25 with which it forms two hydrogen bonds (one to its side-chain Oγ and the other to the main-chain amine). In addition, in our current work using MD simulations along with analysis of protein structure networks (40), we have observed that these residues (Thr-251 and Glu-25) contribute to the stabilization of the allosteric site of Pfk-2 in the presence of K+ by increasing the number of interactions with residues of the monovalent cation-binding site (work in progress). As a consequence, a further two hydrogen bonds, normally formed by backbone groups from Pro-24 and Gly-26 to the O3′ oxygen of the ribose of the allosteric ATP, are severely weakened in the subunit A (increasing in length from 3.05 Å to 3.4 Å). In subunit B a similar trend is observed, although in this case all of the equivalent distances are longer and effectively no hydrogen bonds exist in either case. These hydrogen bonds are only present when the ATP is in the conformation in which its adenine group basepairs with the catalytic ATP. In other words, the release of these restraints on the ribose group may well facilitate the structural rearrangement that allows the allosteric ATP to occupy the second conformation in which it stacks over Tyr-23. In fact, there are several reports using ab initio quantum mechanics, indicating that the hydrogen-bonded nucleobases and the adenine-tyrosine stacking must be somehow isoenergetic on both the gas phase and when solvated by water (41–45). Therefore, a destabilization of the interaction between the adenine groups from the allosteric and catalytic ATP such as observed in the binding site of Pfk-2 bound to Cs, would inevitably lead to a preference for the stacking interaction between Tyr-23 and ATP.
Figure 5.
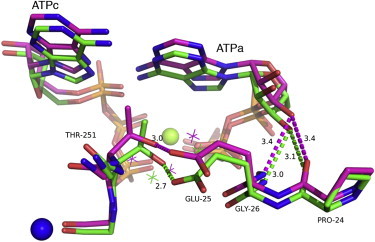
Structural superposition of the monovalent cation site in the presence and absence of Cs+. The backbone of the monovalent cation site is represented in green (Pfk-2-MgATP complex; PDBid 3CQD, chain B) and magenta sticks (Pfk-2-MgATP-Cs complex PDBid 3UMP). The green sphere corresponds to Mg2+ coordinated by the phosphates of the ATPc and ATPa. Upon binding of Cs+ (blue sphere), Thr-251 drawn the side chain of Glu-25 resulting in increments of the hydrogen bonds distances between the backbone groups of Pro-24 and Gly-26 and the O3′ oxygen of the ribose of the allosteric ATP from 3.1 to 3.4 Å on average. This change is also accompanied by rearrangements of the water network observed in the presence (magenta asterisks) and absence of Cs+ (green asterisks).
This sequence of structural changes is apparently triggered by cation binding to a series of backbone carbonyl groups. As suggested by Page and Di Cera (1), the backbone atoms possess useful features for allosteric regulation through cation complexation. Peptide bonds are planar and extremely stable due to resonance. Furthermore, the high dipole moment of the carbonyl moiety provides a strong electrostatic interaction with the ion. In this way, upon interaction via the carbonyl O atoms, binding of potassium leads to a long-range effect that is propagated to the allosteric site of Pfk-2 through a series of subtle knock-on effects. However, it remains to be fully understood how favoring the Tyr-Ade conformation could lead to the observed strengthening of the inhibitory effect.
Finally, considering that the cation-binding site described here is conserved among ribokinases and Pfk-2, we exhaustively searched the PDB for other ribokinase family members that could be potentially regulated by cations, based on the presence of a similar cation binding pocket in the crystal structure. We identified six other structures showing a cation in a structurally equivalent site (Fig. 6). Surprisingly, in addition to the monovalent cations Cs+, Na+, and K+, an Mg2+ ion has been assigned to this position for two members of the family. Unfortunately, to the best of our knowledge, no studies about the effect of cation binding on these activities have been reported. The sequence alignment derived from structural superposition of the loops that form the cation site shows no strong conservation pattern except for Ala and Gly residues (marked with asterisk symbols in Fig. 6), probably because in most cases the metal coordination does not involve contributions from side chains.
Figure 6.
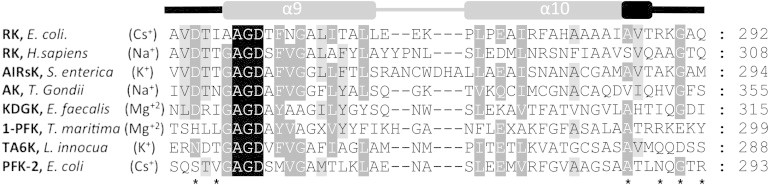
Multiple sequence alignment of the region forming the cation-binding site in members of the ribokinase family. The structures were obtained from the Protein Data Bank as follows: ribokinases (RK) from Escherichia coli (PDBid 1GQT) and Homo sapiens (PDBid 2FV7), aminoimidazole riboside kinase (AIRsK) from Salmonella enterica (PDBid 1TZ6), adenosine kinase (AK) from Toxoplasma gondii (PDBid 2ABS), putative 2-keto-3-deoxygluconate kinase (KDGK) from Enterococcus faecalis (PDBid 3KTN), putative 1-phosphofructokinase (1-PFK) from Termotoga maritima (PDBid 2AJR), putative tagatose-6- phosphate kinase (TA6K) from Listeria innocua (PDBid 3Q1Y), and Pfk-2 from E. coli (PDBid 3UMP). A schematic representation of the secondary structural elements formed by this region in Pfk-2 is shown above the alignment. The loops contributing to the metal binding site in the family are depicted in black. Asterisks indicate the positions of the residues that coordinate the monovalent cation in Pfk-2.
These results suggest that the presence of a cation-binding site is a conserved feature in the ribokinase family that provides different regulatory responses considering the structural characteristics of each active site.
Acknowledgments
This work was supported by Fondecyt 1090336 and 1130510, Conicyt, Chile, and the CEPID and INCT programs of Fundação de Amparo á Pesquisa do Estado de São Paulo (FAPESP) and Científico e Tecnológico (CNPq), Brazil. We acknowledge the National Laboratory for High Performance Computing at the Center for Mathematical Modeling (PIA ECM-02.-Conicyt, Chile). César A. Ramírez-Sarmiento was supported by a Conicyt doctoral fellowship.
The atomic coordinates and structure factors (codes 3UMO and 3UMP) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Footnotes
Andrés Caniuguir’s present address is Departamento de Obstetricia y Ginecología, Facultad de Medicina, Pontificia Universidad Católica de Chile, Santiago, Chile.
Supporting Material
References
- 1.Page M.J., Di Cera E. Role of Na+ and K+ in enzyme function. Physiol. Rev. 2006;86:1049–1092. doi: 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
- 2.Di Cera E. A structural perspective on enzymes activated by monovalent cations. J. Biol. Chem. 2006;281:1305–1308. doi: 10.1074/jbc.R500023200. [DOI] [PubMed] [Google Scholar]
- 3.Maj M.C., Singh B., Gupta R.S. Pentavalent ions dependency is a conserved property of adenosine kinase from diverse sources: identification of a novel motif implicated in phosphate and magnesium ion binding and substrate inhibition. Biochemistry. 2002;41:4059–4069. doi: 10.1021/bi0119161. [DOI] [PubMed] [Google Scholar]
- 4.Parducci R.E., Cabrera R., Guixé V. Evidence for a catalytic Mg2+ ion and effect of phosphate on the activity of Escherichia coli phosphofructokinase-2: regulatory properties of a ribokinase family member. Biochemistry. 2006;45:9291–9299. doi: 10.1021/bi060026o. [DOI] [PubMed] [Google Scholar]
- 5.Park J., Gupta R.S. Adenosine kinase and ribokinase—the RK family of proteins. Cell. Mol. Life Sci. 2008;65:2875–2896. doi: 10.1007/s00018-008-8123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miallau L., Hunter W.N., Leonard G.A. Structures of Staphylococcus aureus D-tagatose-6-phosphate kinase implicate domain motions in specificity and mechanism. J. Biol. Chem. 2007;282:19948–19957. doi: 10.1074/jbc.M701480200. [DOI] [PubMed] [Google Scholar]
- 7.Andersson C.E., Mowbray S.L. Activation of ribokinase by monovalent cations. J. Mol. Biol. 2002;315:409–419. doi: 10.1006/jmbi.2001.5248. [DOI] [PubMed] [Google Scholar]
- 8.Li J., Wang C., Zang J. Crystal structure of Sa239 reveals the structural basis for the activation of ribokinase by monovalent cations. J. Struct. Biol. 2012;177:578–582. doi: 10.1016/j.jsb.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 9.Kotlarz D., Buc H. Regulatory properties of phosphofructokinase 2 from Escherichia coli. Eur. J. Biochem. 1981;117:569–574. doi: 10.1111/j.1432-1033.1981.tb06375.x. [DOI] [PubMed] [Google Scholar]
- 10.Guixé V., Babul J. Effect of ATP on phosphofructokinase-2 from Escherichia coli. A mutant enzyme altered in the allosteric site for MgATP. J. Biol. Chem. 1985;260:11001–11005. [PubMed] [Google Scholar]
- 11.Cabrera R., Baez M., Babul J. The crystal complex of phosphofructokinase-2 of Escherichia coli with fructose-6-phosphate: kinetic and structural analysis of the allosteric ATP inhibition. J. Biol. Chem. 2011;286:5774–5783. doi: 10.1074/jbc.M110.163162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabrera R., Ambrosio A.L.B., Babul J. Crystallographic structure of phosphofructokinase-2 from Escherichia coli in complex with two ATP molecules. Implications for substrate inhibition. J. Mol. Biol. 2008;383:588–602. doi: 10.1016/j.jmb.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 13.Babul J. Phosphofructokinases from Escherichia coli. Purification and characterization of the nonallosteric isozyme. J. Biol. Chem. 1978;253:4350–4355. [PubMed] [Google Scholar]
- 14.Cornish-Bowden A. Butterworths; London; Boston: 1979. Fundamentals of Enzyme Kinetics. [Google Scholar]
- 15.Guixé V., Rodríguez P.H., Babul J. Ligand-induced conformational transitions in Escherichia coli phosphofructokinase 2: evidence for an allosteric site for MgATP2- Biochemistry. 1998;37:13269–13275. doi: 10.1021/bi980576p. [DOI] [PubMed] [Google Scholar]
- 16.Kabsch W. XDS. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCoy A.J., Grosse-Kunstleve R.W., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams P.D., Grosse-Kunstleve R.W., Terwilliger T.C. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project N The CCP4 suite: programs for protein crystallography. Acta Crystallogr. Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Laskowski R.A., Moss D.S., Thornton J.M. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 21.Davis I.W., Murray L.W., Richardson D.C. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32(Web Server issue):W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Case D.A., Darden T.A., Kollman P.A. University of California; San Francisco: 2010. AMBER 11. [Google Scholar]
- 23.Lindorff-Larsen K., Piana S., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meagher K.L., Redman L.T., Carlson H.A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003;24:1016–1025. doi: 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- 25.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 26.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres J.C., Guixé V., Babul J. A mutant phosphofructokinase produces a futile cycle during gluconeogenesis in Escherichia coli. Biochem. J. 1997;327:675–684. doi: 10.1042/bj3270675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sigrell J.A., Cameron A.D., Mowbray S.L. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 A resolution: insights into a new family of kinase structures. Structure. 1998;6:183–193. doi: 10.1016/s0969-2126(98)00020-3. [DOI] [PubMed] [Google Scholar]
- 29.Hsin K., Sheng Y., Walkinshaw M.D. MESPEUS: a database of the geometry of metal sites in proteins. J. Appl. Cryst. 2008;41:963–968. [Google Scholar]
- 30.Woo H.-J., Roux B. Calculation of absolute protein-ligand binding free energy from computer simulations. Proc. Natl. Acad. Sci. USA. 2005;102:6825–6830. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernèche S., Roux B. Energetics of ion conduction through the K+ channel. Nature. 2001;414:73–77. doi: 10.1038/35102067. [DOI] [PubMed] [Google Scholar]
- 32.Noskov S.Y., Bernèche S., Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 33.Yu H., Noskov S.Y., Roux B. Two mechanisms of ion selectivity in protein binding sites. Proc. Natl. Acad. Sci. USA. 2010;107:20329–20334. doi: 10.1073/pnas.1007150107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noskov S.Y., Roux B. Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. J. Gen. Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noskov S.Y., Roux B. Control of ion selectivity in LeuT: two Na+ binding sites with two different mechanisms. J. Mol. Biol. 2008;377:804–818. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett C.H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 1976;22:245–268. [Google Scholar]
- 37.Aaqvist J. Ion-water interaction potentials derived from free energy perturbation simulations. J. Phys. Chem. 1990;94:8021–8024. [Google Scholar]
- 38.Cabrera R., Babul J., Guixé V. Ribokinase family evolution and the role of conserved residues at the active site of the PfkB subfamily representative, Pfk-2 from Escherichia coli. Arch. Biochem. Biophys. 2010;502:23–30. doi: 10.1016/j.abb.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 39.Richey B., Cayley D.S., Record M.T., Jr. Variability of the intracellular ionic environment of Escherichia coli. Differences between in vitro and in vivo effects of ion concentrations on protein-DNA interactions and gene expression. J. Biol. Chem. 1987;262:7157–7164. [PubMed] [Google Scholar]
- 40.Brinda K.V., Vishveshwara S. A network representation of protein structures: implications for protein stability. Biophys. J. 2005;89:4159–4170. doi: 10.1529/biophysj.105.064485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rutledge L.R., Campbell-Verduyn L.S., Wetmore S.D. Characterization of nucleobase-amino acid stacking interactions utilized by a DNA repair enzyme. J. Phys. Chem. B. 2006;110:19652–19663. doi: 10.1021/jp061939v. [DOI] [PubMed] [Google Scholar]
- 42.Churchill C.D.M., Rutledge L.R., Wetmore S.D. Effects of the biological backbone on stacking interactions at DNA-protein interfaces: the interplay between the backbone···π and π···π components. Phys. Chem. Chem. Phys. 2010;12:14515–14526. doi: 10.1039/c0cp00550a. [DOI] [PubMed] [Google Scholar]
- 43.Šponer J., Leszczynski J., Hobza P. Structures and energies of hydrogen-bonded DNA base pairs. A nonempirical study with inclusion of electron correlation. J. Phys. Chem. 1996;100:1965–1974. [Google Scholar]
- 44.Rutledge L.R., Durst H.F., Wetmore S.D. Computational comparison of the stacking interactions between the aromatic amino acids and the natural or (cationic) methylated nucleobases. Phys. Chem. Chem. Phys. 2008;10:2801–2812. doi: 10.1039/b718621e. [DOI] [PubMed] [Google Scholar]
- 45.Florián J., Šponer J., Warshel A. Thermodynamic parameters for stacking and hydrogen bonding of nucleic acid bases in aqueous solution. Ab Initio/Langevin Dipoles Study. J. Phys. Chem. B. 1999;103:884–892. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.