

Abstract
Giant unilamellar vesicles or GUVs are systems of choice as biomimetic models of cellular membranes. Although a variety of procedures exist for making single walled vesicles of tens of microns in size, the range of lipid compositions that can be used to grow GUVs by the conventional methods is quite limited, and many of the available methods involve energy input that can damage the lipids or other molecules present in the growing solution for embedment in the membrane or in the vesicle interior. Here, we show that a wide variety of lipids or lipid mixtures can grow into GUVs by swelling lipid precursor films on top of a dried polyvinyl alcohol gel surface in a swelling buffer that can contain diverse biorelevant molecules. Moreover, we show that the encapsulation potential of this method can be enhanced by combining polyvinyl alcohol-mediated growth with inverse-phase methods, which allow (bio)molecule complexation with the lipids.
Introduction
Cellular membranes, that envelop the cytoplasm of the cell, its nucleus, and its various organelles, are self-assembled lipid bilayers that host also a variety of proteins and other biomolecules. For typical cell sizes on the order of ten micrometers, the lipid membranes span a few thousands of square micrometers, holding on the order of tens of billion lipids. The fact that self-assembly can drive so many individual molecules into a well defined and robust liquid structure of five nanometers thickness has long fascinated researchers in Physics, Chemistry, and Biology, who have studied lipid membrane structure and function.
Lipid bilayers self-assembled from a single or a reduced number of lipid species are often used as biomimetic models of cell membranes. Several membrane structures have become available for biophysical studies on the intrinsic properties of lipid bilayers as well as their interactions with other biomolecules: unilamellar and multilamellar vesicles of different sizes (1), supported bilayers (2) and nanodiscs (3) to only name a few.
Among these model biomembranes, giant unilamellar vesicles (GUVs) have played a prominent role (4). Due to their large dimensions, in the range of typical cell sizes, GUVs can be easily observed and micromanipulated under an optical microscope, thus allowing for the direct observation of relevant biophysical phenomena at a single membrane level (5).
The importance of GUVs has triggered a host of efforts seeking GUV preparation methods that can provide a fast and easy route to form vesicles from a variety of lipid compositions and under different buffer conditions (4). Methods for GUV formation typically involve a lipid bilayer preassembly step by evaporation of a lipid-containing organic solvent at a solid-liquid interface. Depending on the lipid composition, vesicles are sometimes formed spontaneously when the precursor lipid film is simply exposed to a buffer solution. This method is known as gentle hydration (6,7). A disadvantage of this method is that vesicle formation usually requires long timescales on the order of several days. Moreover, it works only with restricted lipid compositions and ionic conditions. Electromagnetic radiation (8) and electrical or mechanical stresses tend to promote the formation of unilamellar vesicles. Indeed, the most widely used vesicle formation method is known as electroformation, where an alternating electrical field is applied across a buffer-filled chamber bounded by conductive slides or crossed by metal wires (9). Electroformation is reasonably fast, taking several hours. However, growing vesicles in physiological buffers or from lipid mixtures containing charged lipids is still a challenging task, despite the recent improvements brought by electroformation at high field frequencies (10,11) or using a flow chamber with exchange of the buffer (12). Another important difficulty of preparation methods based on hydration is that encapsulation of proteins or other (bio)molecules, especially in buffers of physiological ionic strength, is generally inefficient and variable among liposomes. Alternatives are the rapid solvent exchange method (13) or the evaporation method (14). However, the rapid solvent evaporation method requires a large amount, typically one milligram, of lipid per sample.
There is currently broad interest in the use of vesicles as a platform to build either protocells or artificial cells that mimic aspects of bacterial or mammalian cells (15). Examples include the reconstitution of cytoskeletal protein networks (16–19), membrane-protein interactions (20,21), cell adhesion (22), and gene transcription and translation (23). In all these applications, biomolecules need to be encapsulated inside GUVs in physiological buffers. This is still a challenging task due to the high salt levels present in physiological buffers and to the relatively low vesicle formation speed that hinders a precise control of the biological activity of the proteins. Recently several new strategies have been proposed, which were designed to improve preparation speed, encapsulation efficiency, and/or applicability to charged lipids. One class of methods uses water-in-oil emulsion droplets as templates for bilayer vesicles. Bilayers can either be formed by transferring emulsion droplets stabilized by a lipid monolayer through an oil/water interface (24–26) or by forming double emulsion droplets with an oil shell and extracting the oil (27). Biomolecule encapsulation into emulsion droplets is highly efficient and reproducible even at physiological salt conditions. However, the emulsion-based methods generally require advanced equipment such as microfluidic devices. Moreover, traces of the oil phase can often be detected in the self-assembled membrane (4,28). Microfluidic inkjet encapsulation methods also improve biomolecule encapsulation and preparation speed compared to film hydration methods (29,30), but again specialized equipment is needed and residual oil can be left in the membrane.
To minimize damage of the molecules of interest (31), one is restricted so far to film hydration methods. Lipid film hydration methods have a strong potential for further development. It is important for instance to realize that membrane swelling from a preordered bilayer film requires water penetration into the membrane stacks, a process that is partially hindered when the lipids are deposited on glass or other solid substrates. A larger exposure of the preordered lipid films to hydration is achieved in a recently discovered agarose swelling method (32). Here, the organic solution containing the lipids is spread on a thin dried film of agarose, a naturally occurring polysaccharide. Upon addition of the buffer solution, GUVs are rapidly formed at the interface between the swollen agarose gel and the buffer. Vesicle formation is much faster than in classical electroformation or gentle swelling. Moreover, this method works with a wide range of lipid compositions and buffer conditions, and it is possible to efficiently encapsulate various biomolecules (32), including cellular proteins (28). However, the vesicle formation efficiency is reduced because lipids are distributed over the whole gel thickness, which are several micrometers. Furthermore, the agarose gel dissolves partly upon swelling and remnants can be detected in/on the membrane (32).
Here, we report a new, to our knowledge, swelling method inspired by the earlier work on swelling from agarose gels, which provides a facile and fast way to prepare vesicles with a wide range of lipid compositions. Instead of agarose, we use polyvinyl alcohol (PVA) gels. Our hypothesis was that PVA gels can optimize the lipid distribution at the interface between the substrate and the hydrating buffer. Moreover, PVA gels are expected to be much less prone to dissolution upon swelling at room temperature than other physical hydrophilic gels (33,34). Indeed, we find that vesicles formed by PVA-assisted swelling are not contaminated by polymer. We first demonstrate the efficiency of GUV formation on a PVA gel and study the growth process by inspecting the distribution of lipids before and after growth. Next, we compare growing efficiencies for different ionic and nonionic amphiphiles and show that the method allows us to encapsulate hydrophilic substances in the vesicles. We conclude by proposing a mechanism for vesicle growth.
Materials and Methods
Lipids
Lipids were purchased from Avanti Polar Lipids (Alabaster, AL) as a powder and dissolved in chloroform. Lipid solutions were stored at −20°C before use. In addition to 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) with two saturated C-16 tails, all other phospholipids used in this work have two C-18 tails with one unsaturation per tail (1,2-dioleoyl-sn-glycero-). We tested a range of headgroups, including neutral zwitterionic headgroups (-phosphocholine (DOPC)), as well as headgroups bearing a negative net charge (-phosphoglycerol (DOPG), -phosphoserine (DOPS)), or a positive net charge (trimethyl ammonium-propane (DOTAP)). DPPC was used for membranes in the gel state at room temperature, because its main transition temperature is 41°C. All other lipids undergo the fluid-gel transition below 0°C. As a fluorescent marker we used 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)(rhodamine-DOPE) in a 0.2 mol % mixture with other lipids. Other lipids used were 1,1′,2,2′-tetramyristoyl cardiolipin, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamineN-(cap biotinyl) (biotin-PE) and the PEGylated 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000].
Proteins
Neutravidin and Alexa Fluor 350-labeled neutravidin were purchased from Invitrogen (Breda, The Netherlands). Streptavidin was purchased from Thermo Scientific (Breda, The Netherlands). Rabbit skeletal muscle G-actin was purified by standard procedures including a gel filtration on a Sephacryl S-200 high-resolution column (GE Healthcare, Munich, Germany) (35) or purchased from Tebu-bio (Heerhugowaard, The Netherlands). Fluorescent actin with a dye/protein molar ratio of 0.6 was prepared by labeling amine groups with AlexaFluor488 carboxylic acid succinimidyl ester (Invitrogen) (36) or purchased from Invitrogen. G-actin labeled with biotin was purchased from Tebu-bio. G-actin was stored at −80°C in G-buffer (2 mM Tris-HCl, 0.2 mM Na2ATP, 0.2 mM CaCl2, 2 mM dithiothreitol (DTT), pH 7.8). Before use, G-actin solutions were thawed, treated with 5 mM DTT to reduce potential oxidized sulfhydryl groups, centrifuged at 120,000 × g for 30 min to remove potential protein aggregates, and finally bath-sonicated for 5 min to disrupt potential actin dimers (37).
PVA
PVA (MW 145000, Merck KGaA, Darmstadt, Germany) was purchased from VWR International (Fontenay-sous-Bois, France). For fluorescent studies, we labeled PVA with [5-(4,6-Dichlorotriazinyl)-aminofluorescein] (DTAF) by elimination of hydrochloride using previously described protocols (38,39). DTAF covalently couples to the alcohol groups of PVA at pH levels above 9. DTAF-labeled PVA was separated from free DTAF by extensive dialysis against MilliQ water using a regenerated cellulose membrane with a molecular mass cutoff of 4000–6000 Da. The average labeling stoichiometry was determined by spectrophotometric measurements of the light absorbance of the dialyzed PVA-DTAF solution at a wavelength of 495 nm, corresponding to the excitation maximum. The emission maximum of DTAF is at 516 nm. We first determined the extinction coefficient of aqueous solutions of DTAF at different concentrations by recording absorbance spectra between wavelengths of 400 to 800 nm with a scan rate of 600 nm min−1 on a UV/Vis Cary 500 Spectrometer (Fig. S1 in the Supporting Material). These measurements were performed using pH adjusted solutions, because of the pH dependence of the fluorescence properties of fluorescein analogs. We determined an extinction coefficient of 82,900 L mol−1 cm−1 (see inset of Fig. S1), and measured a DTAF concentration of 8.72 10−5 mol L−1 in the solution of DTAF-labeled PVA. The polymer concentration (determined by drying 1 g of the PVA-DTAF solution) was 2.97 0.07% (w/w). Thus, the degree of labeling was 0.43 mol DTAF per mol PVA chain, or 1.30 10−4 mol DTAF per mol repeating unit, implying that approximately every second PVA chain bears one fluorescent label. Both labeled and unlabeled polymers can be dissolved when exposed at temperatures higher than 50°C for longer than 30 min.
Other materials
In some experiments we formed polymersomes from the triblock copolymer, PMOXA10-PDMS87-PMOXA10 (MW 8154, PDI 2.2). Its synthesis is described elsewhere (40). All other chemicals were purchased from Sigma Aldrich (St. Louis, MO) and used without further purification unless specified otherwise.
Electroformation method
Electroformation was performed following the method of Angelova et al. (9). Briefly, 10 μL of a lipid solution (1 mg mL−1 in chloroform) were spread on an indium tin oxide (ITO)-coated glass slide. After drying of the lipid film under vacuum for 30 min, a chamber was formed with a second ITO slide and Sigillum wax (Vitrex, Copenhagen, Denmark) as a sealing agent. This chamber was filled with sucrose solution for neutral lipids and with a phosphate buffered saline (PBS) buffer solution (0.01 M phosphate buffer, 0.0027 M KCl and 0.137 M NaCl, pH 7.4) for lipid mixtures containing charged lipids. The osmolarity of the buffer was measured with an osmometer (Osmomat030, Gonotec, Berlin, Germany) and adjusted to 280 mOsm kg−1. An alternating electric field was applied across the chamber for 3–12 h. The amplitude and the frequency of the field were 1 V and 10 Hz for neutral lipids and up to 5 V and 500 Hz for charged lipids. Successful formation was checked by observing the growing chamber by phase contrast microscopy. The obtained GUVs were transferred to an Eppendorf tube, diluted 5 times with PBS solution, and left undisturbed for 15 min before observation. Table S1 summarizes the growing conditions for different lipids.
Gel-assisted GUV formation
A 5% (w/w) solution of PVA was prepared by stirring PVA in water or 280 mM sucrose solution while heating at 90°C. For fluorescent PVA substrates, one part of the DTAF-labeled PVA was mixed with 140 volume parts of unlabeled PVA. To estimate the amount of residual free fluorophore, we immersed PVA-DTAF-coated glass slides, chemically cross-linked with glutaraldehyde, in water for 1 h and measured the absorbance of the solution after adjusting the pH. We found a maximum amount of free fluorophore of 10%, providing an estimate for the uncertainty in fluorescence measurements. PVA-coated substrates were prepared by spreading 100–300 μL of PVA solution on a microscope coverslip (30 mm in diameter, Menzel-Gläser), which was then dried for 30 min in an oven at 50°C. 10–20 μL of lipids dissolved in chloroform (1 mg mL−1) were spread on the dried PVA film and placed under vacuum for 30 min to evaporate the solvent. Before use, the coverslips were cleaned with UV/Ozone for 15 min to prevent dewetting of the PVA film. A chamber was formed with Vitrex and filled with sucrose for neutral lipids or PBS buffer for charged lipids as described for the electroformation method in the previous section. GUV formation was followed using phase contrast microscopy. When the desired vesicle sizes were reached, typically in <1 h, GUVs were transferred into an Eppendorf tube using a pipette. The solution was diluted 5 times with PBS buffer and left undisturbed for 15 min before use. Table S1 summarizes the growing conditions for different lipids.
Encapsulation of proteins by gel-assisted GUV formation
For encapsulation of proteins a similar protocol was used. Instead of spreading the PVA and lipid solutions (3.75 mg mL−1) by hand, PVA was spin-coated on the coverslips at 1200 rpm for 30 s (DELTA 10 BM, SUSS MicroTec). A typical lipid mixture was composed of 94.8:0.2:5.0 DOPC/RhodB-PE/PEG-PE. In experiments containing biotinylated lipids 1% of DOPC was substituted by biotin-PE. 150 μL of lipid mixture was spin-coated on the dried PVA-coated slide at 1200 rpm for 300 s. We used either open-top formation chambers, assembled by placing a 0.12 mm thick spacer (secure-seal spacer, Invitrogen) on the coated slide, or closed chambers, assembled by placing a 0.5 mm thick spacer (Coverwell, 13 mm in diameter, Invitrogen) and another cover slide on the coated slide. Lipid swelling was initiated by introducing a buffer containing the proteins to be encapsulated (I-buffer). This I-buffer contains all necessary elements for the polymerization of actin (25 mM imidazole-HCl (pH 7.4), 1 mM DTT, 0.1 mM MgATP, 50 mM KCl, 2 mM MgCl2), 280 mM sucrose for osmotic pressure matching, 0.5% (v/v) glycerol, and an oxygen-scavenging mixture of 2 mM trolox, 2 mM protocatechuic acid, and 0.1 μM protocatechuate 3,4-dioxygenase that minimizes blinking and photobleaching (41). The lipid film was allowed to swell for 45 min, at a temperature of 4°C where actin polymerization is minimized. The GUVs were harvested by pipetting at least 2 chamber volumes of a glucose solution (O-buffer) into the formation chamber, causing the GUVs to flow into the adjacent observation chamber or into an open-top observation chamber assembled by placing a 0.5 mm thicker spacer on a glass slide and closed by a cover slide after GUVs harvest. The glass slides were passivated by a casein solution (2 mg mL−1) to prevent liposome adhesion and rupture. Actin polymerization was initiated by bringing the observation chamber to room temperature. The O-buffer osmolarity was adjusted to be at least 20 mOsm kg−1 higher than the I-buffer osmolarity (Osmomat030, Gonotec GmbH) to prevent bursting of vesicles. The actin concentration in all experiments was 23.8 μM (equivalent to 1 mg/mL), including 20 mol % or 30 mol % of AlexaFluor488-labeled actin and 0.25 mol % of biotinylated actin (1:400 molar ratio to actin). Streptavidin at a 1:25 molar ratio to the total actin concentration was included as a network cross-linker.
Inverse-phase precursor films for gel-assisted encapsulation
We adapted the inverse phase precursor method, which was previously combined with electroformation (42), to prepare lipid films containing biotinylated lipids complexed with neutravidin. By dispersing small water droplets containing hydrophilic moieties in the organic solvent containing the phospholipids, this method enables to prepare vesicles decorated with neutravidin or chitosan on their membrane. If neutravidin is used, it enables specific anchoring of encapsulated molecules to the membrane. An inverse emulsion was prepared from a mixture of lipid in chloroform and buffer solution as described previously (42). Briefly, a volume of 2 μL neutravidin in PBS (48 mg mL−1) was added to 60 μL of a lipid solution as described previously. In control experiments without biotin-PE lipids, pure PBS buffer was added. The mixture was pipetted up and down several times with a 500 μL glass syringe in a vial until the mixture became opalescent, signifying that inverted micelles have formed. 40 μL of this precursor emulsion was spin-coated on the PVA-coated slide (prepared as described previously) at 100 rpm for 100 s. The slide was then dried under vacuum for 100 min at room temperature. The remaining steps were the same as in the PVA swelling method, except that the lipid swelling time was 90 min instead of 45 min.
Optical microscopy
Vesicle contours were imaged by phase contrast microscopy using an inverted TE 2000 microscope (Nikon, Japan) equipped with a 60× WI/1.2 NA Plan Apo DIC objective, 100× NA 1.4 Plan Apo objective, or 40× Ph2/NA 0.60 Plan Fluor objective. Images were recorded with a digital camera (Hamamatsu EM-CCD, Japan) with a pixel depth of 16 bits. Three-dimensional (3D) fluorescent imaging was performed using confocal laser scanning microscopy (CLSM) with a Nikon C1 scanhead. Images were captured using EZ-C1 software (Nikon, version 3.50). The DTAF-labeled PVA film was excited using an argon-ion laser (Melles-Griot) at 488 nm, whereas rhodamine-labeled lipids were excited using a helium-neon laser (Melles-Griot) at 543 nm. Quantitative analysis of fluorescent intensities in the confocal images was performed using ImageJ, using appropriate rescaling of the acquired signals to the same standard levels. In all measurements the background noise measured from blank samples containing no fluorophores was subtracted.
For dried DTAF-PVA films, some bleaching of the fluorophores was observed when obtaining confocal z-stacks, typical confocal scans were performed by starting above the PVA gel and finishing on the glass surface. Hydration of the films caused an increase of the fluorescence intensity of both the DTAF-PVA and rhodamine lipids. This intensity increase can be explained by the change in refractive indices of the films upon hydration. In solution, the fluorescence of DTAF increases in the presence of PVA, as determined by fluorescence spectrophotometry using a Horiba Jobin Yvon FluoroMax-4 spectrofluorometer (see Fig. S2), a comparable increase is found when DTAF is directly coupled to PVA. Additionally, in the swollen DTAF-PVA films, the fluorescence spectrum of DTAF was broadened compared to free DTAF in solution (dashed line in Fig. S2), as measured by CSLM. As a result, some emission bleeding of the green channel into the red channel occurred. We checked that this did not influence the qualitative measurements of the lipid distribution by performing additional scans of labeled GUVs grown on unlabeled PVA (see Fig. S3) and also compared the bleeding profile of labeled PVA with our samples, which is exactly the same up to experimental errors (see Fig. 2 and Fig. S4).
Figure 2.
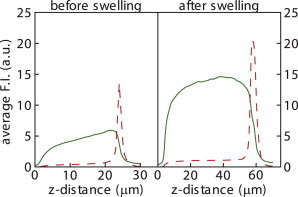
CLSM z profiles of a DTAF-labeled PVA film (solid line) and a fluorescent DPPC film (dashed line). Lipids and gel are well separated before and after swelling with PBS buffer solution at room temperature. Residual red fluorescence inside the gel is due to emission bleeding of the DTAF, as explained in Materials and Methods. A z distance of zero microns corresponds to the bottom of the labeled PVA gel.
Results and Discussion
Fully hydrolyzed high-molecular weight PVA was used to form a dry but swellable polymer film on a glass support. A thin film of lipids was then spread on the gel surface and the lipid-gel system was hydrated in an aqueous solution. Within 2 min, numerous unilamellar vesicles can be observed, as shown in Fig. 1. This observation is in stark contrast with hydration of a lipid stack deposited directly on glass, where the majority of bilayer structures correspond to multilamellar vesicles or cylinders and only a minor fraction correspond to unilamellar vesicles. Compared to the standard electroformation method, vesicle formation from PVA-lipid films has the advantage that GUVs are formed much faster, as shown in Fig. 1. Furthermore, the gel-assisted swelling method avoids the risk of lipid degradation, which may occur during electroformation (31,43).
Figure 1.

Phase contrast images of DOPC-GUVs, obtained by (A) electroformation and by (B) PVA-assisted swelling. Variations in contrast are due to differences in composition of inner and outer solutions. Scale bars 20 μm.
The speed of vesicle formation on swelling PVA gels is comparable to that observed previously on an agarose substrate (32). However, the agarose-growing method leads to encapsulation of some agarose polymer, as well as to polymer decoration of the membrane (28). To test whether PVA is present in the GUVs, we performed confocal microscopy of DOPC GUVs grown on fluorescently labeled PVA films. As shown in Fig. S5, no traces of fluorescent PVA can be found either on the vesicle membranes or inside the vesicles. This visual observation is confirmed by quantitative image analysis; weak fluorescence in the green channel is due to some free DTAF, as discussed in the Materials and Methods section. The absence of vesicle contamination by PVA can be primarily traced to the PVA gel structure, which is much less prone to dissolution upon swelling at room temperature than other physical hydrophilic gels (33,34). Moreover, the lipid distribution in the PVA film prevents contamination of the vesicles by PVA, as discussed below.
Lipid distribution
To visualize the spatial distribution of the lipids with respect to PVA in the lipid-PVA films, we measured the relative fluorescence intensities of the rhodamine-labeled lipids and the fluorescein-labeled PVA of a 20 μm × 20 μm patch as a function of height (z direction) above the glass substrate. We first focused on the lipid distribution before and after gel swelling for DPPC, a lipid that is in a gel state at room temperature. As shown in Fig. 2, the lipid bilayer is localized at the gel surface and does not penetrate the polymer film, independently of the degree of swelling of the gel.
In the original gel-assisted swelling method of Horger (32), it was shown that the lipids completely penetrated the agarose film, which is tens of micrometers thick. The agarose and the lipids thus intimately mix over the full film thickness. Our results show that this is not a requirement to promote giant vesicle growth (see also Fig. S3). Moreover, the reduced mixing of lipids with polymer in PVA gels compared to agarose gels reduces the probability of vesicle contamination by gel residues.
As discussed previously, GUVs are formed rapidly from films composed of lipids which are in a fluid state at room temperature. Confocal imaging shows that several layers of vesicles can be observed on the gel surface (see Fig. 3 inset B and Fig. S3). Small vesicles are typically located at the gel surface, while larger ones are further away. During the growing process, small vesicles often fuse into a larger one that may later detach from the gel. However, although many vesicles detach, many stay on the surface of the PVA film. As the 3D confocal image shows (Fig. S6), holes in the fluorescently labeled lipid film can be observed directly underneath the vesicles, likely corresponding to the partial consumption of the lipid film to build the vesicle above the hole.
Figure 3.
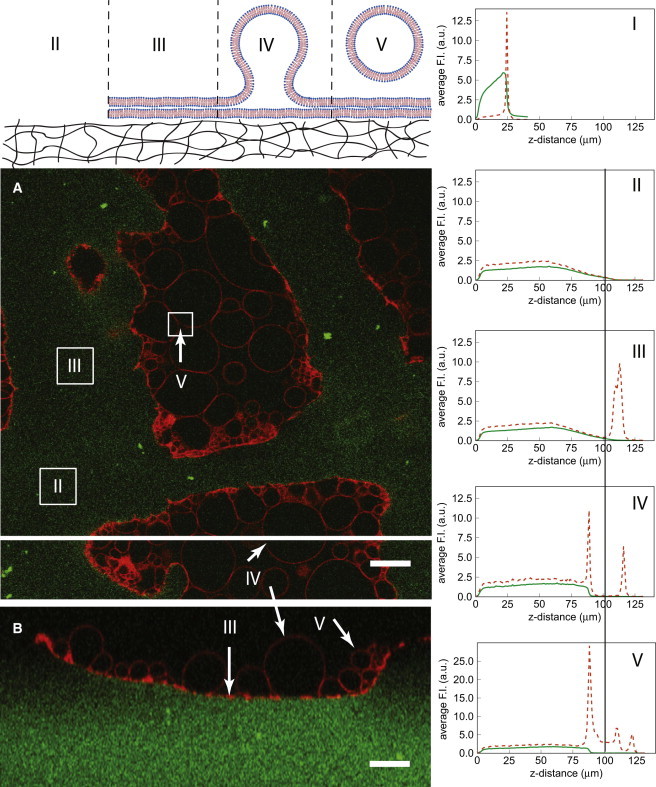
Heterogeneity of lipid distribution on a swollen gel interface. Inset A shows the XY section of a DTAF-labeled PVA gel (green) and DOPC-GUVs labeled with 0.2 mol % Rhodamine (red) recorded with CLSM at a z-position of 31 μm. Inset B is a XZ section at the position of the white line. Graph I is a z profile of the same sample in the dry state before hydration with buffer solution, other graphs correspond to the different labeled regions of the micrograph, which are further sketched in the diagram above. Average intensity of a given z was determined for a 20 μm × 20 μm square and the average intensity variations plotted with z. The scan was performed starting at the top of the sample; a z distance of zero microns corresponds to the bottom of the labeled PVA gel. Scale bar 20 μm.
After swelling, the PVA surface appears very corrugated with valleys and mountains. Any XY plane through this interfacial region, as exemplified by Fig. 3 in inset A, will then display areas inside the gel and areas outside the gel where lipid can be observed. In this geometry the gel interface is almost perpendicular to the confocal section, allowing to precisely screen the GUVs for fluorescent PVA traces and to reconstruct a 3D representation as in Fig. S6. Depth profiles of the PVA and lipid fluorescence intensity reveal the heterogeneity of the lipid distribution on the gel film, which can be related to the lipid deposition process or to the swelling of the gel. Fig. 3 summarizes all the observed cases. The vertical line represents the z-position where the confocal micrograph shown in inset A was recorded. The green channel represents the fluorescently labeled PVA, the red channel the fluorescently labeled lipid. The figure inset I displays for reference a z profile from the same sample before swelling. Inset II shows gel surface regions after swelling without any traces of lipids, whereas inset III corresponds to a lipid film still attached to the gel without any vesicles. In some regions GUVs still attached to the gel can be observed, as displayed in inset IV. The last inset V corresponds to the more rare case where a detached vesicle is still in the neighborhood of the gel interface above a membrane. Inset B in Fig. 3 corresponds to a vertical cross section, which better views the cases III to V.
In summary, our results show that GUVs free of polymer contamination grow during gel swelling from a heterogeneous lipid film at the gel surface. Given that the lipid solution spread on a PVA gel does not penetrate into the gel, we tested the effect of lipid/PVA mixing by forcing lipids into the gel. This was achieved by spreading a PVA solution mixed with small multilamellar vesicles. In this case, no vesicle formation could be observed upon hydration, thus confirming that gel/lipid mixing is not required for vesicle growth on PVA.
GUV formation: dealing with the hard cases
As shown in the preceding section, GUVs can be rapidly formed from the zwitterionic lipid DOPC by PVA-assisted swelling. In this section, we will show that the PVA growing method also provides a valuable tool to form vesicles from other types of lipids, which are difficult to form by other film swelling methods. Lipids with charged headgroups are a clear challenge for electroformation. Recently, a method to grow charged lipid vesicles under physiological buffer conditions at high frequencies was proposed (10,11). However, even under such conditions, a number of difficult issues remain, related to the growth of vesicles from purely charged lipids, to the vesicle yield, and to the detachment of the vesicles from the growing substrate. We have for instance compared the growth of DOPC-GUVs containing DOTAP, a cationic lipid, by electroformation and by PVA swelling. For these lipid mixtures, vesicles are obtained in a high number by electroformation, but they cannot be detached from the ITO substrate. With the PVA swelling method, even 100% DOTAP vesicles can be formed and transferred, albeit with a smaller yield than in the case of zwitterionic lipids. Fig. S7 shows a free floating GUV composed from DOTAP grown by PVA swelling. The detachment yield can be improved by gentle sonication.
Examples of other systems grown by the PVA swelling method are displayed in Fig. 4. Images in the left column show vesicles formed by growing on pure PVA gel, whereas images in the right column show vesicles grown on a PVA gel containing sucrose. In some cases, the presence of sucrose in the dry gel improves vesicle formation. The figure shows GUVs grown from DOPC, DOPG, DOPS, and the triblock copolymer PDMS-PMOXA-PDMS. In all cases, GUVs formed on a PVA gel that contains sucrose detach better during the growing process. Furthermore, the formed vesicles are less prone to adhere to each other in the growing chamber. Similar results can be obtained by making the PVA gel in a PBS solution, although with somewhat less efficiency of vesicle detachment (compare Fig. S8). Interestingly, in the case of DOPS on pure PVA, tubular vesicles could be observed in the first few seconds of the swelling, which disappear over time to form spherical vesicles. None of the other lipid cases showed this behavior. Tubular vesicles were also observed for the triblock copolymer (PMOXA-PDMS-PMOXA) on pure PVA, but here they do not lead to spherical polymersomes, a structure that could only be observed on PVA containing sucrose. Nonetheless, the formation of polymersomes yielded in a fewer number of GUVs compared with phospholipids.
Figure 4.
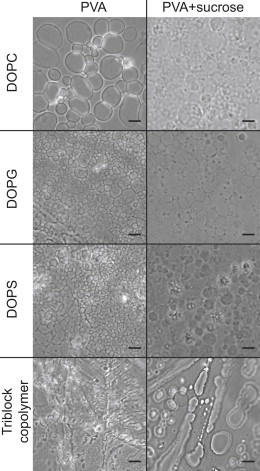
Phase contrast images of GUVs formed from different amphiphiles. Left column: growth on pure PVA gel. Right column: growth on a PVA gel containing sucrose. Amphiphiles: DOPC, DOPG, DOPS, and triblock copolymer (PDMS10-PMOXA87-PDMS10). Swelling solution: PBS. Scale bars 20 μm.
We also investigated lipids with more than one charge per headgroup. A particularly relevant example is cardiolipin, a lipid containing four lipid tails and two negatively charged headgroups. Although GUVs containing up to 50% cardiolipin in DOPC/cardiolipin mixtures can be grown by electroformation, we find that the structures obtained systematically display defects, similar to those observed by Fedotenko et al. (I. Fedotenko, A. Weinberger, T. Tanasescu, F. Favarger, C. Stefaniu, I. Tashi, G. Brezesinski, C. Marques, and A. Zumbuehl, unpublished), which may be caused by headgroup degradation from the applied electric field (Fig. 5 A). The PVA-assisted swelling method allows us to form defect-free GUVs within minutes, even from 100% cardiolipin (Fig. 5, B and C).
Figure 5.
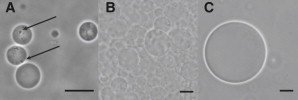
(A) Vesicles of 1:1 DOPC/cardiolipin mixtures grown by electroformation. Arrows show vesicle defects. (B) Cardiolipin vesicles on the PVA gel after growing. (C) Cardiolipin vesicles after dispersion in PBS. Images were recorded in phase contrast mode. Swelling solution: sucrose. Scale bars 20 μm.
The PVA swelling method can also be performed at temperatures different from room temperature. For instance, one can obtain DPPC vesicles by swelling with preheated buffer in a heated chamber at 50°C or DOPC vesicles by swelling in the fridge at 4°C. For high temperatures, the only limitation is the preservation of gel integrity. Indeed, depending on PVA molecular mass, some of the polymer can dissolve upon heating, thus leading to lower yields and GUV contamination. The PVA used in this work, in the range of 145 kDa, does not dissolve at room temperature but partially dissolves if exposed for more than half an hour at temperatures above 50°C. When lower molecular mass PVA is used, around 16 kDa, partial dissolution is already observed at room temperature. It is also possible to successfully grow GUVs composed from Sphingomyelin and Cholesterol in a range where this lipid mixture is in the liquid-ordered phase, however a complete study of the conditions under which GUVs from domain-forming ternary mixtures can be grown by the gel method is beyond the scope of this work.
Careful tuning of the system composition, such as the introduction of a small fraction of PEGylated lipids (45), or better spreading of the precursor film by spin-coating (12), have been reported in the literature to improve the yield of GUVs containing charged headgroups and to facilitate detachment from the growing surface. Similar results have been observed in our case for gel-assisted growth.
A common problem with some lipids is that vesicles do not detach easily from the growing surface after formation. We found that detachment can be improved by several different strategies. Sonication for 1–2 s in a standard ultrasound bath, for instance, can better detach the GUVs without breaking them. Furthermore, if a sugar such as sucrose is added to the gel, a larger number of vesicles spontaneously detach during the formation process. This can be due to the extra osmotic pressure from the sugar dissolving from the gel that pushes away the membranes (46).
In summary, we have shown that the PVA swelling method can successfully deal with different classes of lipids, which are otherwise difficult to grow, the method being in particular well adapted for charged lipids.
Encapsulation
The PVA gel formation method is advantageous for encapsulating many molecular species of biological relevance that are often prone to react at room temperature, e.g., cross-linking or degradation due to peroxidation caused by electric fields (43). We first tested the ability of this method to encapsulate the molecules required to reconstitute a simple biomimetic cytoskeleton inside a GUV. As shown in Fig. 6, A and B, actin filaments were successfully incorporated into vesicles and bundles formed if the swelling solution contained additional cross-linker. Similar actin structures have been shown by swelling on agarose gels (28). However, in this case the vesicles are contaminated by agarose, which adheres to the membrane.
Figure 6.
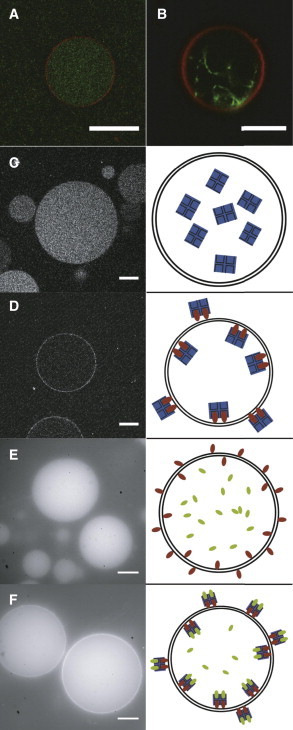
CLSM images of DOPC-GUVs grown by (A and B) PVA swelling at 4°C from 94.8:0.2:5.0 DOPC/RhodB-PE/PEG-PE mixtures and (C–F) DOPC-GUVs grown by a combination of PVA swelling and the inverse-phase precursor method (42). (A) Encapsulated actin filaments in the interior of the GUV. (B) Actin bundles inside the GUV, formed by adding of streptavidin with an actin/streptavidin ratio of 25:1 as a cross-linker. (C) In the absence of biotinylated lipids, AlexaFluor350-labeled neutravidin is homogeneously distributed in the vesicle interior. (D) In the presence of 1% biotinylated lipids, AlexaFluor350-labeled neutravidin binds to the membrane. (E) In vesicles swollen in a solution containing 500 nM fluorescently labeled biotin, the biotin is homogeneously distributed inside the vesicle interior in the absence of membrane-bound neutravidin. (F) Vesicles were formed as in (E) but with neutravidin on the membrane surface. The encapsulated biotin-fluorescein binds to the membrane-anchored neutravidin, resulting in a bright ring of fluorescent-biotin. The right column displays sketches of the systems in the left images: The membrane is represented by black lines, biotinylated lipids by red rods, fluorescein-biotin by green rods, and neutravidin by a blue square. Scale bars 20 μm.
It is often desirable to control the anchoring of encapsulated molecules to the vesicle membrane. A convenient way to achieve anchoring is to use biotinylated lipids, which enables anchoring to biotinylated species by neutravidin. We can localize neutravidin specifically on the membrane by combining the PVA swelling method with the inverse-phase precursor method, which allows complexation of lipids with neutravidin. Inverse-phase precursor films were already successfully used for the encapsulation of large hydrophilic molecules during electroformation (42). As shown in panel D of Fig. 6, GUVs grown from a precursor film containing neutravidin have a neutravidin functionalized membrane. In the absence of biotinylated lipids, the neutravidin is in the vesicle interior, as shown in panel C. The neutravidin is still functional, as shown by encapsulation of fluorescently labeled biotin. When neutravidin is on the membrane, fluorescent biotin forms a bright ring, signifying binding to neutravidin (panel F). In the absence of membrane-bound neutravidin, fluorescent biotin is found throughout the vesicle (panel E).
A growth scenario
To understand how the efficiency of encapsulation of water-soluble (bio)molecules in the vesicles can be optimized, one needs to gain a deeper insight into the mechanisms of vesicle formation during gel swelling. When a film of DOPC is spread from a chloroform solution on a glass slide and subsequently hydrated in an aqueous solution, one can observe the spontaneous formation of bilayer structures with a majority of multilamellar vesicles (47). The left drawing of Fig. 7 shows the possible pathways for water penetration into the stacks of preordered phospholipid bilayers on glass. There are two main water transport modes. First, the water can access the interlamellar region from the edges and swell the stack; a feature easily observed using differential interference contrast microscopy (48). A second pathway for water transport is by direct membrane permeation, because phospholipid bilayers display a finite water permeability in the order of 160 μm s−1 (49). Direct permeation can generate swollen structures of tens of micrometers in a few seconds.
Figure 7.
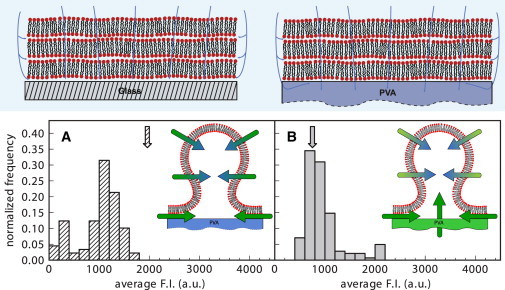
Top panel schematically depicts water penetration pathways through a bilayer stack deposited on glass and PVA gel. Lower panel are CLSM measurements of the average internal fluorescence intensity of GUVs grown on a PVA gel. (A) Vesicles swollen with a solution containing fluorescein. The average solution intensity is denoted by the arrow. (B) Vesicles swollen with a nonfluorescent solution on PVA gel loaded with fluorescein. The arrow indicates the fluorescent intensity of the solution after the fluorophore diffused from the gel. Histograms built from 89 vesicles for (A) and 142 vesicles for (B). Vesicles smaller than 5 μm were neglected.
The right drawing of Fig. 7 shows the pathways of water penetration during swelling of a preordered bilayer stack on the surface of a dry PVA gel. The most prominent difference from the case of swelling on glass is related to water-uptake by the PVA gel: indeed a strong chemical potential gradient exists between the outer solution and the dry gel, which drives water across the bilayer stack. As a consequence of gel swelling, the capillary forces driving water at the membrane-gel interface are modified. This effect is further amplified by gel stretching and by gel surface corrugation, which may increase the number of membrane defects for water access. These different solution transport mechanisms are likely to display different degrees of selectivity with respect to aqueous solutions containing other molecules or nanoparticles. We argue that the differences previously observed in encapsulation efficiencies as a function of molecular size and growing methods (28,32,50–52) are due to the respective importance of different water penetration pathways in the final aqueous content of GUVs.
To discriminate between the effects of different water penetration pathways on encapsulation efficiency, we performed two comparative experiments. In the first, we formed vesicles by swelling lipids on a PVA gel with a solution containing fluorescein. In the second, we formed vesicles by swelling a PVA gel preloaded with fluorescein with a nonfluorescent solution. Fig. 7 A displays the fluorescence intensity of vesicles grown from a fluorescent solution whose average fluorescence intensity is indicated by the arrow. The fluorophore content of the vesicles is roughly 50% of that of the swelling solution, comparable to encapsulation efficiencies reported in the literature (28,32). This result suggests that on average roughly half of the water content inside the GUVs is due to solution transport across the membrane, which is impermeable to the fluorophore. Fig. 7 B shows fluorescence levels from GUVs grown on a fluorophore-loaded gel. The arrow indicates the average fluorescence intensity of the swelling solution after the swelling process took place. Solution volumes used in both experiments were comparable, but the total amount of fluorophore in the second experiment was ∼2.5 times smaller than in the first one, which corresponds to the ratio of the average fluorescence intensities of the two solutions. As the histogram in Fig. 7 B shows, the encapsulation efficiency are higher when the PVA gel is preloaded with fluorophore. As sketched in the inset, we propose that this is due to a direct transport of fluorophore from the gel reservoir to the growing vesicle and not only from side penetration through defects.
Our findings thus suggest that membrane permeability is an important parameter determining vesicle growth by gel swelling. Indeed, the water permeability of DOPC is circa 160 μm s−1 (49), whereas the water permeability for typical polymersomes was reported to be the order of 2.5 μm s−1 (53). This is likely an important factor, which can explain why the yield of polymersomes grown by swelling is lower than the yield of liposomes. Furthermore, permeability plays a role in the final composition of solutions encapsulated in GUVs, by determining the relative importance of the different possible water penetration pathways.
Conclusions
Gel-assisted formation of GUVs on PVA allows for an easy and rapid growth of polymer-free GUVs in high yields without the need for special equipment. The process works for many different lipid mixtures, covering a wide range of lipids with one or more headgroups of zwitterionic, cationic or anionic nature. Due to the ability of PVA to swell at low temperatures close to the freezing point of water, the method is well adapted for the encapsulation of biomolecules such as actin, which can be encapsulated in monomer form at low temperature and subsequently polymerized by warming the vesicles to room temperature. Importantly, no input of energy is required to form GUVs, which minimizes the risk of degradation of the amphiphiles and of the proteins for membrane embedment or vesicle encapsulation.
Furthermore, we found experimentally that the gel formation process can be combined with inverse-phase precursor films to improve the encapsulation efficiency or localize (bio)molecules specifically to the membrane surface. Our method provides also a natural way to improve encapsulation efficiency of small hydrophilic molecules by predissolving them in the PVA gel.
Our results point to different penetration pathways for the aqueous solution that swells the membrane stacks. Part of the swelling originates at the defects in the membrane, where water can invade the interlamellar spacing. However, a significant amount of water also penetrates by direct permeation across the bilayers. This confirms the importance of bilayer permeability for water in the formation of GUVs. For polymersomes too, osmotic imbalance generated by added sucrose improves conditions for vesicle formation.
PVA-assisted formation of GUVs provides a new route to prepare GUVs in a membrane laboratory. Although we have prepared small-scale samples, the method has potential for large-scale production and can be easily automated. Current work in our group, exploring new lipids and buffer compositions, points to an application range of this method beyond the one reported here.
Acknowledgments
The authors thank S. Z. Omar and I. Moroz for valuable input.
This work was performed in the framework of the International Research Training Group (IRTG) Soft Matter Science and Foundation for Fundamental Research on Matter (FOM). A.W. thanks the Région Alsace for grant support, F.-C.T. and G.H.K. the Netherlands Organisation for Scientific Research (NWO), and W.M. the Swiss National Science Foundation and NCCR Nano Sciences for funding the work leading to the paper.
Supporting Material
References
- 1.Lasic D. Elsevier Science; 1993. Liposomes: from physics to applications. [Google Scholar]
- 2.Tanaka M., Sackmann E. Polymer-supported membranes as models of the cell surface. Nature. 2005;437:656–663. doi: 10.1038/nature04164. [DOI] [PubMed] [Google Scholar]
- 3.Bayburt T.H., Sligar S.G. Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci. 2003;12:2476–2481. doi: 10.1110/ps.03267503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walde P., Cosentino K., Stano P. Giant vesicles: preparations and applications. ChemBioChem. 2010;11:848–865. doi: 10.1002/cbic.201000010. [DOI] [PubMed] [Google Scholar]
- 5.Dimova R., Aranda S., Lipowsky R. A practical guide to giant vesicles. Probing the membrane nanoregime via optical microscopy. J. Phys. Condens. Matter. 2006;18:S1151–S1176. doi: 10.1088/0953-8984/18/28/S04. [DOI] [PubMed] [Google Scholar]
- 6.Reeves J.P., Dowben R.M. Formation and properties of thin-walled phospholipid vesicles. J. Cell. Physiol. 1969;73:49–60. doi: 10.1002/jcp.1040730108. [DOI] [PubMed] [Google Scholar]
- 7.Akashi K., Miyata H., Kinosita K., Jr. Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys. J. 1996;71:3242–3250. doi: 10.1016/S0006-3495(96)79517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Billerit C., Jeffries G., Jesorka A. Formation of giant unilamellar vesicles from spin-coated lipid films by localized IR heating. Soft Matter. 2012;8:10823–10826. [Google Scholar]
- 9.Angelova M.I., Dimitrov D.S. Liposome electroformation. Faraday Discuss. 1986;81:303–311. [Google Scholar]
- 10.Montes L.-R., Alonso A., Bagatolli L.A. Giant unilamellar vesicles electroformed from native membranes and organic lipid mixtures under physiological conditions. Biophys. J. 2007;93:3548–3554. doi: 10.1529/biophysj.107.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pott T., Bouvrais H., Méléard P. Giant unilamellar vesicle formation under physiologically relevant conditions. Chem. Phys. Lipids. 2008;154:115–119. doi: 10.1016/j.chemphyslip.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Estes D.J., Mayer M. Giant liposomes in physiological buffer using electroformation in a flow chamber. Biochim. Biophys. Acta. 2005;1712:152–160. doi: 10.1016/j.bbamem.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Buboltz J.T., Feigenson G.W. A novel strategy for the preparation of liposomes: rapid solvent exchange. Biochim. Biophys. Acta. 1999;1417:232–245. doi: 10.1016/s0005-2736(99)00006-1. [DOI] [PubMed] [Google Scholar]
- 14.Moscho A., Orwar O., Zare R.N. Rapid preparation of giant unilamellar vesicles. Proc. Natl. Acad. Sci. USA. 1996;93:11443–11447. doi: 10.1073/pnas.93.21.11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noireaux V., Maeda Y.T., Libchaber A. Development of an artificial cell, from self-organization to computation and self-reproduction. Proc. Natl. Acad. Sci. USA. 2011;108:3473–3480. doi: 10.1073/pnas.1017075108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takiguchi K., Yamada A., Yoshikawa K. Chapter 3 - Construction of cell-sized liposomes encapsulating actin and actin-cross-linking proteins. Methods Enzymol. 2009;464:31–53. doi: 10.1016/S0076-6879(09)64003-9. [DOI] [PubMed] [Google Scholar]
- 17.Shaklee P.M., Semrau S., Schmidt T. Protein incorporation in giant lipid vesicles under physiological conditions. ChemBioChem. 2010;11:175–179. doi: 10.1002/cbic.200900669. [DOI] [PubMed] [Google Scholar]
- 18.Merkle D., Kahya N., Schwille P. Reconstitution and anchoring of cytoskeleton inside giant unilamellar vesicles. ChemBioChem. 2008;9:2673–2681. doi: 10.1002/cbic.200800340. [DOI] [PubMed] [Google Scholar]
- 19.Osawa M., Anderson D.E., Erickson H.P. Reconstitution of contractile FtsZ rings in liposomes. Science. 2008;320:792–794. doi: 10.1126/science.1154520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walde P., Ichikawa S. Enzymes inside lipid vesicles: preparation, reactivity and applications. Biomol. Eng. 2001;18:143–177. doi: 10.1016/s1389-0344(01)00088-0. [DOI] [PubMed] [Google Scholar]
- 21.Sunami T., Sato K., Yomo T. Femtoliter compartment in liposomes for in vitro selection of proteins. Anal. Biochem. 2006;357:128–136. doi: 10.1016/j.ab.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 22.Fenz S.F., Sengupta K. Giant vesicles as cell models. Integr. Biol. (Camb) 2012;4:982–995. doi: 10.1039/c2ib00188h. [DOI] [PubMed] [Google Scholar]
- 23.Noireaux V., Libchaber A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. USA. 2004;101:17669–17674. doi: 10.1073/pnas.0408236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pautot S., Frisken B., Weitz D. Production of unilamellar vesicles using an inverted emulsion. Langmuir. 2003;19:2870–2879. [Google Scholar]
- 25.Abkarian M., Loiseau E., Massiera G. Continuous droplet interface crossing encapsulation (cDICE) for high throughput monodisperse vesicle design. Soft Matter. 2011;7:4610–4614. [Google Scholar]
- 26.Pontani L.-L., van der Gucht J., Sykes C. Reconstitution of an actin cortex inside a liposome. Biophys. J. 2009;96:192–198. doi: 10.1016/j.bpj.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shum H.C., Lee D., Weitz D.A. Double emulsion templated monodisperse phospholipid vesicles. Langmuir. 2008;24:7651–7653. doi: 10.1021/la801833a. [DOI] [PubMed] [Google Scholar]
- 28.Tsai F.-C., Stuhrmann B., Koenderink G.H. Encapsulation of active cytoskeletal protein networks in cell-sized liposomes. Langmuir. 2011;27:10061–10071. doi: 10.1021/la201604z. [DOI] [PubMed] [Google Scholar]
- 29.Stachowiak J.C., Richmond D.L., Fletcher D.A. Unilamellar vesicle formation and encapsulation by microfluidic jetting. Proc. Natl. Acad. Sci. USA. 2008;105:4697–4702. doi: 10.1073/pnas.0710875105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ota S., Yoshizawa S., Takeuchi S. Microfluidic formation of monodisperse, cell-sized, and unilamellar vesicles. Angew. Chem. Int. Ed. Engl. 2009;48:6533–6537. doi: 10.1002/anie.200902182. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y., Berry C.K., Raphael R.M. Peroxidation of polyunsaturated phosphatidyl-choline lipids during electroformation. Biomaterials. 2007;28:1298–1306. doi: 10.1016/j.biomaterials.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Horger K.S., Estes D.J., Mayer M. Films of agarose enable rapid formation of giant liposomes in solutions of physiologic ionic strength. J. Am. Chem. Soc. 2009;131:1810–1819. doi: 10.1021/ja805625u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hassan C., Peppas N. Structure and applications of poly (vinyl alcohol) hydrogels produced by conventional cross-linking or by freezing/thawing methods. J. Adv. Polymer. Sci. 2000;153:37–65. [Google Scholar]
- 34.Peppas N., Hilt J., Langer R. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Adv. Mater. 2006;18:1345–1360. [Google Scholar]
- 35.Pardee J.D., Spudich J.A. Purification of muscle actin. Methods Enzymol. 1982;85 doi: 10.1016/0076-6879(82)85020-9. Pt B:164–181. [DOI] [PubMed] [Google Scholar]
- 36.Gentry B.S., van der Meulen S., Koenderink G.H. Multiple actin binding domains of Ena/VASP proteins determine actin network stiffening. Eur. Biophys. J. 2012;41:979–990. doi: 10.1007/s00249-012-0861-1. [DOI] [PubMed] [Google Scholar]
- 37.Carlier M.F., Pantaloni D., Korn E.D. Polymerization of ADP-actin and ATP-actin under sonication and characteristics of the ATP-actin equilibrium polymer. J. Biol. Chem. 1985;260:6565–6571. [PubMed] [Google Scholar]
- 38.Dai W., Barbari T.A. Characterization of mesh size asymmetry in hydrogel membranes using confocal microscopy. J. Membr. Sci. 2000;171:45–58. [Google Scholar]
- 39.Wu L., Brazel C.S. Modifying the release of proxyphylline from PVA hydrogels using surface crosslinking. Int. J. Pharm. 2008;349:144–151. doi: 10.1016/j.ijpharm.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 40.Nardin C., Hirt T., Meier W. Polymerized ABA triblock copolymer vesicles. Langmuir. 2000;16:1035–1041. [Google Scholar]
- 41.Aitken C.E., Marshall R.A., Puglisi J.D. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mertins O., da Silveira N.P., Marques C.M. Electroformation of giant vesicles from an inverse phase precursor. Biophys. J. 2009;96:2719–2726. doi: 10.1016/j.bpj.2008.12.3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ayuyan A.G., Cohen F.S. Lipid peroxides promote large rafts: effects of excitation of probes in fluorescence microscopy and electrochemical reactions during vesicle formation. Biophys. J. 2006;91:2172–2183. doi: 10.1529/biophysj.106.087387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reference deleted in proof.
- 45.Yamashita Y., Oka M., Yamazaki M. A new method for the preparation of giant liposomes in high salt concentrations and growth of protein microcrystals in them. Biochim. Biophys. Acta. 2002;1561:129–134. doi: 10.1016/s0005-2736(02)00338-3. [DOI] [PubMed] [Google Scholar]
- 46.Tsumoto K., Matsuo H., Yoshimura T. Efficient formation of giant liposomes through the gentle hydration of phosphatidylcholine films doped with sugar. Colloids Surf. B Biointerfaces. 2009;68:98–105. doi: 10.1016/j.colsurfb.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 47.Hope M., Bally M., Cullis P. Generation of multilamellar and unilamellar phospholipid vesicles. Chem. Phys. Lipids. 1986;40:89–107. [Google Scholar]
- 48.Rodriguez N., Pincet F., Cribier S. Giant vesicles formed by gentle hydration and electroformation: a comparison by fluorescence microscopy. Colloids Surf. B Biointerfaces. 2005;42:125–130. doi: 10.1016/j.colsurfb.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 49.Mathai J.C., Tristram-Nagle S., Zeidel M.L. Structural determinants of water permeability through the lipid membrane. J. Gen. Physiol. 2008;131:69–76. doi: 10.1085/jgp.200709848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dominak L.M., Keating C.D. Polymer encapsulation within giant lipid vesicles. Langmuir. 2007;23:7148–7154. doi: 10.1021/la063687v. [DOI] [PubMed] [Google Scholar]
- 51.Dominak L.M., Keating C.D. Macromolecular crowding improves polymer encapsulation within giant lipid vesicles. Langmuir. 2008;24:13565–13571. doi: 10.1021/la8028403. [DOI] [PubMed] [Google Scholar]
- 52.Dominak L.M., Omiatek D.M., Keating C.D. Polymeric crowding agents improve passive biomacromolecule encapsulation in lipid vesicles. Langmuir. 2010;26:13195–13200. doi: 10.1021/la101903r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Discher B.M., Won Y.Y., Hammer D.A. Polymersomes: tough vesicles made from diblock copolymers. Science. 1999;284:1143–1146. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.