

Abstract
CLC-K chloride channels are expressed in the kidney and the inner ear, where they are involved in NaCl reabsorption and endolymph production, respectively. These channels require the beta subunit barttin for proper function. Mutations in ClC-Kb and barttin, lead to Bartter’s syndrome. Block of CLC-K channels by acid pH was described in a previous work, and we had identified His-497 as being responsible for the acidic block of CLC-K channels. Here, we show that ClC-K currents are blocked also by alkaline pH with an apparent pK value of ∼8.7 for ClC-K1. Using noise analysis, we demonstrate that alkaline block is mediated by an allosteric reduction of the open probability. By an extensive mutagenic screen we identified K165, a highly conserved residue in the extracellular vestibule of the channel, as the major element responsible for the alkaline pH modulation. Deprotonation of K165 underlies the alkaline block. However, MTS modification of the K165C mutant demonstrated that not only the charge but also the chemical and sterical properties of lysine 165 are determinants of CLC-K gating.
Introduction
CLC-K channels belong to the family of CLC proteins comprising both Cl− channels and Cl−/H+ antiporters. Members of this family are found in several tissues and organs where anion transport is important.
In humans, CLC-K channels comprise ClC-Ka and ClC-Kb (in rodents they are called ClC-K1 and ClC-K2). They were isolated from kidney by sequence homology with other CLC proteins (1,2). The human CLC-Ks have 90% identity to each other and 80% identity to rodent CLC-Ks (1). The combined action of the Na-K-ATPase, the apical Na+-K+-2Cl− cotransporter NKCC2, the apical K+ channel ROMK, and the basolateral chloride channel ClC-Kb/barttin, leads to NaCl reabsorption in the thick ascending limb (TAL) of Henle’s loop (3,4). CLC-Ks were identified also in the inner ear in the basolateral membrane of the marginal cells of the stria vascularis and in the dark cells of the vestibular organ (5). Both isoforms contribute to the maintenance of the high K+ concentration and the positive potential of the endolymph (5–7). Both in the kidney and in the inner ear, CLC-Ks coassemble with the β-subunit, barttin, which affects the trafficking and the function of these channels (5,8–10). Mutations in the NKCC2 transporter, the ROMK channel, or the ClC-Kb channel cause different forms of the same renal disease, Bartter’s syndrome, which demonstrates the common role of these proteins in NaCl reabsorption (11–13). Another form of Bartter’s syndrome that includes deafness arises from mutations of barttin or from simultaneous mutations of ClC-Ka and ClC-Kb (8,14).
The regulation of CLC-K channels by various extracellular ligands has been the subject of several previous publications (15–22). External Ca2+ and protons have been found to modulate these channels in the physiological concentration range (5,23–25). Currents are activated by extracellular Ca2+, whereas high [H+]ext (acidic pH) completely blocks CLC-Ks. Previously, a detailed biophysical analysis of the Ca2+ and proton effect on the human ClC-Ka showed that Ca2+ and protons affect the open probability of the channel via independent mechanisms and binding sites (25). An extensive mutagenic screen performed on ClC-Ka allowed us to identify four acidic residues, E259, E261, D278, and E281, that likely form an intersubunit Ca2+-binding site (25,26). Modulation by protons is a characteristic found in most CLC proteins, both in Cl−/H+ antiporters and in CLC channels. Since protons are one of the substrates of the antiporters, it is not surprising that the Cl−/H+ exchange is influenced by varying the H+ concentration (27–31). In most CLC channels, a highly conserved glutamate, the gating glutamate, has been found to be responsible for proton modulation (32–44). For example, in the ClC-0 channel, extracellular protons bind the gating glutamate and increase the open probability (36,38). Interestingly, intracellular acidic pH also activates ClC-0. Intracellular protons binding to the same glutamate lead to a shift of the voltage dependence of the open probability to more negative voltages (32,40–42). The effect of external protons on the gating is seen at acidic pH, whereas ClC-0 currents do not change from neutral to alkaline conditions (36). The gating of the ClC-1 channel, similar to that of ClC-0, is affected both by external and internal pH (33,45). Interestingly, the pH regulation of the ClC-2 channel is quite different from that of ClC-0. First, intracellular pH has only small effects on the open probability (44), but the channel shows a biphasic response to extracellular pH, with block both at alkalinization and at acidification (34,37,39,43,44). Activation and inhibition arise from the protonation of two different residues: the protonation of the gating glutamate activates the channel (39), whereas the protonation of an off-pore extracellular histidine inactivates the channel (43).
Similar to the case for ClC-2, CLC-K channels are also blocked by acidic pH (5,24,25,46), and in fact, they share the histidine residue (H497, corresponding to H532 of ClC-2), which was found to be responsible for H+-induced block of ClC-Ka (25). In contrast, CLC-K channels lack the gating glutamate, whose deprotonation underlies the inactivation of ClC-2 currents at alkaline pH. Here, we found that despite this, CLC-K channels are strongly blocked by alkaline pH via a reduction of the open probability. By an extensive mutagenic screen and cysteine modifications, we identified a pore lysine, K165, whose deprotonation likely mediates this block.
Materials and Methods
Molecular biology
Mutants of ClC-Ka and ClC-K1 were obtained by recombinant PCR as described previously (47). All new constructs were sequenced and coexpressed with the barttin mutant Y98A (5). The cRNA of CLC-K constructs was prepared using the AmpliCAP SP6 Message Maker kit (Biospa, Italy) after linearization with MluI, whereas the cRNA of Y98 barttin was transcribed by the mMessage mMachine T7 kit (Life Technologies, Italy) after linearization with NotI. All wild-type (WT) cDNA constructs were kindly provided by T. J. Jentsch.
Electrophysiology
The cRNA of CLC-K and barttin constructs was coinjected in defolliculated Xenopus oocytes, which were incubated at 18°C in the maintaining solution containing (in mM) 90 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2, and 10 Hepes (pH 7.5). One to five days after the injection, voltage-clamp measurements were performed at room temperature using the custom acquisition program GePulse (available at http://users.ge.ibf.cnr.it/pusch/programs-mik.htm) and a Turbo TEC-03X amplifier (npi electronics, Tamm, Germany). The standard bath solution contained (in mM) 112 NaCl, 10 Ca-Gluconate2, 1 MgSO4, and 10 HEPES (pH 7.3) (osmolarity, 227 mOsm). HEPES was replaced by MES buffer in solutions at pH <7, Bis-tris propane in solutions at pH 9, and CAPS in solutions at pH >9.
The membrane was kept at a holding potential corresponding to the resting membrane potential (∼−30 mV). To evaluate CLC-K currents at different voltages, a stimulation protocol was applied as follows (IV-pulse protocol). A prepulse to −100 mV for 100 ms was followed by voltages ranging from −140 to 80 mV with 20-mV increments for 200 ms. Pulses ended with a tail to 60 mV for 100 ms. The effect of the different pH values was monitored by applying 200-ms pulses to 60 mV once per second. The records were taken under continuous perfusion with the desired solution until steady state was reached. The stability of the currents was verified by applying the standard bath solution at the end of the experiment, while endogenous currents were estimated by using a solution containing (in mM) 100 NaI, 5 MgSO4, and 10 Hepes (pH 7.3) that inhibits only CLC-K channels (20). The effect of pH was quantified by the ratio of the current measured at a specific pH to that in standard bath solution. Leak currents (i.e., residual currents in iodide) were subtracted.
Modification of K165C ClC-K1 by methanethiosulfonate reagents
Methanethiosulfonate (MTS) reagents (Biotium, Hayward, CA) and dithiothreitol (DTT) were dissolved in the standard bath solution immediately before use and kept on ice during the experiments. For 2-hydroxyethyl methanethiosulfonate (MTSEH), a 1 M stock solution was prepared in dimethyl sulfoxide (DMSO) and stored at −80°C. A typical experiment involving MTS modification was performed in several stages. Before modification, ClC-K1 K165C currents were measured at various pH values (from 7.3 to 11). Then the oocyte was removed from the set-up and incubated in a solution containing 1 mM MTS reagent. Incubation time was 10 min for 2-aminoethyl methanethiosulfonate (MTSEA), 15 min for MTSEH, and 60 min for 2-(trimethylammonium) ethyl methanethiosulfonate (MTSET). Finally, the oocyte was placed back in the recording chamber and currents were measured at various pH values. For experiments with MTSEA and MTSEH, the recordings ended with the perfusion of 10 mM DTT, followed by a washout using the standard solution. For experiments with MTSET, which did not induce a change of currents, modification efficiency was verified by an additional treatment with 1 mM MTSEA.
The experiments to determine the reaction rate of K165C ClC-K1 with MTSEA or MTSEH were performed under continuous perfusion of 1 mM MTSEA or MTSEH. The current was monitored by repetitive 200-ms pulses to 60 mV once per second. The speed of perfusion was estimated by the speed of iodide block and found to be faster than ∼2 s, significantly faster than the speed of modification by MTS reagents at 1 mM.
MTS modification was quantified as the ratio of the current measured in standard bath solution after MTS treatment to that measured before incubation. Leak currents were subtracted.
Noise analysis
Patch-clamp measurements were performed in the inside-out configuration. The intracellular (bath) solution contained (in mM) 100 N-methyl-D-glucamine-Cl (NMDG-Cl), 2 MgCl2, 1 EGTA, and 10 HEPES (pH 7.3). An intracellular solution in which Cl− was replaced by glutamate was used to evaluate endogenous and leak currents. The extracellular solution contained (in mM) 92 tetraethylammonium chloride (TEA-Cl), 10 CaCl2, and 10 HEPES (pH 7.3) or, alternatively, 92 TEA-Cl, 10 CaCl2, and 10 CAPS (pH 10). Pipettes were pulled from borosilicate glass capillaries (Hilgenberg, Malsfeld, Germany) and had resistances of 1–2 MOhm in the recording solutions. To estimate the single-channel current by noise analysis, the following stimulation protocol was applied 50–100 times: a prepulse to 60 mV was followed by a pulse to −100 mV for 500 ms. Pulses ended with a tail at 60 mV for 200 ms. Currents were recorded at 50 kHz after filtering at 10 kHz with an eight-pole Bessel filter. Data analysis was performed at two potentials, −100 mV and +60 mV. The mean current, I, was first calculated. The variance, σ2, was estimated from the averaged squared difference of consecutive traces with the background variance at 0 mV subtracted. The variance-mean plot was assembled by binning as described previously (48). Finally the variance-mean plot was fitted by σ2 = iI – I2/N, with the single-channel current, i, and the number of channels, N, as free parameters (49).
Results and Discussion
Alkaline pH blocks CLC-K channels
In our previous work (25), we examined how high [H+]ext affects CLC-K channels, and found that the protonation of an extracellularly facing histidine, H497, is responsible for the block of CLC-K-mediated currents at acidic pH (25). Here, we investigated the effect of alkalinization on the currents of human ClC-Ka and ClC-Kb and of rodent ClC-K1, extending the pH range examined to include values from pH 5 to pH 11. Interestingly, alkaline pH blocks all CLC-K channels, with remaining currents at pH 11 being <∼6% of the currents recorded in control conditions (pH 7.3, 10 mM Ca2+) for ClC-Ka and ClC-K1 (Fig. 1 A), and with a strong block of ClC-Kb also (Fig. S1 A in the Supporting Material). The observation that ClC-Ka, ClC-Kb, and ClC-K1 show the highest current levels at different pH values (Fig. 1 A and Fig. S1 A) could be due to different degrees of block by acidic pH, alkaline pH, or both. The lines in Fig. 1 A are derived from a quantitative model discussed below.
Figure 1.
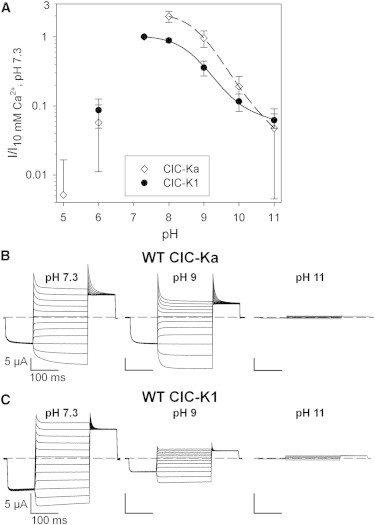
Effect of alkaline pH on WT ClC-Ka and WT ClC-K1. (A) Mean currents of ClC-Ka (n ≥ 11; solid circles) and ClC-K1 (open diamonds; n ≥ 7) at 60 mV recorded at pH values between 5 and 11 were normalized to the current measured in standard bath solution and plotted versus pH. The lines represent the best fit of WT ClC-Ka currents (dashed line; fit parameters, pK = 8.9, pL = 11.3, and R = 0.02) and WT ClC-K1 (solid line; fit parameters, pK = 8.7, pL = 11.8, and R = 0.07) obtained by Eq. 1, as described in Results. Error bars indicate the SD. (B and C) Typical current traces of WT ClC-Ka (B) and WT ClC-K1 (C) evoked by the IV-pulse protocol (see Materials and Methods) at pH 7.3, 9, and 11.
The drastic block by alkaline pH is further highlighted by the current traces evoked by the IV-pulse protocol (see Materials and Methods) at different pH values from oocytes expressing ClC-Ka (Fig. 1 B), ClC-K1 (Fig. 1 C), or ClC-Kb (Fig. S1 B). These data show that low [H+]ext reduces the current density without dramatically modifying the kinetics of the currents.
An important question is whether alkaline pH has a direct effect on the ion permeation or whether it acts indirectly by modulating the open probability. Because preliminary single-channel recordings performed on WT ClC-K1 at pH 7.3 had a very flickery behavior (data not shown), we estimated the single-channel conductance at neutral and alkaline pH using nonstationary noise analysis. This approach also allowed us to discriminate small changes of current by varying external pH. These inside-out patch-clamp experiments were performed on ClC-K1 because of its higher functional expression compared to human CLC-Ks. Using extracellular (pipette) solutions at pH 7.3 or pH 10, repetitive pulses to 60 mV were applied after a prepulse to −100 mV. Mean current and variance were evaluated (Fig. 2, A and B, left), and the single-channel current was estimated by the best fit of a parabola to the variance as a function of the mean current (Fig. 2, A and B, right), as described in Materials and Methods. The data cover only the initial linear part of the parabola due to the small open probability. Nevertheless, the fit provides a robust estimate of the single-channel current. Results are summarized in Fig. 2 C: in depolarizing conditions (+60 mV), the single-channel current at pH 10 is ∼60% of the current at pH 7.3; similarly, at −100 mV, the current is ∼70% of that at pH 7.3. This relatively small reduction of the conductance at pH 10 is not able to explain the reduction of macroscopic currents (at pH 10, ClC-K1 currents are ∼12% of those at pH 7.3). Thus, we conclude that alkaline pH acts mainly by an allosteric reduction of the open probability.
Figure 2.
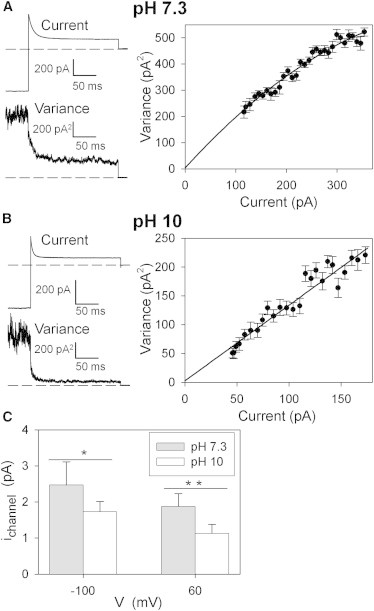
(A and B) Nonstationary noise analysis of WT ClC-K1 at pH 7.3 (A) and pH 10 (B). (Left) Mean current (upper) and variance (lower) are shown as a function of time. (Right) Variance (symbols) is plotted versus the mean current and fitted with a parabola (line), as described in Materials and Methods. (C) Bars represent the mean single-channel current at different pH values and potentials. ∗P < 0.05; ∗∗P < 0.005 (unpaired Student’s t-test). Error bars indicate the SD.
Scanning mutagenesis identifies a pore lysine as the most likely candidate to mediate the effect of alkaline pH
Since histidine 497, responsible for the acidic block of CLC-K currents (25), is not involved in the alkaline inhibition of these channels (data not shown), other residue(s) had to be implicated in this process. To identify these residues, we used the same strategy employed successfully in our previous work (25): based on the crystal structure of the homologous bacterial EcClC (38,50), we chose and mutated all titratable residues of ClC-Ka for which deprotonation at alkaline pH is plausible (C, H, K, R, and Y) and which are accessible from the extracellular side of the pore (Fig. 3 A). In general, the mutations inserted neutralized charged residues and/or replaced titratable with nontitratable residues. The 29 selected residues are colored in pink and light blue in the surface representation of the two subunits of a homology model of ClC-Ka (26) (Fig. 3). All constructs were coexpressed with barttin in Xenopus oocytes and tested for their response to alkaline pH. Alkaline pH inhibited all ClC-Ka mutants tested, but few of them showed a weakened sensitivity (Fig. 4, A–C). Our focus was on mutants in which a titratable residue was replaced by a nontitratable residue and whose response to alkaline pH was strongly modified. In case such mutants did not express, we also considered mutants in which a titratable residue was exchanged with another titratable amino acid and which exhibited a slight alteration of the pH dependence. Based on these criteria, the mutants chosen were K355A, Y520A, K268Q, and K165R. These mutants were examined in further detail, and when the data were not conclusive because of low current expression, the mutations were inserted in the background of ClC-K1, which shows larger expression than ClC-Ka. The pH sensitivity of K355A seems to be shifted, given that the currents recorded at pH 8 and 9 are fourfold larger than those at pH 7.3 (Fig. 4 B), which renders unlikely a direct involvement of K355 in alkaline block. Since also niflumic acid (at 200 μM) is more effective on K355A than on WT, with a twofold larger potentiation compared to WT (see Fig. 3 of Zifarelli et al. (21)), it seems that mutating K355 to alanine renders ClC-Ka more sensitive to potentiation factors, suggesting that the mutant somehow alters the gating properties of the channel. Y520A ClC-Ka (Figs. 4 C and S2) is only slightly affected at pH 10, but is blocked at pH 11. Y520 corresponds to Y512 of ClC-0 and Y445 of EcClC, a residue that is accessible to the intracellular solution and is involved in the coordination of pore chloride ions in the bacterial CLC homolog (47,50–52). In ClC-Ka and in ClC-K1 (data not shown), the mutant induces the appearance of very large currents that lack typical WT kinetics (Fig. S2 B). Thus, the relative insensitivity up to pH 10 likely reflects an indirect, unspecific effect of the mutant on gating. This phenotype, combined with the exposition of the residue to the intracellular (but not extracellular) solution, safely excludes deprotonation of Y520 as the process underlying the effects of alkaline external pH. The small currents of K268Q ClC-Ka made the effect of alkaline pH very difficult to evaluate properly (Fig. 4 B). However, in the context of ClC-K1, currents of mutant K268Q were similar to the WT in magnitude and pH sensitivity (Fig. S3, A and B), excluding this residue also as responsible for mediating the block at alkaline pH.
Figure 3.
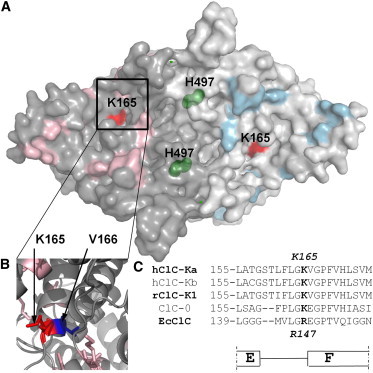
Mapping the mutants on the homology model of ClC-Ka. (A) Surface representation of the ClC-Ka model (26) based on the bacterial EcClC homodimer (Protein Data Bank accession no. 1KPK (50)) viewed from the extracellular side. The two subunits are colored gray and light gray. The residues of ClC-Ka selected for mutation are shown in pink and light blue in the two subunits. The two residues that are involved in CLC-K modulation by pH, H497 and K165, are colored in green and red, respectively. Several residues of ClC-Ka (K355, R351, H357, H365, and H386) are not included in the homology model, because they are contained in portions of the channel that are not present in EcClC. (B) Enlargement of the region containing K165 is shown in cartoon representation. K165 (R147 in EcClC) and the neighboring V166 (E148 in EcClC) are represented as sticks and colored in red and blue, respectively. (C) Alignment of sequences around K165.
Figure 4.
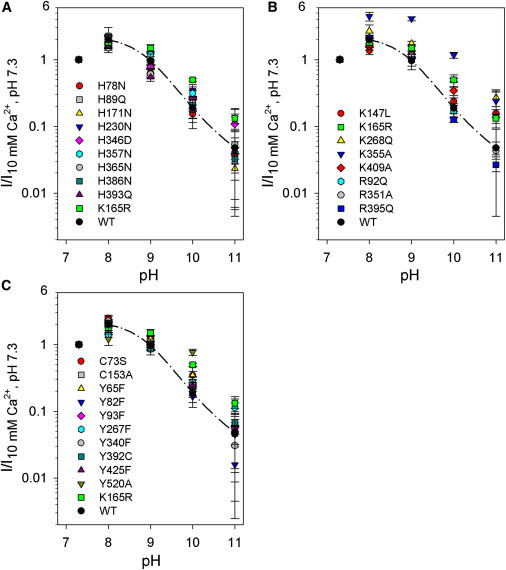
Effect of alkaline pH on all ClC-Ka mutants. To facilitate the data representation, the mutants were grouped by chemical properties of the mutated residues. The diagrams collect the data resulting from the mutations of histidine (A), lysine and arginine (B), and cysteine and tyrosine (C). The currents acquired at 60 mV were normalized to those measured at pH 7.3 (n ≥ 3). Mutants R184G and H480N did not yield functional expression. Data and fit for WT ClC-Ka are the same as in Fig. 1A. Data for K165R ClC-Ka are shown in all three groupings. Some variability in the response to alkaline pH is caused by different levels of functional expression of the mutants. Error bars indicate the SD.
K165, the only other residue from our experimental data (Fig. 4 B and Fig. S4) that emerged as possibly responsible for the channel’s sensitivity to alkaline pH, is highly conserved in CLC proteins (53). This lysine is located in the E-F loop in the extracellular pore vestibule of the channel, next to V166, the residue corresponding to the gating glutamate that is shared by all CLC proteins except CLC-K channels (Fig. 3, A–C). The effect of alkaline pH on the K165R mutant is weakened compared to the WT (Fig. S4, A and B). The slight impairment is caused by the conservative mutation of lysine to arginine, which does not prevent deprotonation at this site. A stronger effect could be expected by substituting a nontitratable residue for K165. Unfortunately, the other mutations inserted in this position of ClC-Ka (K165A, K165C, K165H, and K165Q) did not show functional expression.
The lysine of ClC-1 and ClC-0 that corresponds to K165 of ClC-Ka has been the subject of previous studies (54,55). Lin and Chen (55) found that the nonfunctional K165C/C212S ClC-0 channel could be activated by reaction with MTSEA, which confers a positive charge and changes the side chain of the cysteine to one that is lysinelike. The other MTS reagents tested, MTSET and 2-sulphonatoethyl MTS (MTSES), did not induce currents (55). Since ClC-0 is ∼39% identical to ClC-K channels (2), we hypothesized that the MTSEA reagent could also reactivate the dormant K165C ClC-Ka. Before testing this idea, we verified that MTSEA does not affect WT ClC-Ka currents by interacting with native cysteine residues (data not shown). When we applied MTSEA to K165C ClC-Ka-expressing oocytes, however, the currents elicited were too small to allow an easily quantitative investigation (data not shown). Also, as found for ClC-0, K165C ClC-Ka was not activated by the positively charged MTSET and the negatively charged MTSES (data not shown).
MTS-modified K165C ClC-K1 confirms the involvement of K165 in CLC-K modulation by alkaline pH
To obtain measurable currents from a nonconservative mutant of the K165 residue, we tried to insert the mutation K165C in the background of the rat WT ClC-K1 channel, which has higher functional expression than WT ClC-Ka. Fortunately, the mutant K165C ClC-K1 channel shows functional expression even without activation by MTSEA. The currents are much smaller than those of WT ClC-K1 (Fig. 1 C), and the typical current kinetics of the channel are almost absent at pH 7.3 and just perceptible at pH 8 and 9 (Fig. 5 A). It is important to note that K165C ClC-K1 shows a modified sensitivity to alkaline pH compared to WT. For WT ClC-K1, maximal currents are measured at pH 7.3, and at pH 9, for example, they are only ∼36% of those in control conditions (Fig. 5 B). Instead, for K165C, currents peak at pH 9, where they are more than twofold larger than those at pH 7.3. The current at pH 11 is comparable to that at pH 7.3 (Fig. 5, A and B), strongly suggesting that K165 is indeed involved in alkaline pH block (the lines in Fig. 5 B were derived from a quantitative model, discussed below). To investigate in more detail the pH sensitivity of K165C ClC-K1, we used the MTS reagents on this mutant. The MTS modification allowed us to obtain higher functional expression of the mutant and enabled us to study the effect of varying the charge and the titratability of the side chain at position 165. We used the positive, titratable MTSEA, the neutral MTSEH, and the positive, nontitratable MTSET. None of these reagents had an effect on WT ClC-K1 currents (data not shown).
Figure 5.
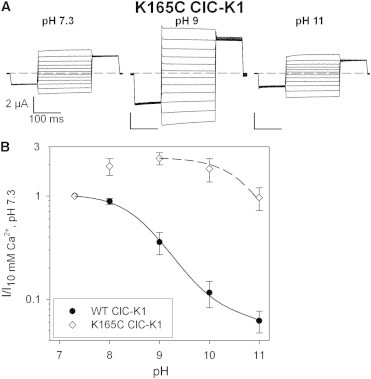
K165C ClC-K1 shows reduced sensitivity to alkaline pH. (A) Voltage-clamp traces in response to the IV-pulse protocol (see Materials and Methods) from the same oocyte under three different pH conditions. The small currents of K165C recorded before MTSEA activation were obtained by injecting ∼2.5 ng of cRNA, whereas only ∼0.1 ng of WT ClC-K1 cRNA was injected. (B) Effect of [H+]ext on WT ClC-K1 (solid circles; n = 10) and K165C ClC-K1 (open diamonds; n ≥ 23). Currents at 60 mV were normalized to the value recorded in standard bath solution (pH 7.3) and plotted versus pH. The dashed line represents the best fit of Eq. 2 to the K165C ClC-K1 currents (pL = 10.8). Data and fit for WT ClC-K1 are the same as in Fig. 1A. Error bars indicate the SD.
Because MTSEA and MTSEH modification cause an increase of the currents mediated by K165C ClC-K1, we could directly follow their modification of C165 (Fig. S5). MTSET was excluded from these measurements, because MTSET modification of K165C ClC-K1 was not accompanied by a change of the current level. From such experiments, in which the MTS reagent was continuously applied, we could estimate the time constant, τ, of modification (Fig. S5). From τ, we calculated the reaction rate, k, as k = τ−1/1 mM. For MTSEA, we obtained τ = (4.9 ± 1.0) s and k = (206 ± 40) s−1 M−1 and for MTSEH, τ = (8.9 ± 1.8) s and k = (112 ± 23) s−1 M−1 (all errors are expressed as the mean ± SD). These parameters provide a hint of the external accessibility of a residue. The reaction rate of K165C CLC-K1with MTSEA is smaller than that found by Lin and Chen (55) for the analogous mutation K165C in ClC-0 (4.6 × 103 s−1 M−1), indicating that this residue is less accessible from the extracellular side in ClC-K1 than in ClC-0.
MTSEA treatment increases currents approximately fivefold (Fig. 6, A and B). Moreover, the MTSEA-modified K165C ClC-K1 partially recovers the time-dependent relaxations that characterize WT ClC-K1 (Fig. 6 A, right). It is interesting that the pH sensitivity of the MTSEA-modified mutant is different from that of both the unmodified K165C and the WT (Fig. 6 C). Slight akalinization beyond pH 7.3 reduces currents to a relative level of ∼0.5 compared to the currents at pH 7.3, with a further reduction at pH 11. This phenotype indicates the presence of two distinct pH-dependent processes, which will be described in a quantitative model below. The strong qualitative effect of MTSEA modification on the pH dependence of K165C strengthens the conclusion that K165 is involved in at least one of these processes. MTSEA modification was reversible upon addition of 10 mM DTT: after perfusion with DTT, the current amplitude was restored to the phenotype before MTSEA treatment (Fig. 6 B). MTSEA converts C165 to a lysinelike residue with respect to charge and pK.
Figure 6.
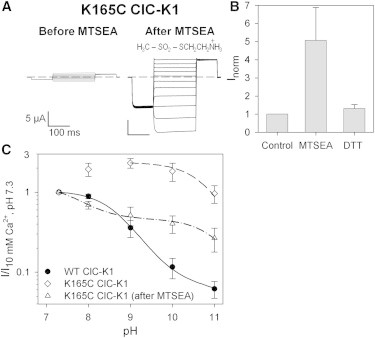
MTSEA-modified K165C ClC-K1 partially recovers the WT sensitivity to alkaline pH. (A) Current traces of K165C ClC-K1 evoked by the IV-pulse protocol (see Materials and Methods) from the same oocyte. The currents were recorded in the standard bath solution before (left) and after (right) incubation in 1 mM MTSEA. The chemical formula of MTSEA is shown. (B) Bars represent the normalized currents in standard bath solution under three different conditions: before MTSEA treatment (control), after incubation in 1 mM MTSEA (n = 6), and after a subsequent wash in 10 mM DTT (n = 3). Currents were normalized versus the current recorded at pH 7.3 before incubation in MTSEA. Error bars indicate the SD. (C) Mean currents at 60 mV from WT ClC-K1 (solid circles; n = 10), untreated K165C ClC-K1 (open diamonds; n ≥ 23), and K165C ClC-K1 after incubation in 1 mM MTSEA (open triangles; n ≥ 5) were normalized to the current at pH 7.3. The dot-dashed line represents the best fit of MTSEA-modified K165C currents obtained by Eq. 1, as described in Results. The fit parameters are pK = 7.6, pL =11.1, and R = 0.37. Data and fit for WT ClC-K1 are the same as in Figs. 1 A and 5B, those for K165C ClC-K1 are the same as in Fig. 5B. Error bars indicate the SD.
We next tested the neutral MTSEH. As for MTSEA, MTSEH modification increases current levels of K165C approximately threefold (Fig. 7, A and B). This indicates that the open probability is determined not only by the charge at position 165, but also by the general chemical and steric properties of the residue at this position. It is important to note that the sensitivity to alkaline pH is strongly reduced compared to that of WT ClC-K1 (Fig. 7 C), displaying a reduction of currents only at values beyond pH 10. The insensitivity of the MTSEH-modified K165C mutant at pH values ≤10 is again consistent with the idea that deprotonation of residue K165 underlies the major part of the alkaline pH dependence of CLC-K channels. Since MTSEH is not titratable, the reduction at pH 11 likely reflects an additional process of deprotonation, unrelated to K165. DTT only partially reversed the MTSEH modification (Fig. 7 B).
Figure 7.
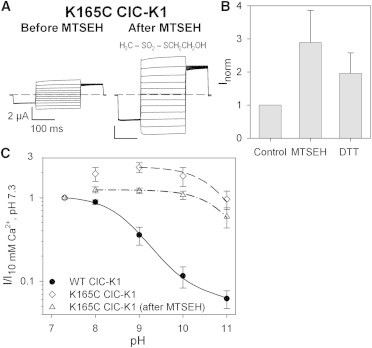
MTSEH-modified K165C ClC-K1 loses most of its sensitivity to alkaline pH. (A) Current responses to the IV-pulse protocol from an oocyte expressing K165C ClC-K1 before (left) and after (right) incubation in 1 mM MTSEH. The chemical formula of MTSEH is shown. (B) Mean values of currents before MTSEH treatment (Control), after incubation in 1 mM MTSEH (n = 16), and after a subsequent wash in 10 mM DTT (n = 10) were normalized to those recorded in standard bath solution before MTSEH perfusion. Error bars indicate the SD. (C) Mean currents measured at 60 mV at pH values ranging from 7.3 to 11 from WT ClC-K1 (solid circles; n = 10), untreated K165C ClC-K1 (open diamonds; n ≥ 23), and K165C ClC-K1 after incubation in 1 mM MTSEH (open triangles; n ≥ 15, except at pH 10, where n = 8) normalized to the current at pH 7.3. The dot-dashed line represents the best fit of Eq. 2 to the MTSEH-modified K165C currents with pL = 10.9. Data and fit for WT ClC-K1 are the same as in Figs. 1A, 5B, and 6C; those for K165C ClC-K1 are the same as in Figs. 5B and 6C. Error bars indicate the SD.
Finally, we examined the effect of the positive and nontitratable MTSET reagent. MTSET treatment did not induce an increase in the current amplitude, raising the possibility that it does not react. To test this, we applied MTSEA after incubation with MTSET. Our expectation was that if MTSET did not react with C165, MTSEA treatment would increase currents about fivefold. Instead, MTSEA increased the current only ∼1.6-fold, proving that most channels had reacted with MTSET (Fig. 8 A). The fact that modification by a positively charged reagent has, in the case of MTSET, no effect on the current level shows again that a charge at position 165 is not the only important parameter in determining open probability. The pH dependence of MTSET-modified K165C is very similar to that found after modification with MTSEH: currents are barely affected by pH values between 7.3 and 10, and at pH 11, MTSET-modified K165C currents are ∼59% of those at pH 7.3 (Fig. 8 B). However, keeping in mind that the MTSET-modified K165C has a low level of current, these data have to be considered cautiously. Nevertheless, we can conclude that without the titratability of K165, the channel loses most of its sensitivity to alkaline pH.
Figure 8.
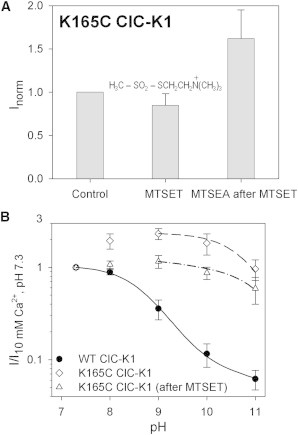
MTSET does not increase K165C current levels but strongly decreases sensitivity to alkaline pH. (A) Normalized current amplitudes in standard solution before modification by MTSET (Control), after incubation in 1 mM MTSET (n = 8), and after a subsequent treatment in 1 mM MTSEA (n = 3). Currents are normalized to those recorded in control conditions before treatment with MTS reagents. The chemical formula of MTSET is shown in the middle of the diagram. Error bars indicate the SD. (B) Voltage-clamp measurements at 60 mV of WT ClC-K1 (solid circles; n = 10), untreated K165C ClC-K1 (open diamonds; n ≥ 23), and K165C ClC-K1 after incubation in 1 mM MTSET (open triangles; n = 8) were normalized to the current in standard bath solution and plotted versus pH. The dot-dashed line represents the best fit of Eq. 2 to the MTSET-modified K165C currents with pL = 11.1. Data and fit for WT ClC-K1 are the same as in Figs. 1A, 5B, 6C, and 7C; those for K165C ClC-K1 are the same as in Figs. 5B, 6C, and 7C. Error bars indicate the SD.
Modeling alkaline pH modulation of CLC-K channels
The mutagenesis of residue K165 and the various MTS modifications of K165C clearly establish that deprotonation of K165 underlies the reduction in open probability at pH values ≤10. However, an additional pH-dependent process is revealed by the behavior of mutant K165C and by recordings of the mutant modified by nontitratable MTS reagents: at pH values >10, currents decrease in all circumstances.
To test these hypotheses in a quantitative manner, we invoke the simplest model incorporating these two pH-dependent processes: protonation/deprotonation of K165 (site K in Scheme 1), and protonation/deprotonation of an unknown site, U.
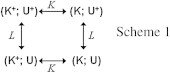
The model is composed of four states, as shown in Scheme 1. Deprotonation of site K and deprotonation of the unknown site are considered as independent processes governed by the dissociation constants K and L, respectively.
Protonated and deprotonated states of site K are associated with open probabilities and , respectively (states (K+; U+) and (K; U+)), whereas deprotonation of site U is hypothesized to shut the channel completely. The latter assumption is difficult to test, because it would require exploration of pH values >11. However, it is also of little relevance for the pH range considered here.
With these assumptions, for Scheme 1, the open probability is given by the equation
| (1) |
where is the extracellular proton concentration. Moreover, we apply the definitions , , and the ratio .
The lines in Fig. 1 A represent fits of Eq. 1 with the parameters reported in the legends. Eq. 1 describes well the sigmoidal pH dependence of WT ClC-K1 and ClC-Ka (Fig. 1 A), with pK = 8.7, pL = 11.8, and R = 0.07 for CLC-K1, i.e., deprotonation of K165 is predicted to reduce the open probability to a value of 7% compared to the protonated state. For the unmodified K165C and the mutant modified by MTSEH and MTSET, no biphasic pH dependence is visible and the data can be well described by a simple blocking deprotonation process:
| (2) |
(Figs. 5 B, 7 C, and 8 B), with pL ranging from 10.8 to 11.1, similar to the value found for WT ClC-K. The most interesting case regards the pH dependence of MTSEA-modified K165C. The biphasic pH dependence is very well described by Eq. 1, with pK = 7.6, pL = 11.1, and R = 0.37 (Fig. 6 C). Again pL is similar to the other cases, whereas pK is about one pH unit more acidic than for WT ClC-K1. Most notably, R is significantly larger than for WT, suggesting that deprotonation of the MTSEA moiety has only a relatively small effect on the open probability.
Conclusions
The kidney plays a pivotal role in pH regulation. In particular, the thick ascending limb of Henle’s loop, where ClC-Kb is expressed, effectively participates in the maintenance of the acid-base balance (56). Thus the pH dependence of CLC-K channels is probably of physiological relevance and represents an interesting topic of study. Here, we reveal that CLC-K channels are inhibited at alkaline pH, resulting in an overall biphasic pH dependence with a block of the currents both at extremely acidic and at alkaline pH values. A similar behavior was found in the ClC-2 channel (34,37,39,43,44). Inhibition by acidic pH results from the protonation of a homologous histidine in both channels (25,43). In contrast, a different mechanism underlies the inhibition of these channels by alkaline pH. ClC-2 is modulated via deprotonation of the gating glutamate (39,43,44). However, as this residue is a valine in CLC-K channels, other mechanisms must underlie the block of CLC-K currents at alkaline pH.
To determine whether low [H+]ext affects the ion permeation of the channel or acts on the open probability, we estimated the conductance of WT ClC-K1 at pH 7.3 and 10 by noise analysis. At pH 10, the single-channel conductance was only slightly smaller, showing that the alkaline block mainly depends on an allosteric decrease of the open probability. Even if the reduction of conductance is not responsible for alkaline block, it is significant. Thus, we might speculate that deprotonation of K165 could weakly affect Cl− permeation across the channel. However, it has to be kept in mind that the noise analysis is complicated by the fact that CLC proteins have two pores, each with a proper gate and an additional common gate. Therefore, a consistent apparent variability of the single-channel current (maximally by a factor of 2) can be caused indirectly by effects on the relative contributions of single/double pore openings to the noise, thus reflecting a gating rather than a conductance effect. Further investigation is required to resolve this question.
Using the homology model of ClC-Ka (26), we identified a pore lysine K165 that is involved in the modulation of the channel by alkaline pH. Lysine 165 is highly conserved in CLC proteins (50,53) and positioned next to valine 166, which corresponds to the conserved gating glutamate in CLC-Ks. Based on our extensive analysis of the pH dependence of point mutants of K165 and the cysteine-modified K165C variants in the background of ClC-K1, we can conclude that deprotonation of K165, with a pK of ∼8.7, leads to dramatic inhibition of CLC-K channels. It is interesting that the apparent pK of K165 is closer to the physiological pH range than that of free lysine (pK ∼10.5), suggesting that the ability of K165 to inactivate CLC-Ks in alkaline conditions could have a physiological relevance.
Furthermore, modification of K165C with nontitratable MTS reagents uncovered an additional pH-dependent inhibitory process occurring at more alkaline pH values (pH ≥11). In the absence of an investigation at pH values >11, we can only speculate about the causes of this further process. Plausible hypotheses are that this block could be due to deprotonation of an unknown residue or to a direct block of the pore by OH− ions. Finally, our results demonstrate that a positive charge at position 165 is not the only determinant of the open probability, but that the general chemical and sterical properties of the residue are equally important. In fact, modification of K165C by the uncharged and nontitratable MTSEH leads to an increase in currents similar to that observed with modification by MTSEA, whereas modification by the positive MTSET does not increase the current amplitude. Previous studies have demonstrated the involvement of K165 in mechanisms of gating of CLC proteins. The mutation to alanine of the homologous residue in ClC-1, K231, completely modifies the channel kinetics. In fact at hyperpolarizing potentials, K231A shows activating currents instead of the peculiar deactivating kinetics observed in the WT (54). The mutant K165R of ClC-0 exhibits inwardly rectifying currents, which demonstrates that the conservative mutation of lysine to arginine at position 165 is enough to modify deeply the gating of ClC-0 (57). The less conservative mutant, K165C, in ClC-0 lacks functional expression. However, it can be activated by MTSEA modification (55). Taking advantage of this, Lin and Chen (55) constructed homodimers and heterodimers of this mutant and applied MTSEA modification to these to understand how this residue is involved in the gating of ClC-0 (55). They concluded that the mutation of K165 affects both slow and fast gating of the channel (55). An interesting finding in their study was that single-channel recordings of unmodified K165C-K165 heterodimers in the background of the C212S mutant of ClC-0 (55) displayed openings of a single pore (presumably the K165 protopore). Since by definition the slow gate acts on both pores simultaneously, this observation by Chen and colleagues suggests that the drastic reduction of the open probability of the K165C pore reflects an effect on the fast gate, and that the slow gate is only indirectly affected. Matters are more complex for CLC-K channels, because in these channels, the gating glutamate is substituted by a valine (V166), and little is known about the mechanisms of CLC-K channel gating. In fact, the alkaline pH effect might provide a tool to gain more insight into these mechanisms. Nevertheless, this residue is important for the gating of both CLC channels bearing the gating glutamate (ClC-0) and those lacking it (CLC-Ks). It will be interesting to find out the details of the gating processes influenced by K165 in CLC-K channels. Finally, another study (58) highlighted that the homologous lysine in ClC-5 (K210) can be substituted by many different residues without gross impairment of function, suggesting, however, that K210 plays a role in the determination of the anionic specificity of this transporter and stressing that this residue plays different roles in different CLC proteins.
Our results also indicate that deprotonation of K165 in WT ClC-K1 does not completely shut the channel. From our quantitative model, we obtained a residual open probability of 7% of the value for the protonated lysine. The model also nicely described the pH dependence of the MTSEA-modified K165C mutant, for which the open probability of the protonated and deprotonated forms is predicted to differ by less than a factor of 3.
In summary, we identify here the residue responsible for alkaline block close to the physiological pH range. In addition, we reveal that this residue is involved in the gating of CLC-K channels. This information is quite surprising: CLC-Ks lack the gating glutamate, but the same region of the protein determines their gating machinery.
Acknowledgments
This work was supported by Telethon Italy (grant GGP12008), the Italian Ministry of Education (progetto PRIN), and the Compagnia San Paolo.
We thank T. J. Jentsch for all wild-type clones and F. Quartino, A. Barbin, and D. Magliozzi for technical assistance.
Supporting Material
References
- 1.Kieferle S., Fong P., Jentsch T.J. Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc. Natl. Acad. Sci. USA. 1994;91:6943–6947. doi: 10.1073/pnas.91.15.6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uchida S., Sasaki S., Marumo F. Molecular cloning of a chloride channel that is regulated by dehydration and expressed predominantly in kidney medulla. J. Biol. Chem. 1993;268:3821–3824. [PubMed] [Google Scholar]
- 3.Jentsch T.J., Maritzen T., Zdebik A.A. Chloride channel diseases resulting from impaired transepithelial transport or vesicular function. J. Clin. Invest. 2005;115:2039–2046. doi: 10.1172/JCI25470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jentsch T.J., Neagoe I., Scheel O. CLC chloride channels and transporters. Curr. Opin. Neurobiol. 2005;15:319–325. doi: 10.1016/j.conb.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Estévez R., Boettger T., Jentsch T.J. Barttin is a Cl− channel β-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature. 2001;414:558–561. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- 6.Rickheit G., Maier H., Jentsch T.J. Endocochlear potential depends on Cl− channels: mechanism underlying deafness in Bartter syndrome IV. EMBO J. 2008;27:2907–2917. doi: 10.1038/emboj.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zdebik A.A., Wangemann P., Jentsch T.J. Potassium ion movement in the inner ear: insights from genetic disease and mouse models. Physiology (Bethesda) 2009;24:307–316. doi: 10.1152/physiol.00018.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birkenhäger R., Otto E., Hildebrandt F. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 2001;29:310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- 9.Fischer M., Janssen A.G., Fahlke C. Barttin activates ClC-K channel function by modulating gating. J. Am. Soc. Nephrol. 2010;21:1281–1289. doi: 10.1681/ASN.2009121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scholl U., Hebeisen S., Fahlke C. Barttin modulates trafficking and function of ClC-K channels. Proc. Natl. Acad. Sci. USA. 2006;103:11411–11416. doi: 10.1073/pnas.0601631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon D.B., Bindra R.S., Lifton R.P. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 12.Simon D.B., Karet F.E., Lifton R.P. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat. Genet. 1996;13:183–188. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 13.Simon D.B., Karet F.E., Lifton R.P. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 14.Schlingmann K.P., Konrad M., Waldegger S. Salt wasting and deafness resulting from mutations in two chloride channels. N. Engl. J. Med. 2004;350:1314–1319. doi: 10.1056/NEJMoa032843. [DOI] [PubMed] [Google Scholar]
- 15.Liantonio A., Accardi A., Pusch M. Molecular requisites for drug binding to muscle CLC-1 and renal CLC-K channel revealed by the use of phenoxy-alkyl derivatives of 2-(p-chlorophenoxy)propionic acid. Mol. Pharmacol. 2002;62:265–271. doi: 10.1124/mol.62.2.265. [DOI] [PubMed] [Google Scholar]
- 16.Liantonio A., Picollo A., Camerino D.C. Activation and inhibition of kidney CLC-K chloride channels by fenamates. Mol. Pharmacol. 2006;69:165–173. doi: 10.1124/mol.105.017384. [DOI] [PubMed] [Google Scholar]
- 17.Liantonio A., Picollo A., Camerino D.C. Molecular switch for CLC-K Cl− channel block/activation: optimal pharmacophoric requirements towards high-affinity ligands. Proc. Natl. Acad. Sci. USA. 2008;105:1369–1373. doi: 10.1073/pnas.0708977105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liantonio A., Pusch M., Conte Camerino D. Investigations of pharmacologic properties of the renal CLC-K1 chloride channel co-expressed with barttin by the use of 2-(p-chlorophenoxy)propionic acid derivatives and other structurally unrelated chloride channel blockers. J. Am. Soc. Nephrol. 2004;15:13–20. doi: 10.1097/01.asn.0000103226.28798.ea. [DOI] [PubMed] [Google Scholar]
- 19.Picollo A., Liantonio A., Pusch M. Mechanism of interaction of niflumic acid with heterologously expressed kidney CLC-K chloride channels. J. Membr. Biol. 2007;216:73–82. doi: 10.1007/s00232-007-9034-z. [DOI] [PubMed] [Google Scholar]
- 20.Picollo A., Liantonio A., Pusch M. Molecular determinants of differential pore blocking of kidney CLC-K chloride channels. EMBO Rep. 2004;5:584–589. doi: 10.1038/sj.embor.7400169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zifarelli G., Liantonio A., Pusch M. Identification of sites responsible for the potentiating effect of niflumic acid on ClC-Ka kidney chloride channels. Br. J. Pharmacol. 2010;160:1652–1661. doi: 10.1111/j.1476-5381.2010.00822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gradogna A., Pusch M. Molecular pharmacology of kidney and inner ear CLC-K chloride channels. Front. Pharmacol. 2010;1:130. doi: 10.3389/fphar.2010.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uchida S., Sasaki S., Marumo F. Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J. Clin. Invest. 1995;95:104–113. doi: 10.1172/JCI117626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waldegger S., Jeck N., Seyberth H.W. Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Arch. 2002;444:411–418. doi: 10.1007/s00424-002-0819-8. [DOI] [PubMed] [Google Scholar]
- 25.Gradogna A., Babini E., Pusch M. A regulatory calcium-binding site at the subunit interface of CLC-K kidney chloride channels. J. Gen. Physiol. 2010;136:311–323. doi: 10.1085/jgp.201010455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gradogna A., Fenollar-Ferrer C., Pusch M. Dissecting a regulatory calcium-binding site of CLC-K kidney chloride channels. J. Gen. Physiol. 2012;140:681–696. doi: 10.1085/jgp.201210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iyer R., Iverson T.M., Miller C. A biological role for prokaryotic ClC chloride channels. Nature. 2002;419:715–718. doi: 10.1038/nature01000. [DOI] [PubMed] [Google Scholar]
- 28.Accardi A., Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- 29.Friedrich T., Breiderhoff T., Jentsch T.J. Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J. Biol. Chem. 1999;274:896–902. doi: 10.1074/jbc.274.2.896. [DOI] [PubMed] [Google Scholar]
- 30.Picollo A., Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- 31.Zifarelli G., Pusch M. Conversion of the 2 Cl−/1 H+ antiporter ClC-5 in a NO3−/H+ antiporter by a single point mutation. EMBO J. 2009;28:175–182. doi: 10.1038/emboj.2008.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanke W., Miller C. Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 1983;82:25–45. doi: 10.1085/jgp.82.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rychkov G.Y., Pusch M., Bretag A.H. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J. Physiol. 1996;497:423–435. doi: 10.1113/jphysiol.1996.sp021778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jordt S.E., Jentsch T.J. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 1997;16:1582–1592. doi: 10.1093/emboj/16.7.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saviane C., Conti F., Pusch M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 1999;113:457–468. doi: 10.1085/jgp.113.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen M.F., Chen T.Y. Different fast-gate regulation by external Cl− and H+ of the muscle-type ClC chloride channels. J. Gen. Physiol. 2001;118:23–32. doi: 10.1085/jgp.118.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arreola J., Begenisich T., Melvin J.E. Conformation-dependent regulation of inward rectifier chloride channel gating by extracellular protons. J. Physiol. 2002;541:103–112. doi: 10.1113/jphysiol.2002.016485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dutzler R., Campbell E.B., MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 39.Niemeyer M.I., Cid L.P., Sepúlveda F.V. A conserved pore-lining glutamate as a voltage- and chloride-dependent gate in the ClC-2 chloride channel. J. Physiol. 2003;553:873–879. doi: 10.1113/jphysiol.2003.055988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Traverso S., Zifarelli G., Pusch M. Proton sensing of CLC-0 mutant E166D. J. Gen. Physiol. 2006;127:51–65. doi: 10.1085/jgp.200509340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zifarelli G., Murgia A.R., Pusch M. Intracellular proton regulation of ClC-0. J. Gen. Physiol. 2008;132:185–198. doi: 10.1085/jgp.200809999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zifarelli G., Pusch M. The role of protons in fast and slow gating of the Torpedo chloride channel ClC-0. Eur. Biophys. J. 2010;39:869–875. doi: 10.1007/s00249-008-0393-x. [DOI] [PubMed] [Google Scholar]
- 43.Niemeyer M.I., Cid L.P., Sepúlveda F.V. Voltage-dependent and -independent titration of specific residues accounts for complex gating of a ClC chloride channel by extracellular protons. J. Physiol. 2009;587:1387–1400. doi: 10.1113/jphysiol.2008.167353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sánchez-Rodríguez J.E., De Santiago-Castillo J.A., Arreola J. Sequential interaction of chloride and proton ions with the fast gate steer the voltage-dependent gating in ClC-2 chloride channels. J. Physiol. 2012;590:4239–4253. doi: 10.1113/jphysiol.2012.232660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennetts B., Parker M.W., Cromer B.A. Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. J. Biol. Chem. 2007;282:32780–32791. doi: 10.1074/jbc.M703259200. [DOI] [PubMed] [Google Scholar]
- 46.Waldegger S., Jentsch T.J. Functional and structural analysis of ClC-K chloride channels involved in renal disease. J. Biol. Chem. 2000;275:24527–24533. doi: 10.1074/jbc.M001987200. [DOI] [PubMed] [Google Scholar]
- 47.Accardi A., Pusch M. Conformational changes in the pore of CLC-0. J. Gen. Physiol. 2003;122:277–293. doi: 10.1085/jgp.200308834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinemann S.H., Conti F. Nonstationary noise analysis and application to patch-clamp recordings. Methods Enzymol. 1992;207:131–148. doi: 10.1016/0076-6879(92)07009-d. [DOI] [PubMed] [Google Scholar]
- 49.Pusch M., Steinmeyer K., Jentsch T.J. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys. J. 1994;66:149–152. doi: 10.1016/S0006-3495(94)80753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dutzler R., Campbell E.B., MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 51.Lin C.W., Chen T.Y. Probing the pore of ClC-0 by substituted cysteine accessibility method using methane thiosulfonate reagents. J. Gen. Physiol. 2003;122:147–159. doi: 10.1085/jgp.200308845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Accardi A., Lobet S., Dutzler R. Synergism between halide binding and proton transport in a CLC-type exchanger. J. Mol. Biol. 2006;362:691–699. doi: 10.1016/j.jmb.2006.07.081. [DOI] [PubMed] [Google Scholar]
- 53.Mindell J.A., Maduke M. ClC chloride channels. Genome. Biol. 2001;2:S3003. doi: 10.1186/gb-2001-2-2-reviews3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fahlke C., Yu H.T., George A.L., Jr. Pore-forming segments in voltage-gated chloride channels. Nature. 1997;390:529–532. doi: 10.1038/37391. [DOI] [PubMed] [Google Scholar]
- 55.Lin C.W., Chen T.Y. Cysteine modification of a putative pore residue in ClC-0: implication for the pore stoichiometry of ClC chloride channels. J. Gen. Physiol. 2000;116:535–546. doi: 10.1085/jgp.116.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koeppen B.M. The kidney and acid-base regulation. Adv. Physiol. Educ. 2009;33:275–281. doi: 10.1152/advan.00054.2009. [DOI] [PubMed] [Google Scholar]
- 57.Ludewig U., Jentsch T.J., Pusch M. Inward rectification in ClC-0 chloride channels caused by mutations in several protein regions. J. Gen. Physiol. 1997;110:165–171. doi: 10.1085/jgp.110.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Stefano S., Pusch M., Zifarelli G. Extracellular determinants of anion discrimination of the Cl−/H+ antiporter protein CLC-5. J. Biol. Chem. 2011;286:44134–44144. doi: 10.1074/jbc.M111.272815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.