

Abstract
Isolated receptor tyrosine kinase transmembrane (TM) domains have been shown to form sequence-specific dimers in membranes. Yet, it is not clear whether studies of isolated TM domains yield knowledge that is relevant to full-length receptors or whether the large glycosylated extracellular domains alter the interactions between the TM helices. Here, we address this question by quantifying the effect of the pathogenic A391E TM domain mutation on the stability of the fibroblast growth factor receptor 3 dimer in the presence of the extracellular domain and comparing these results to the case of the isolated TM fibroblast growth factor receptor 3 domains. We perform the measurements in plasma membrane-derived vesicles using a Förster-resonance-energy-transfer-based method. The effect of the mutation on dimer stability in both cases is the same (∼−1.5 kcal/mol), suggesting that the interactions observed in simple TM-peptide model systems are relevant in a biological context.
Introduction
Receptor tyrosine kinases (RTKs) are membrane proteins that are critical for cell growth, differentiation, and survival (1). They are single-pass receptors with extracellular (EC) ligand-binding domains and intracellular domains with kinase activity. Over the last decade, structures of isolated soluble EC and kinase domains have been solved using x-ray crystallography (2–7), and structures of isolated transmembrane (TM)-domain dimers have been determined by NMR (8–11). These structures have greatly enhanced our understanding of RTK function. However, the details behind the signal transduction across the plasma membrane are still unknown, partly due to the lack of full-length RTK structures.
Recent work has shown that the isolated RTK TM domains have a propensity to form dimers in membranes (12–14), suggesting that RTK TM domains play an important active role in RTK signaling. Yet sequence alterations in the TM domains sometimes do not alter RTK signaling (15). Questions therefore arise as to whether the behavior of the isolated TM domains is the same as their behavior within the full-length receptors. In particular, the large glycosylated EC domains may affect the structure of the TM-domain dimer or may prevent the close approach of the two TM helices, despite the fact that their sequences encode for sequence-specific dimerization. Thus, currently it is not known whether studies of isolated TM domains yield knowledge that is relevant to full-length receptors.
Of all RTKs, fibroblast growth factor receptor 3 (FGFR3) is known as the receptor with the largest number of pathogenic mutations in its TM domain (13,16). FGFR3 is a negative regulator of bone growth and is critically important for skeletal development (17). FGFR3 TM-domain mutations are gain-of-function mutations, known to overinhibit bone growth and cause skeletal dysplasias or craniosynostoses (18). In addition, the mutations linked to skeletal disorders have also been identified as somatic mutations in epithelial cancers (19). The very fact that mutations in FGFR3 TM domains cause diseases strongly suggests that FGFR3 TM domain is not a passive membrane anchor. Furthermore, Förster-resonance-energy-transfer (FRET)-based measurements have shown that the isolated FGFR3 TM domains form sequence-specific dimers in lipid bilayers (20). Dimer stabilities of ∼−2.8 kcal/mol were measured in palmitoyl oleoyl phosphatidylcholine (POPC), POPC/palmitoyl oleoyl phosphatidylserine, and POPC/Cholesterol bilayers (21,22).
One of the pathogenic mutations in FGFR3, A391E, is known as the genetic cause for Crouzon syndrome with acanthosis nigricans, characterized by premature ossification of the skull and skin abnormalities (23). The stability of the mutant TM-domain dimer was measured as −4.1 kcal/mol in lipid bilayers, and thus this mutation was shown to increase the dimerization of the isolated FGFR3 TM domain by −1.3 kcal/mol (21). Based on this work, it was hypothesized that this mutation stabilizes the receptor dimeric state in the absence of ligand. The mutant full-length FGFR3 was shown to exhibit higher phosphorylation than the wild-type in live human embryonic kidney (HEK) 293T cells, consistent with the hypothesis (24,25). Despite all this work, however, there are no direct demonstrations that the EC domain does not affect the behavior of the TM domain within the full-length receptor dimer or that the A391E mutation stabilizes the full-length receptor dimer and the TM-domain dimer in the same way.
One way to assess whether the presence of the EC domain alters the nature of the interactions between the TM domains is to compare the magnitude of the effect of the mutation on dimer stability within the contexts of the FGFR3 receptor and the isolated FGFR3 TM domains. Quantitative measurements of dimer stabilities for complex glycosylated proteins are now feasible, without the need for their extraction, purification, and reconstitution (26,27). Such measurements utilize a FRET-based method and are carried out in plasma membrane-derived vesicles that bud off cells as a result of treatments that destroy the actin cytoskeleton (28–31). Here, we use this method to quantify the effect of the A391E mutation on the stability of FGFR3 dimers in the presence of its large glycosylated EC domain, such that this effect can be directly compared to results for the isolated TM-domain dimer in lipid bilayers.
Experimental Methods
Plasmid constructs
The enhanced yellow fluorescent protein (EYFP) plasmid was received from Dr. M. Betenbaugh (Johns Hopkins University, Baltimore, MD) and pRSET-mCherry was obtained from Dr. R. Tsien (University of California, San Diego, CA). The plasmid encoding human wild-type FGFR3 in the pcDNA 3.1(+) vector was a gift from Dr. D. J. Donoghue (University of California, San Diego, CA). All of the plasmids used for mammalian expression were constructed with the pcDNA 3.1(+) vector (Invitrogen, Carlsbad, CA). All primers were purchased from Invitrogen.
The receptors used for these experiments were truncated FGFR3 receptors, with the intracellular domains substituted with fluorescent proteins such that FRET can be used for detection of dimerization. In particular, the wild-type plasmid encoded the 22-amino-acid signal peptide of FGFR3 (MGAPACALALCVAVAIVAGASS), the EC and TM domains of FGFR3, a (GGS)5 flexible linker, and a fluorescent protein, either EYFP or mCherry (a FRET pair) (see Fig. 1). Details about the construction of this plasmid are given in Chen et al. (26). The A391E mutation was engineered in the wild-type plasmid for this work using a QuickChange mutagenesis kit (Roche, Madison, WI).
Figure 1.

The receptor constructs used in the experiments. The wild-type FGFR3 construct consisted of 1), the signal peptide (SP), the extracellular domain (EC), the transmembrane domain (TM), a (GGS)5 unstructured linker, and a fluorescent protein, either EYFP or mCherry (a FRET pair). The A391E mutation in the TM domain is shown in red.
Cell culture and transfection
HEK293T cells were a kind gift from Dr. Denis Wirtz (Johns Hopkins University). HEK293T cells were chosen for this study because the activities of wild-type and mutant FGFR3 have been studied previously in this cell line (24,25,32). The cells were cultured at 37°C with 5% CO2 for 24 h, and 3 × 105 cells were seeded in each well of a six-well plate. Transfection was carried out using Fugene HD transfection reagent (Roche Applied Science, Basel, Switzerland) according to the manufacturer’s protocol. Cells in each well were cotransfected with 3 μg DNA encoding either 1), the wild-type FGFR3 construct tagged with EYFP or mCherry, or 2), the mutant FGFR3 construct tagged with EYFP or mCherry. No expression of FGFR3 in HEK 293T cells was detected via immunostaining or Western blots unless the cells were transfected (33–35), demonstrating that HEK 293T cells do not express endogenous FGFR3.
Production of mammalian plasma membrane vesicles
Here, we used the vesiculation buffer developed by Scott (28), as this is the only buffer that vesiculates HEK293T cells before their detachment from the substrate. As discussed previously, the original vesiculation procedure was modified by the addition of glycine to quench the formaldehyde in the buffer after vesiculation is triggered (36). We have shown that the interactions of wild-type FGFR3 and GpA in plasma membrane-derived vesicles produced by this method are not affected by the formaldehyde presence (37,38). In particular, we demonstrated that the measured FRET efficiencies and dimeric fractions are the same in Chinese hamster ovary (CHO) cells and A431 vesicles produced with this method and with an osmotic vesiculation buffer we recently developed that contains salts but no active chemicals (37,38).
Vesiculation was performed as described previously (28,30). HEK293T cells were rinsed once with phosphate-buffered saline (PBS; pH 7.4) containing 0.75 mM calcium chloride and 0.5 mM magnesium chloride (CM-PBS), and incubated with 1 mL of vesiculation buffer for 2 h at 37°C. The vesiculation buffer consisted of CM-PBS with 25 mM formaldehyde, 0.5 mM 1,4-dithiotreitol (DTT), and a protease inhibitor cocktail (complete mini EDTA-free tabs, Roche Applied Science). To quench the formaldehyde after vesiculation, glycine solution in PBS was added to the vesiculation buffer to a final concentration of 0.125 M. A large number of vesicles were produced after 1.5 h, and the vesicles were transferred into four-well Nunc (Penfield, NY) Lab-Tek II chambered coverslips for imaging.
Fluorescence Image Acquisition
Vesicles were imaged using a Nikon (Melville, NY) Eclipse confocal laser scanning microscope using a 60× water-immersion objective. All the images were collected and stored at 512 × 512 resolution. Three different scans were performed for each vesicle: 1), excitation at 488 nm, with a 500–530-nm emission filter (donor scan); 2), excitation at 488 nm, with a 565–615-nm emission filter (FRET scan); and 3), excitation at 543 nm, with a 650-nm longpass filter (acceptor scan). Gains of 8.0 and pixel dwell time of 1.68 μs were used for the three scans. To minimize the bleaching of fluorescent proteins, ND8 filters were used during excitation with the 488-nm laser.
Results
To measure the dimerization propensities of wild-type FGFR3 and the A391 mutant, we cotransfected HEK293T cells with 3 μg DNA encoding either 1) the wild-type FGFR3 construct tagged with EYFP or mCherry, or 2), the mutant FGFR3 construct tagged with EYFP or mCherry (see Fig. 1). The dimerization of the wild-type construct has been characterized previously in a different cell line, CHO cells (26). Here, five independent experiments were performed in a human cell line, HEK293T cells, and in each experiment, the wild-type and the mutant were studied in parallel.
After transfection, vesicles were produced by incubating HEK293T cells with the vesiculation buffer, as described in Materials and Methods and in previous publications (see Fig. 2). Each vesicle was imaged in three different scans, the donor, FRET, and acceptor scans. The donor scan served to excite EYFP at 488 nm and to collect EYFP emission in the range 500–530 nm. The acceptor scan was used to excite the acceptor at 543 nm, and its emission spectrum was collected in the range >650 nm. For the FRET scan, EYFP was excited and the emission of mCherry was collected in the range 565–615 nm. The imaged vesicles exhibited uniform fluorescence intensities, which allowed us to determine the concentrations of the fluorescent proteins in the membrane using solutions of purified EYFP and mCherry of known concentration (26,31). The fluorescent protein solutions were prepared as described previously (31). The solutions were imaged in the microscope using the same settings as above, to allow direct comparison of solution and vesicle intensities.
Figure 2.
(A) A vesicle budding off from a HEK293T cell. (B) A vesicle co-expressing EYFP and mCherry-tagged FGFR3. Vesicles in the size range 10–40 μm in diameter were imaged and analyzed as described in Materials and Methods.
Each vesicle was analyzed using a Matlab program to determine the fluorescence intensity across the membrane (26,31), which was fitted to a Gaussian function and the background intensity was approximated as an error function. The donor, acceptor, and FRET intensities for each vesicle were used to determine 1), the donor concentration, 2), the acceptor concentration, and 3), the FRET efficiency in each vesicle, as described in detail elsewhere (26,31). Briefly, for each vesicle, the concentration of the acceptor-labeled proteins, CA, and the concentration of the donor-labeled proteins, CD, per unit area were obtained using the equations
| (1) |
| (2) |
The parameters iD and iA in Eqs. 1 and 2 are given by the slopes of the fluorescence-intensity-versus-concentration linear calibration curves for the soluble donor and acceptor, respectively. The parameters βD and βA are the donor and acceptor bleed-through coefficients, obtained by imaging the purified EYFP and mCherry solutions as described elsewhere (31). The intensities ID, IFRET, and IA are the intensities measured for each vesicle in the donor, FRET, and acceptor channels, as described above. The gauge factor, GF, was obtained by analyzing vesicles loaded with a soluble linked EYFP-mCherry construct (31). As discussed previously (31), the gauge factor relates the sensitized emission of the acceptor to the quenching of the donor.
The FRET efficiency for each vesicle is calculated as
| (3) |
The raw FRET efficiencies are plotted as a function of acceptor concentration in Fig. 3. About 500 vesicles were imaged for each construct in five independent experiments. Each data point in Fig. 3 corresponds to a single vesicle. Each independent experiment yielded ∼100 data points for the wild-type and 100 data points for the mutant. The results from the independent experiments were indistinguishable (p > 0.1).
Figure 3.
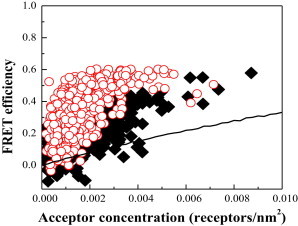
FRET data for wild-type FGFR3 and the A391 mutant constructs as a function of acceptor concentration. Each of the ∼1000 data points represents a single vesicle for which the FRET efficiency, donor concentration, and acceptor concentration are determined using the quantitative-imaging FRET method. Black solid squares and red open circles indicate the FRET efficiencies measured for the wild-type and mutant constructs, respectively. Data scatter in this type of experiment is due to random noise in image acquisition, and is reducible by collecting a large number of data points (36). The solid line shows the so-called called random FRET, which occurs if there are no specific interactions between the membrane proteins; this phenomenon is well described by the model of Wolber and Hudson (40).
In Fig. 3, the FRET efficiencies for the mutant are higher than the FRET efficiencies measured for the wild-type. To calculate and compare the dimerization propensities, however, we also need to take into account the donor concentration for each point. Furthermore, we need to take into account that unlike soluble proteins, membrane proteins are confined to two-dimensional membranes. Thus, the fluorescent proteins can come into close proximity to each other (within 100 Å) and FRET can occur even in the absence of specific protein-protein interactions. This so-called random FRET is well accounted for by a model developed by Wolber and Hudson (40), as previously described (41–43), and is shown with the solid line in Fig. 3. The model of Wolber and Hudson has been verified experimentally in previous work (41). In particular, it has been demonstrated that this random contribution can be measured directly for simple transmembrane helices labeled with organic dyes in lipid bilayers using a fluorescence-lifetime approach, and that the experimental data follow the theoretical prediction (41). In addition, the proximity FRET measured for fluorophores attached to lipids in vesicles is also very well predicted by the model of Wolber and Hudson (44). This random-FRET contribution therefore needs to be subtracted from the measured FRET efficiency to obtain the FRET efficiency due to sequence-specific interactions, ED.
Next, we calculate the dimeric fraction (i.e., the fraction of receptors that are dimeric) in each vesicle, using the equation (43,45)
| (4) |
where xA is the fraction of proteins tagged with the acceptor and is the FRET efficiency in a dimer containing a donor and an acceptor. Here, as in previous work (36), the fluorescent proteins are attached to the TM domains via a 15-amino-acid (GGS)5 flexible linker (46). The attachment of the fluorescent proteins to the TM domains via the (GGS)5 flexible linker has been used in previous studies of GpA dimerization (36), where for this attachment was determined as = 0.63 ± 0.04, corresponding to a 48.5-Å separation distance between the two fluorophores in the dimer. With xA and known, the dimeric fractions in each vesicle were calculated from Eq. 4 and then averaged within bins of total protein concentration of width 5 × 10−4 molecules/nm2. The averaged dimeric fractions, along with the standard deviations, are shown in Fig. 4, A and B, for the wild-type and the mutant, respectively. Fig. 4 C compares the averaged dimeric fractions and their standard errors.
Figure 4.
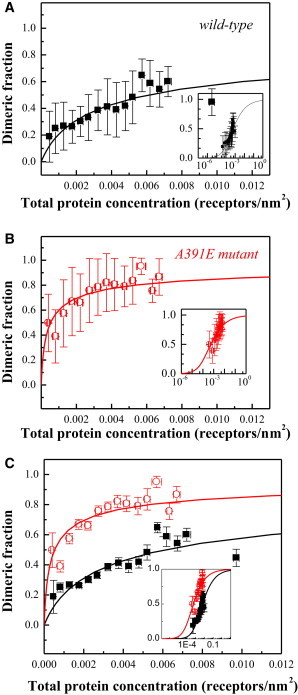
(A and B) Binned dimeric fractions as a function of the total receptor concentration, and their standard deviations, for wild-type (A) and the A391E mutant (B). The data were fitted to the dimerization model given by Eqs. 5–7 (solid lines), yielding the dimerization constants, KD. The free energies of dimerization were calculated as −3.0 kcal/mol for the wild-type and −4.5 kcal/mol for the A391E mutant. (C) Comparison of dimerization propensities of wild-type FGFR3 (black solid squares) and the A391E mutant (red open circles). In (C) we show binned data with their standard errors. The dimerization constant increases 13 times due to the mutation, and the stability of the FGFR3 dimer increases by −1.5 kcal/mol. (Insets) Data and model predictions are replotted on a semilog scale.
A two-state model describing receptor dimerization was used to fit the experimental dimeric fractions and calculate the dimerization constants. The reaction scheme describing dimerization is given by Eq. 5,
| (5) |
where the dimerization constant is
| (6) |
and the total concentration is given by
| (7) |
Using Eqs. 5–7, we can predict the dimeric fraction, f = 2[D]/[T], as a function of the total concentration, [T], for any value of the dimerization constant, KD. Then, the value of KD is optimized using a Matlab code such that the prediction gives the best fit to the single-vesicle dimeric fractions (before binning) while optimizing for the dimerization constant, KD. The optimal KD values were 145 ± 30 and 1890 ± 380 nm2 for the wild-type and the mutant, respectively. The errors correspond to the 95% confidence intervals. The fits are shown in Fig. 4 by solid lines, allowing a direct comparison between the binned data and the fits.
The statistical significance of the mutation-induced increase in dimerization can be determined using the standard errors for each data point (shown in Fig. 4 C) and chi-squared analysis as described (34). The analysis yielded a chi-squared value of 56.26 and a p-value of ≪0.001, indicative of very high statistical significance. Thus, the statistical analysis demonstrates that the mutation induces a significant, measurable increase in dimeric fractions.
Defining the standard state as Kst = 1 nm2, we can calculate the free energy of dimer formation according to
| (8) |
Using Eq. 8, we obtain −3.0 ± 0.1 kcal/mol for the wild-type and −4.5 ± 0.1 kcal/mol for the mutant. Thus, the mutation stabilizes the dimer by −1.5 ± 0.2 kcal/mol.
Discussion
Here we measured the difference in dimerization between a wild-type FGFR3 construct and an A391E mutant FGFR3 construct, with kinase domains substituted with fluorescent proteins for FRET detection. As the genes encoding for these proteins were introduced into mammalian cells, the RTK constructs were produced, folded, glycosylated, and trafficked to the plasma membrane by the endogenous cellular machinery. Upon vesiculation, the RTKs were localized in the membrane such that their interaction strength could be quantified in the vesicles without the need for their extraction, purification, and reconstitution (16,26). Thus, the methodology used here is unique and useful, as it allows us to calculate dimerization free energies for complex glycosylated proteins in the plasma membrane.
Yet, in these experiments we are limited to membrane proteins that can be correctly processed by the cellular machinery of the cell line that we use. For instance, we discovered that a mutant RTK construct with a deleted EC domain does not localize to the membranes, despite the fact that the signal peptide is present in the gene, and despite the fact that a similar truncated wild-type construct exhibited proper membrane localization in CHO cells (26). The vesicles in the case of the mutant lacking the EC domain exhibited uniform fluorescence throughout the vesicle interior, and no membrane fluorescence. This behavior has been observed for water-soluble fluorescent proteins (31) and is indicative of a membrane insertion deficiency via a mechanism that is currently unknown. Thus, we could not quantify the effect of the mutation in the absence of the EC domain in plasma membrane-derived vesicles. However, the results in the presence of the EC domain, shown here, can be directly compared to previous results for the isolated FGFR3 TM domains in model lipid bilayers (21). Although both experiments utilized FRET for detection of interactions, these two experiments are in fact very different: In one case, synthetic TM helices labeled with organic dyes were used and their interactions were characterized in lipid bilayers. In the other case, glycosylated receptors linked to fluorescent proteins at their C terminus were expressed in mammalian cells and their interactions were measured in vesicles derived from the plasma membrane. The exact value of the dimerization free energy, ΔG, depends on the definition of the standard free energy in each environment and is expected to depend on the exact protein sequence used in each experiment. It will further depend on the degree of macromolecular crowding in the plasma membrane-derived vesicles. Thus, it is not clear whether the dimer stability measurements in the two environments can be directly compared. The values of ΔΔG, on the other hand, do not depend on the exact definition of the standard free energy, as in both environments ΔΔG represents the effect of the mutation on dimerization. Therefore, the values of ΔΔG that we measure in vesicles and in plasma membrane-derived vesicles can be compared directly without the need for any assumptions. The two very different experiments yielded the same contribution of the A391E mutation to dimer stability: −1.3 ± 0.2 and −1.5 ± 0. 2 kcal/mol. It therefore appears that the presence of the large EC domain does not prevent the Glu391-mediated interactions that lead to the increase in dimer stability.
The TM domains of wild-type FGFR3 and the A391E mutant have been shown to form sequence-specific dimers (21). In a published model of the wild-type TM domain dimer, which has been experimentally verified using NMR (47), the two wild-type TM helices pack tightly into a knob-into-hole configuration, and the dimer is stabilized via van der Waals interactions. Although there is no structure for the mutant dimer, the wild-type dimer structure is consistent with the idea that each Glu residue in the A391E mutant can form a hydrogen bond with the neighboring helix (21). Therefore, the increase in dimer stability that we measure in the presence and in the absence of the extracellular domain is likely due to the formation of hydrogen bonds between the TM helices in the membrane.
The environment that the proteins explore in the model lipid bilayers is distinctly different from the one they explore in the plasma membrane-derived vesicles, because the vesicles contain diverse lipids and proteins that are found in the plasma membrane of mammalian cells. Questions may therefore arise as to why the strength of the hydrogen bonds is the same in the two different environments. There are a few measurements of the contribution of side-chain hydrogen bonds to the stability of tertiary and quaternary membrane protein structures, all of them in model lipid bilayers, and they show that the side-chain hydrogen-bond strength is very modest, ∼1 kcal/mol (21,39,48,49). This strength is much weaker than expected based on measurements in organic solvent and on calculations (50,51), and it is similar to the values observed in soluble proteins (52–56). To rationalize these results, it has been argued that the membrane protein itself can provide “a rich source of competitive hydrogen bonds and a polarizable environment that can weaken hydrogen bonds,” thus likely eliminating the effect of the environment on hydrogen-bond strength (39). As the hydrogen-bond strength has been shown to be the same in lipid bilayers and in water, it is not surprising that the strength is the same in model bilayers and in plasma membrane-derived vesicles.
Until structures of full-length receptor dimers become available, we will likely not know with certainty whether the TM helices in full-length RTKs interact intimately or whether their close approach is hindered by the large soluble RTK domains. Questions therefore arise as to whether the numerous studies of isolated RTK TM domains lead to better understanding of RTK signaling. Here we use thermodynamic measurements as a tool to gain some insight into this problem. The fact that we observe the same effect of the A391E mutation on the stability of both isolated TM domain dimers and glycosylated FGFR3 dimers suggests that the interactions that we observe in the simple TM peptide model systems are highly relevant in a biological context.
Acknowledgments
We thank Drs. Edwin Li and Lijuan He, and Mr. Jesse Placone for many helpful discussions.
This work was supported by National Institutes of Health grants GM68619 and GM95930.
References
- 1.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 2.Plotnikov A.N., Schlessinger J., Mohammadi M. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 3.Plotnikov A.N., Hubbard S.R., Mohammadi M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101:413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 4.Bouyain S., Longo P.A., Leahy D.J. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. USA. 2005;102:15024–15029. doi: 10.1073/pnas.0507591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgess A.W., Cho H.S., Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 6.Cho H.S., Leahy D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science. 2002;297:1330–1333. doi: 10.1126/science.1074611. [DOI] [PubMed] [Google Scholar]
- 7.Cho H.S., Mason K., Leahy D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 8.Bocharov E.V., Mineev K.S., Arseniev A.S. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J. Biol. Chem. 2008;283:6950–6956. doi: 10.1074/jbc.M709202200. [DOI] [PubMed] [Google Scholar]
- 9.Bocharov E.V., Mayzel M.L., Arseniev A.S. Spatial structure and pH-dependent conformational diversity of dimeric transmembrane domain of the receptor tyrosine kinase EphA1. J. Biol. Chem. 2008;283:29385–29395. doi: 10.1074/jbc.M803089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bocharov E.V., Mayzel M.L., Arseniev A.S. Left-handed dimer of EphA2 transmembrane domain: helix packing diversity among receptor tyrosine kinases. Biophys. J. 2010;98:881–889. doi: 10.1016/j.bpj.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mineev K.S., Bocharov E.V., Arseniev A.S. Spatial structure of the transmembrane domain heterodimer of ErbB1 and ErbB2 receptor tyrosine kinases. J. Mol. Biol. 2010;400:231–243. doi: 10.1016/j.jmb.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Finger C., Escher C., Schneider D. The single transmembrane domains of human receptor tyrosine kinases encode self-interactions. Sci. Signal. 2009;2:ra56. doi: 10.1126/scisignal.2000547. [DOI] [PubMed] [Google Scholar]
- 13.Li E., Hristova K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry. 2006;45:6241–6251. doi: 10.1021/bi060609y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He L., Hristova K. Physical-chemical principles underlying RTK activation, and their implications for human disease. Biochim. Biophys. Acta. 2012;1818:995–1005. doi: 10.1016/j.bbamem.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kashles O., Szapary D., Schmidt A. Ligand-induced stimulation of epidermal growth factor receptor mutants with altered transmembrane regions. Proc. Natl. Acad. Sci. USA. 1988;85:9567–9571. doi: 10.1073/pnas.85.24.9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Passos-Bueno M.R., Wilcox W.R., Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum. Mutat. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Deng C., Wynshaw-Boris A., Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 18.Vajo Z., Francomano C.A., Wilkin D.J. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr. Rev. 2000;21:23–39. doi: 10.1210/edrv.21.1.0387. [DOI] [PubMed] [Google Scholar]
- 19.van Rhijn B.W.G., van Tilborg A.A.G., Radvanyi F. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur. J. Hum. Genet. 2002;10:819–824. doi: 10.1038/sj.ejhg.5200883. [DOI] [PubMed] [Google Scholar]
- 20.Li E., You M., Hristova K. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Förster resonance energy transfer suggest weak interactions between fibroblast growth factor receptor 3 transmembrane domains in the absence of extracellular domains and ligands. Biochemistry. 2005;44:352–360. doi: 10.1021/bi048480k. [DOI] [PubMed] [Google Scholar]
- 21.Li E., You M., Hristova K. FGFR3 dimer stabilization due to a single amino acid pathogenic mutation. J. Mol. Biol. 2006;356:600–612. doi: 10.1016/j.jmb.2005.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.You M., Li E., Hristova K. The achondroplasia mutation does not alter the dimerization energetics of fibroblast growth factor receptor 3 transmembrane domain. Biochemistry. 2006;45:5551–5556. doi: 10.1021/bi060113g. [DOI] [PubMed] [Google Scholar]
- 23.Meyers G.A., Orlow S.J., Jabs E.W. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat. Genet. 1995;11:462–464. doi: 10.1038/ng1295-462. [DOI] [PubMed] [Google Scholar]
- 24.Chen F., Degnin C., Hristova K. The A391E mutation enhances FGFR3 activation in the absence of ligand. Biochim. Biophys. Acta. 2011;1808:2045–2050. doi: 10.1016/j.bbamem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen F., Sarabipour S., Hristova K. Multiple consequences of a single amino acid pathogenic RTK mutation: the A391E mutation in FGFR3. PLoS ONE. 2013;8:e56521. doi: 10.1371/journal.pone.0056521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L., Placone J., Hristova K. The extracellular domain of fibroblast growth factor receptor 3 inhibits ligand-independent dimerization. Sci. Signal. 2010;3:ra86. doi: 10.1126/scisignal.2001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Placone J., Hristova K. Direct assessment of the effect of the Gly380Arg achondroplasia mutation on FGFR3 dimerization using quantitative imaging FRET. PLoS ONE. 2012;7:e46678. doi: 10.1371/journal.pone.0046678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott R.E. Plasma membrane vesiculation: a new technique for isolation of plasma membranes. Science. 1976;194:743–745. doi: 10.1126/science.982044. [DOI] [PubMed] [Google Scholar]
- 29.Scott R.E., Maercklein P.B. Plasma membrane vesiculation in 3T3 and SV3T3 cells. II. Factors affecting the process of vesiculation. J. Cell Sci. 1979;35:245–252. doi: 10.1242/jcs.35.1.245. [DOI] [PubMed] [Google Scholar]
- 30.Scott R.E., Perkins R.G., Maercklein P.B. Plasma membrane vesiculation in 3T3 and SV3T3 cells. I. Morphological and biochemical characterization. J. Cell Sci. 1979;35:229–243. doi: 10.1242/jcs.35.1.229. [DOI] [PubMed] [Google Scholar]
- 31.Li E., Placone J., Hristova K. Quantitative measurements of protein interactions in a crowded cellular environment. Anal. Chem. 2008;80:5976–5985. doi: 10.1021/ac800616u. [DOI] [PubMed] [Google Scholar]
- 32.Chen F.H., Hristova K. The physical basis of FGFR3 response to fgf1 and fgf2. Biochemistry. 2011;50:8576–8582. doi: 10.1021/bi200986f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He L., Horton W.A., Hristova K. Physical basis behind achondroplasia, the most common form of human dwarfism. J. Biol. Chem. 2010;285:30103–30114. doi: 10.1074/jbc.M109.094086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He L., Shobnam N., Hristova K. FGFR3 heterodimerization in achondroplasia, the most common form of human dwarfism. J. Biol. Chem. 2011;286:13272–13281. doi: 10.1074/jbc.M110.205583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He L.J., Shobnam N., Hristova K. Specific inhibition of a pathogenic receptor tyrosine kinase by its transmembrane domain. Biochim. Biophys. Acta. 2011;1808:253–259. doi: 10.1016/j.bbamem.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen L., Novicky L., Hristova K. Measuring the energetics of membrane protein dimerization in mammalian membranes. J. Am. Chem. Soc. 2010;132:3628–3635. doi: 10.1021/ja910692u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del Piccolo N., Placone J., Hristova K. Production of plasma membrane vesicles with chloride salts and their utility as a cell membrane mimetic for biophysical characterization of membrane protein interactions. Anal. Chem. 2012;84:8650–8655. doi: 10.1021/ac301776j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarabipour S., Hristova K. Glycophorin A transmembrane domain dimerization in plasma membrane vesicles derived from CHO, HEK 293T, and A431 cells. Biochim. Biophys. Acta. 2013;1828:1829–1833. doi: 10.1016/j.bbamem.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowie J.U. Membrane protein folding: how important are hydrogen bonds? Curr. Opin. Struct. Biol. 2011;21:42–49. doi: 10.1016/j.sbi.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolber P.K., Hudson B.S. An analytic solution to the Förster energy transfer problem in two dimensions. Biophys. J. 1979;28:197–210. doi: 10.1016/S0006-3495(79)85171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Posokhov Y.O., Merzlyakov M., Ladokhin A.S. A simple “proximity” correction for Förster resonance energy transfer efficiency determination in membranes using lifetime measurements. Anal. Biochem. 2008;380:134–136. doi: 10.1016/j.ab.2008.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.You M., Li E., Hristova K. Förster resonance energy transfer in liposomes: measurements of transmembrane helix dimerization in the native bilayer environment. Anal. Biochem. 2005;340:154–164. doi: 10.1016/j.ab.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merzlyakov M., Hristova K. Förster resonance energy transfer measurements of transmembrane helix dimerization energetics. Methods Enzymol. 2008;450:107–127. doi: 10.1016/S0076-6879(08)03406-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wimley W.C., White S.H. Determining the membrane topology of peptides by fluorescence quenching. Biochemistry. 2000;39:161–170. doi: 10.1021/bi991836l. [DOI] [PubMed] [Google Scholar]
- 45.Merzlyakov M., Chen L., Hristova K. Studies of receptor tyrosine kinase transmembrane domain interactions: the EmEx-FRET method. J. Membr. Biol. 2007;215:93–103. doi: 10.1007/s00232-007-9009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evers T.H., van Dongen E.M.W.M., Merkx M. Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry. 2006;45:13183–13192. doi: 10.1021/bi061288t. [DOI] [PubMed] [Google Scholar]
- 47.Bocharov E., Mineev K., Arseniev A. Structural aspects of transmembrane domain interactions of receptor tyrosine kinases. Biophys. J. 2011;100(Suppl. 1):207a. [Google Scholar]
- 48.Joh N.H., Min A., Bowie J.U. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao Z., Bowie J.U. Shifting hydrogen bonds may produce flexible transmembrane helices. Proc. Natl. Acad. Sci. USA. 2012;109:8121–8126. doi: 10.1073/pnas.1201298109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ben-Tal N., Sitkoff D., Honig B. Free energy of amide hydrogen bond formation in vacuum, in water, and in liquid alkane solution. J. Phys. Chem. B. 1997;101:450–457. [Google Scholar]
- 51.Rose G.D., Wolfenden R. Hydrogen bonding, hydrophobicity, packing, and protein folding. Annu. Rev. Biophys. Biomol. Struct. 1993;22:381–415. doi: 10.1146/annurev.bb.22.060193.002121. [DOI] [PubMed] [Google Scholar]
- 52.Takano K., Yamagata Y., Yutani K. Contribution of hydrogen bonds to the conformational stability of human lysozyme: calorimetry and X-ray analysis of six Ser → Ala mutants. Biochemistry. 1999;38:6623–6629. doi: 10.1021/bi9901228. [DOI] [PubMed] [Google Scholar]
- 53.Myers J.K., Pace C.N. Hydrogen bonding stabilizes globular proteins. Biophys. J. 1996;71:2033–2039. doi: 10.1016/S0006-3495(96)79401-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takano K., Scholtz J.M., Pace C.N. The contribution of polar group burial to protein stability is strongly context-dependent. J. Biol. Chem. 2003;278:31790–31795. doi: 10.1074/jbc.M304177200. [DOI] [PubMed] [Google Scholar]
- 55.Fersht A.R., Matouschek A., Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J. Mol. Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 56.Albeck S., Unger R., Schreiber G. Evaluation of direct and cooperative contributions towards the strength of buried hydrogen bonds and salt bridges. J. Mol. Biol. 2000;298:503–520. doi: 10.1006/jmbi.2000.3656. [DOI] [PubMed] [Google Scholar]