

Abstract
The preparation and characterization of nitric oxide (NO)-releasing silica particles formed following the synthesis of N-diazeniumdiolate-modified aminoalkoxysilanes are reported. Briefly, an aminoalkoxysilane solution was prepared by dissolving an appropriate amount of aminoalkoxysilane in a mixture of ethanol, methanol, and sodium methoxide (NaOMe) base. The silane solution was reacted with NO (5 atm) to form N-diazeniumdiolate NO donor moieties on the amino-alkoxysilanes. Tetraethoxy- or tetramethoxysilane (TEOS or TMOS) was then mixed with different ratios of N-diazeniumdiolate-modified aminoalkoxysilane (10 – 75 mol%, balance TEOS or TMOS). Finally, the silane mixture was added into ethanol in the presence of an ammonia catalyst to form NO donor silica nanoparticles via a sol–gel process. This synthetic approach allows for the preparation of NO delivery silica scaffolds with remarkably improved NO storage and release properties, surpassing all macromolecular NO donor systems reported to date with respect to NO payload (11.26μmol·mg−1), maximum NO release amount (357000 ppb·mg−1), NO release half-life (253 min), and NO release duration (101 h). The N-diazeniumdiolate-modified silane monomers and the resulting silica nanoparticles were characterized by 29Si nuclear magnetic resonance (NMR) spectroscopy, UV-visible spectroscopy, chemiluminescence, atomic force microscopy (AFM), gas adsorption-desorption isotherms, and elemental analysis.
Introduction
Recent studies have established that nitric oxide (NO), a diatomic free radical endogenously produced in the body, is an important bioregulatory agent involved in multiple physiological processes including vasodilation, neurotransmission, angiogenesis, and phagocytosis.1–4 For example, the non-thrombogenic properties of vascular surfaces are primarily attributed to NO generated from endothelial cells that line the inner walls of all blood vessels.2,5 Nitric oxide derived from endothelial cells regulates blood flow and pressure, and inhibits platelet activation and aggregation under normal conditions.5 Furthermore, the generation of NO by phagocytic cells (e.g., macrophages) has been implicated in fighting invading microorganisms (e.g., bacteria).6–10 Macrophages stimulated by foreign cells produce NO to destroy such pathogens via pathways mediated by NO and its reactive derivatives (e.g., N2O3 and ONOO−).6,7 The astounding pace of discovery on the physiological roles of NO demands methods for stable storage and specific delivery of NO to biological targets.
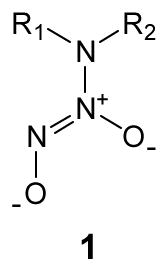
N-Bound diazeniumdiolate ions of structure 1 (where R1 and R2 are typically alkyl side groups), adducts of NO with secondary amines, represent one of the most studied NO donor systems due to their straightforward synthesis and ability to generate NO spontaneously under biological conditions.11,12 Indeed, several small molecules modified with N-diazeniumdiolates have been employed to elucidate their potent effects on diverse NO-mediated disease states and pathophysiological disorders including cardiovascular disease, ischemia-reperfusion injury, and tumor progression.12,13 However, the use of such NO donors as therapeutics has been hindered due to several factors, including poor solubility in physiological milieu, lack of specific targeting, limited ability to deliver therapeutic concentrations of NO, and generation of potentially toxic byproducts (e.g., diamines and nitrosamines), all of which have circumvented their clinical application.14–16
To overcome such drawbacks, effort has been devoted to developing macromolecular NO storage/delivery systems whereby N-diazeniumdiolate NO donors are covalently anchored to host frameworks.16–25 Such scaffolds provide a rational means to store large quantities of NO and readily modulate NO release kinetics. Indeed, nanoparticle-based drug delivery has been studied extensively for increasing drug efficacy via the enhanced permeability/retention (EPR) effect, site-specific targeting, and controlled release of drugs.26–31 Several NO donor micro- and nanoscale materials have been developed using a range of host macromolecules including albumin conjugates,16 polylactic-co-glycolic acid copolymers,17 polyethyleneimine microspheres,18 polymethacrylate particles,19,20 gold clusters,21,22 dendrimers,23 and silica particles.24,25 Of these, silica scaffolds provide an attractive platform as NO delivery vehicles because of their physical and chemical versatility.32–35 The ability to form tunable porous structures of various sizes with tailored surface functionalities has led to the use of silica nanoparticles in bioseparation and drug delivery systems.32 In addition, the particles are easily prepared (i.e., straightforward synthesis and isolation) and non-toxic, setting the stage for their use in nanomedicine.33–35
Zhang et al. synthesized NO-releasing fumed silica (amorphous particles with diameters of 0.2 – 0.3 μm) by grafting amine-functionalized silylation reagents onto the silica surface and then converting the amines to N-diazeniumdiolates.24 The silica particles prepared via this method released 0.56 μmol NO·mg−1.24 More recently, our laboratory reported an alternative strategy for preparing NO-releasing silica particles of variable sizes (d = 20 – 500 nm), NO payloads (0.05 – 1.78 μmol·mg−1), and NO release half-lives (0.1 – 12 h).25 Briefly, the NO-releasing silica nanoparticles were synthesized via the co-hydrolysis and condensation of aminoalkoxysilanes (NO donor precursors) and tetraalkoxysilanes (i.e., tetramethoxy- or tetraethoxysilane) followed by exposure to NO (5 atm, 3 d) in the presence of sodium methoxide (NaOMe) base to form N-diazeniumdiolates (Figure 1A).25 Although the NO release levels of the silica nanoparticles prepared in this manner were significantly greater than small molecule and surface-grafted silica NO donor counterparts, the NO storage was limited due to particle aggregation at high aminoalkoxysilane concentrations,25 attributed to interactions between the amines and adjacent silanols and/or other amines via hydrogen bonding.25,36
Figure 1.

Schematic of N-diazeniumdiolate-modified silica nanoparticles as NO delivery scaffolds synthesized via two different routes: (A) post-formation; and, (B) pre-formation strategies. Represented are example syntheses using tetraethoxysilane (TEOS) and N-(6-aminohexyl)aminopropyltrimethoxysilane (AHAP3) as tetraalkoxy- and aminoalkoxysilane precursors, respectively.
Herein, we report a new synthetic approach for preparing NO-releasing silica nanoparticles with markedly improved NO storage and release capabilities. In contrast to the previous method25 where the amine-functionalized silica particles were first synthesized and then reacted with NO to form N-diaze-niumdiolate moieties (hereafter referred to as “post-formation”), N-diazeniumdiolate-modified silane monomers were prepared prior to particle construction (“pre-formation”) (Figure 1B). This approach enables the synthesis of nanoparticles with higher concentrations of aminoalkoxysilanes and greater NO storage capacity. Indeed, the pre-formation strategy inherently reduces particle aggregation because the aminoalkoxysilanes are first converted to N-diazeniumdiolates, thereby avoiding interaction of amines during silica formation. As such, the approach facilitates better access of NaOMe and NO to the amine sites of the silane precursors and significantly greater yields of NO per mole of aminoalkoxysilane precursor. In this work, the pre-formed N-diazeniumdiolate-modified silanes and the resulting silica particles were characterized by 29Si nuclear magnetic resonance (NMR) spectroscopy, UV-visible spectroscopy, chemiluminescence, atomic force microscopy (AFM), gas adsorption-desorption isotherms, and elemental analysis.
Experimental Section
Reagents and Materials
Tetraethoxysilane (TEOS), tetramethylsilane (TMS), and sodium methoxide (NaOMe) were purchased from Fluka (Buchs, Switzerland). N-(6-Aminohexyl)aminopropyl-trimethoxysilane (AHAP3), N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAP3), (aminoethyl-aminomethyl)phenethyltrimethoxysilane (AEMP3), N-(6-aminohexyl)aminomethyltrimethoxysilane (AHAM3), N-(2-aminoethyl)-11-aminoundecyltrimethoxysilane (AEAUD3), and (3-trimethoxysilyl-propyl)diethylenetriamine (DET3) were purchased from Gelest (Tullytown, PA). Tetramethoxysilane (TMOS) was purchased from Sigma (St. Louis, MO). Methanol (MeOH), ethanol (EtOH), toluene, and ammonia solution (NH4OH, 30 wt% in water) were purchased from Fisher Scientific (Fair Lawn, NJ). Nitric oxide (NO, 99.5%), argon (Ar), and nitrogen (N2) gases were obtained from Linde Gas (Morrisville, NC) or National Welders Supply (Raleigh, NC). Other solvents and chemicals were analytical-reagent grade and used as received. A Millipore Milli-Q UV Gradient A10 System (Bedford, MA) was used to purify distilled water to a final resistivity of 18.2 MΩ·cm and a total organic content of ≤6 ppb.
Synthesis of N-Diazeniumdiolate-Modified Aminoalkoxysilane Monomers
An aminoalkoxy-silane solution was prepared by dissolving 10.3 mmol of AHAP3, AEAP3, AEMP3, AHAM3, or AEAUD3 in 24 mL of EtOH and 6 mL of MeOH in the presence of NaOMe (10.3 mmol; an equimolar amount corresponding to the secondary amine content of the silane precursor). Since DET3 contains two secondary amine moieties, two equimolar amounts of NaOMe (i.e., 20.6 mmol) were used. The solution was then placed into 10 mL vials equipped with a stir bar. The vials were placed in a Parr hydrogenation bottle (200 mL), connected to an in-house NO reactor, and flushed with Ar for 5 min, followed by a series of six charge/discharge cycles with Ar (5 atm, 6 × 10 min) to remove oxygen in the solution. The reaction bottle was then charged with NO to 5 atm and sealed for 3 d at room temperature while stirring. The NO gas was purified before use by passing it through a column packed with KOH pellets for 2 h to remove trace NO degradation products. Prior to removing the N-diazeniumdiolate-modified aminoalkoxysilane solution, unreacted NO was purged from the chamber with Ar for 10 min. Absorption spectra were recorded using a Perkin-Elmer Lambda 40 UV-visible spectrophotometer (Norwalk, CT) in dilute methanolic solutions (100 μM) to confirm the formation of diazeniumdiolates bound on the amine nitrogen of the silane precursors. Once synthesized, the resulting alkoxysilane-derived N-diazeniumdiolates were immediately used to prepare NO-releasing particles in EtOH/MeOH solutions.
Preparation of NO-Releasing Silica Nanoparticles
The silane mixtures were prepared by adding 2.8 mmol of TEOS or TMOS into different concentrations (0.32 – 8.36 mmol corresponding to 10 – 75 mol%, balance TEOS or TMOS) of the N-diazeniumdiolated silane solutions (prepared in the previous step), and mixed for 5 min. The silane solution was then added to 5.6 – 29.1 mL of cold EtOH (adjusting the total volume of alcoholic solvents to 30 mL) and 6 mL of cold ammonia (30 wt% in water), and mixed vigorously for 30 min at 4 °C to minimize any thermal decomposition of N-diazeniumdiolates. The silica precipitate was collected by centrifugation at 4000 rpm for 5 min, washed copiously with cold EtOH, dried under ambient conditions for 1 h, and then stored in a sealed container at −20 °C until use.
Dissociation Kinetic Studies of N-Diazeniumdiolate Derivatives
To study the decomposition kinetics of the N-diazeniumdiolate-derived NO donor species (i.e., silane monomers and silica particles), NO release was monitored in deoxygenated phosphate-buffered saline (PBS; 0.01 M, pH 7.4) at 37 °C using a Sievers NOA 280i chemiluminescence Nitric Oxide Analyzer (Boulder, CO). The instrument was calibrated with air passed through a zero filter (0 ppm NO) and 27.6 ppm of NO standard gas (balance N2, purchased from Linde Gas). Evolved NO was transported to the analyzer by a stream of N2 (70 mL·min−1) passed through the reaction cell.
The dissociation rate constant (kobs) of N-diazeniumdiolated silane monomers was calculated from a linear regression of the ln Cdiaz versus time plots that consistently yielded a good linear relationship (R2 ≥ 0.995), indicating first-order kinetics. The change of N-diazeniumdiolate content (Cdiaz) with time was determined from , where and t[NO]t are the initial N-diazeniumdiolate concentration and the total amount of NO released from time 0 to t, respectively. The “apparent” reaction order (n) of the N-diazeniumdiolate dissociation kinetics derived from the silica scaffolds was also determined from the slope of the lnυ versus lnCdiaz plots fitted with a linear regression (R2 ≥ 0.995), where υ is the dissociation rate of the N-diazeniumdiolate (−dCdiaz/dt, μmol·mg−1·h−1).
Characterization of Functionalized Silica Particles
Imaging of the particles was carried out via atomic force microscopy (AFM), the silica particles were suspended in toluene, deposited on a freshly cleaved mica surface (SPI; West Chester, PA), and dried under ambient conditions for 3 h. Contact mode AFM images were obtained in air using a Molecular Force Probe 3D Atomic Force Microscope (Asylum Research; Santa Barbara, CA) controlled with a MFP-3D software running under Igor Pro (Wavemetrics; Lake Oswego, OR). Triangular silicon nitride cantilevers with a nominal spring constant of 0.12 N·m−1 and resonance frequency of 20 kHz (Veeco; Santa Barbara, CA) were used to acquire height/topography images at a scan rate range of 0.5 – 2 Hz. To determine the particle size of the silica composite materials, multiple areas of the substrate were imaged.
Solid-state cross-polarization/magic angle spinning (CP/MAS) 29Si nuclear magnetic resonance (NMR) spectra were obtained at 20 °C on a Bruker 360 MHz DMX spectrometer (Billerica, MA) equipped with wide-bore magnets (triple-axis pulsed field gradient double-resonance probes). Silica composite particles were packed into 4 mm rotors (double-resonance frequency of 71.548 MHz) and spun at a speed of 8.0 kHz. The chemical shifts were determined in ppm relative to a TMS external standard.
Nitrogen adsorption/desorption isotherms were obtained at −196 °C on an ASAP 2010 Surface Area and Porosimetry Analyzer (Micromeritics Instrument; Norcross, GA). Prior to the measurements, silica samples were degassed at 200 °C in a vacuum for 2 h. The specific surface area (SBET) of the silica was calculated by the Brunauer-Emmett-Teller (BET) method using the adsorption data in a relative pressure (P/P0) range of 0.75 – 0.98.
Elemental (CHN) analyses were performed by Midwest Microlab, LLC (Indianapolis, IN). Specifically, the concentration of amines incorporated in the functionalized silica nanoparticles was determined. Prior to the measurements, samples were calcinated at 70 °C for 3 d to eliminate NO from the particles.
Results and Discussion
Synthesis of N-Diazeniumdiolate-Modified Silane Monomers
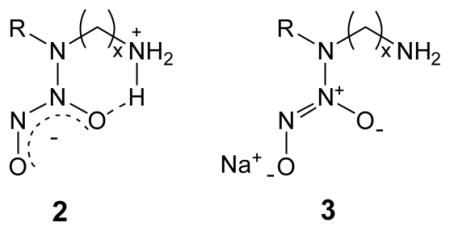
To prepare NO-releasing silica nanoparticles via a “pre-formation” strategy, N-diazeniumdiolate conjugated silane monomers were first synthesized. In initial studies of the N-diazeniumdiolate formation reaction, an aminoalkoxysilane precursor was dissolved in ethanol and reacted with NO at high pressures (5 atm, 3 d). This synthesis represents a typical route used to form N-diazeniumdiolate anions, which are stabilized via a neighboring cationic amine (i.e., intramolecularly stabilized species).11 The zwitterionic NO adducts (2, where R = alkyltrimethoxysilane, x = 2 or 6) formed under these conditions showed poor NO addition efficiencies ranging from 4.5 to 8.1%. Zhang et al. reported that the addition of a strong base such as sodium methoxide (NaOMe) during NO exposure promotes the deprotonation of the secondary amine nitrogen, thereby shifting the reaction equilibrium toward N-diazeniumdiolate formation.24 Furthermore, the sodium ion serves as a countercation to stabilize the anionic N-diazeniumdiolate structure of 3 (see Supporting Information). When NaOMe was employed in this work, the conversion efficiencies increased more than 10-fold with typical yields of 54 – 100% (see below for details). Figure 2 shows structures of sodium-stabilized N-diazeniumdiolates derived from corresponding parent aminoalkoxysilanes of N-(6-aminohexyl)aminopropyltrimethoxysilane (AHAP3), N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAP3), (aminoethylaminomethyl)phenethyltrimethoxysilane (AEMP3), (3-trimethoxysilylpropyl)diethylenetriamine (DET3), N-(6-aminohexyl)aminomethyltrimethoxysilane (AHAM3), and N-(2-aminoethyl)-11-aminoundecyltrimethoxysilane (AEAUD3). The formation of the N-diazeniumdiolate functional group was confirmed via UV-visible spectroscopy in the MeOH solution. These polyamine/NO adducts exhibited λmax ranging from 248 – 253 nm, a characteristic maximum absorption of the N-diazeniumdiolate species (data not shown).10,11 29Si NMR analysis indicated that the presence of NaOMe did not lead to the self-condensation of N-diazeniumdiolate-modified silane precursors during the NO addition reactions (Step 1 in Figure 1B). Tn peaks, characteristic of organosilane polymerization11,23,24 were not observed.
Figure 2.

Structures of sodium-stabilized N-diazeniumdiolate ions derived from corresponding parent aminoalkoxysilanes of (A) N-(6-aminohexyl)aminopropyltrimethoxysilane (AHAP3); (B) N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAP3); (C) (aminoethylaminomethyl)phenethyltrimethoxysilane (AEMP3); (D) (3-trimethoxysilyl)diethylenetriamine (DET3); (E) N-(6-aminohexyl)aminomethyltrimethoxysilane (AHAM3); and, (F) N-(2-aminoethyl)-11-aminoundecyltrimethoxysilane (AEAUD3).
Chemiluminescence NO measurements were employed to obtain kinetic data for the decomposition of N-diazeniumdiolate-functionalized silanes in deoxygenated phosphate-buffered saline (PBS; 0.01 M, pH 7.4) at 37 °C. Our findings illustrate the drastic effects of amine derivatization on both the NO storage and release (i.e., decomposition) properties, including the number of moles of NO generated per mole of the starting N-diazeniumdiolate (CNO), amine to N-diazeniumdiolate conversion efficiency (%Econv), half-life of NO release (t1/2), and rate constant (kobs) observed in N-diazeniumdiolate breakdown (Table 1).
Table 1.
Dissociation characteristics of N-diazeniumdiolate-modified aminoalkoxysilane precursors in 0.01 M phosphate-buffered saline at pH 7.4 and 37 °Ca
| aminoalkoxysilaneb,c | CNOd | % Econve | t1/2 (min) | 10−5 × kobs (s−1)f |
|---|---|---|---|---|
| AHAP3 | 1.99 | ~100 | 9 | 112 |
| AEAP3 | 1.47 | 73 | 114 | 8.86 |
| AEMP3 | 1.15 | 58 | 210 | 4.58 |
| DET3 | 2.17 | 54 | 81 | 21.7 (0.68)g |
| AHAM3 | 1.79 | 90 | 3 | 541 |
| AEAUD3 | 1.34 | 67 | 176 | 5.16 |
Values were obtained using a Sievers chemiluminescence NO analyzer (NOA280i) under deoxygenated conditions.
AHAP3, N-(6-aminohexyl)aminopropyltrimethoxysilane; AEAP3, N-(2-aminoethyl)-3-aminopropyltrimethoxysilane; AEMP3, (aminoethylaminomethyl)phenethyltrimethoxysilane; DET3, (3-trimethoxysilyl)diethylenetriamine; AHAM3, N-(6-aminohexyl)aminomethyltrimethoxysilane; AEAUD3, N-(2-aminoethyl)-11-aminoundecyltrimethoxysilane.
Aminoalkoxysilanes used consist of one primary and one secondary amines except DET3, which contains one primary and two secondary amines.
Moles of NO generated (CNO) per mole of the starting silane-derived N-diazeniumdiolate employed. Theoretical 2.0 per mole of the secondary amine.
Amine to N-diazeniumdiolate conversion efficiency (Econv) = moles of NO released/(2 × moles of secondary amine).
First-order rate constants (kobs) were calculated from a linear regression of the ln Cdiaz versus time plots consistently with R2 ≥ 0.995.
The N-diazeniumdiolate-modified DET3 precursor has two distinct kobs values obtained from 0 to 3 h and 5 to 90 h (in parenthesis), respectively (see Figure 3B).
The NO generation ratio (CNO) and NO addition efficiency (%Econv) of sodium-stabilized N-diazeniumdiolates are significantly affected by the ligand structures bound to the secondary amine nitrogen where the N-diazeniumdiolate NO donor moiety is formed (see Figure 2).11,37–39 For example, N-diazeniumdiolated AEAP3 and AHAM3 exhibited a decreased Econv of 73 and 90%, respectively, compared to Econv of ~100% for N-diazeniumdiolated AHAP3. This result is attributed to an increase in steric hindrance as the parent silanes (i.e., AEAP3 and AHAM3) possess shorter alkyl chain lengths from the secondary amine nitrogens: –(CH2)2NH2 and –CH2Si(OMe)3, respectively. For the AEMP3 silane, the presence of a rigid benzyl ring adjacent to the secondary nitrogen significantly impeded N-diazeniumdiolate formation (Econv = 58%).39 In addition to such steric interference, the electrophilic nature of the benzyl substituent may deteriorate the amine’s nucleophilicity, thereby further hindering N-diazeniumdiolate formation.39 Although the parent DET3 silane has >1 secondary amines, only one N-diazeniumdiolate moiety was formed (CNO = 2.17 and Econv = 54%). This low conversion efficiency may be attributed to both steric hindrance and electrostatic repulsion between two N-diazeniumdiolate groups formed on adjacent secondary amines separated by only a short ethyl (C2) chain.
Dissociation Kinetics of N-Diazeniumdiolate-Modified Silane Monomers
The half-life (t1/2) of the silane bound N-diazeniumdiolates was determined from the NO release plots, at the time point when half the total amount of NO was liberated. The dissociation rate constants (kobs) of N-diazeniumdiolate derivatives studied in this work were also calculated (see Table 1). On the basis of monitoring the amount of NO release, the concentration of N-diazeniumdiolate species (Cdiaz) at any given time may be determined.24 The dissociation kinetics of two representative N-diazeniumdiolates derived from AEAP3 and DET3 are shown in Figure 3. Plots of the natural logarithm of the N-diazeniumdiolate concentration (ln Cdiaz) versus time yielded a linear relationship, with R2 values typically ≥0.995 for all N-diazeniumdiolates examined (indicating first-order kinetic behaviors in all cases).37,38 It is well-known that the decomposition of N-diazeniumdiolates follows proton-derived first-order kinetics.11,38 As the pH of the solution becomes more basic, the decomposition rate of N-diazeniumdiolates decreases.37,38 Although most potential applications for N-diazeniumdiolates would be at physiological pH, the dissociation rate dependence on pH is of interest for practical reasons. Of note, N-diazeniumdiolates stability at high pH represents a useful means for manipulating nanoparticles, for example, without loss of NO release during preparation, modification, and storage.38 The polyamine-derived N-diazeniumdiolated silanes exhibited a wide range of t1/2 (3 to 210 min) and kobs (4.58 × 10−5 to 5.41 × 10−3 s−1), depending on the stability of the N-diazeniumdiolate structures in the reaction media.11,40
Figure 3.

Dissociation kinetic plots of lnCdiaz – time for N-diazeniumdiolate-modified AEAP3 (A) and DET3 (B) precursors in deoxygenated PBS (0.01 M, pH 7.4) at 37 °C. The fitting lines (dashed red) were determined from a linear regression of the corresponding experimental data (solid black). Inserted are plots of total NO concentration released – time for each N-diazeniumdiolate species.
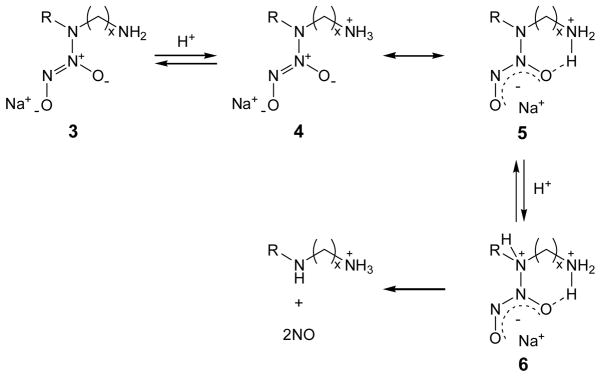
Scheme 1 illustrates the structural stability and dissociation pathway of diamine-based NO adducts (i.e., N-diazeniumdiolated AHAP3, AEAP3, AEMP3, AHAM3, and AEAUD3). Initially, the high basicity of the primary amines (pKa = 9.73 – 11.02)41 on these N-diazeniumdiolate derivatives leads to protonation of the primary nitrogen site in 3 to yield 4.38 In addition to the formation of sodium salt derivatives, N-diazeniumdiolate anions may be stabilized with an intramolecular ammonium cation via hydrogen bonding, yielding a resonance structure 5. The short alkyl length between primary and secondary nitrogens for AEAP3-, AEMP3-, and AEAUD3-derived N-diazeniumdiolates (C2) would facilitate hydrogen bonding, thereby leading to relatively long half-lives (i.e., t1/2 = 114 – 210 min) and slow dissociation rates (i.e., kobs = 4.58 – 8.86 × 10−5 s−1). However, such effect was not observed for N-diazeniumdiolates derived from AHAP3 and AHAM3, where the cationic primary amine is six carbons (C6) removed from the N-diazeniumdiolate group, resulting in fast decomposition kinetics (i.e., t1/2 = 3 and 9 min, and kobs = 1.12 and 5.41 × 10−3 s−1, respectively). The rapid decrease in stability with increasing the chain length is attributed to the decreasing influence of hydrogen bonding in 5.38 Upon protonating the secondary nitrogen of 5 (conjugated with the N-diazeniumdiolate moiety) to form 6, NO is liberated. When comparing of N-diazeniumdiolate-modified AEAUD3 and AEAP3, AEAUD3 is characterized by a longer t1/2 (176 vs. 114 min, respectively). The longer NO release may be the result of the increased hydrophobicity for AEAUD3 imparted by the bulky undecyl (C11) chain between the secondary nitrogen and trimethoxysilane ligand.40
Scheme 1.
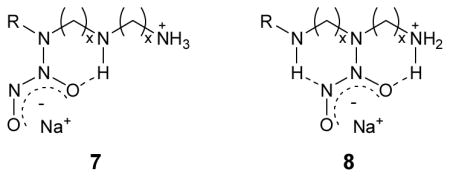
Similarly, structures 7 and 8 (where R = propyltrimethoxysilane, x = 2) are representatives of the N-diazeniumdiolate forms and hydrogen bonding interactions that are possible for triamines such as DET3.40 Two different structures of the N-diazeniumdiolate derived from the parent DET3 silane contribute to the biphasic character (i.e., two distinct linear regions) in the dissociation kinetic plot (see Figure 3B). The more stable structure of 8 engaged in two hydrogen bonding interactions is responsible for the slower dissociation rate (kobs = 6.8 × 10−6 s−1) observed from 5 to 90 h. Based on the total NO amount released from the DET3-based N-diazeniumdiolate derivative, it was estimated that approximately 64 and 36% of dissociation reactions were attributed to structures 7 and 8, respectively.
Synthesis and Characterization of Functionalized Silica Scaffolds
Nitric oxide-releasing silica nanoparticles were synthesized by mixing the N-diazeniumdiolate-modified silane prepared previously with different ratios of a tetraalkoxysilane (i.e., tetraethoxy- or tetramethoxysilane, TEOS or TMOS) in the presence of an ammonia catalyst. Since the sol–gel process for silica polymerization was carried out under strongly basic conditions (pH >12), N-diazeniumdiolate moieties conjugated on the silanes were preserved without serious deterioration (see below for details). Furthermore, the synthesis of N-diazeniumdiolate NO donors prior to silica construction enabled the formation of particles with superior NO storage and release ability. Particle aggregation was reduced due to decreased hydrogen bonding interactions between amines by synthesizing N-diazeniumdiolate-modified silanes prior to silica polymerization.
Control over both the structure and concentration of the N-diazeniumdiolate-modified silane precursors allowed for the preparation of NO donor silica nanoparticles of widely varying size and NO release properties. As summarized in Table 2, the size of the silica nanoparticles was tunable by varying the type and concentration of N-diazeniumdiolated silane derivatives used, and ranged from 21 to 151 nm in diameter. Some example AFM images of NO-releasing silica particles are shown in Figure 4. In general, as the mol% of N-diazeniumdiolated silane was increased, the diameter of the particle decreased (e.g., 46 ± 5 to 21 ± 2 nm in diameter for 35 to 75 mol% N-diazeniumdiolated AHAM3 silica). Recent work suggests that the size of nanodelivery vehicles is particularly important in determining cellular/tissue uptake and accumulation, with 20 to 100 nm (diameter) particles being optimal.26,42,43 Most NO-releasing silica nanoparticles described herein fit this range.
Table 2.
NO release properties of a series of NO-releasing silica nanoparticles with different tetraalkoxy-and N-diazeniumdiolate-modified aminoalkoxysilane precursors synthesized via a “pre-formation” strategya,b
| tetraalkoxy-silanec | aminoalkoxysilane
|
particle size (nm)f | t[NO] (μmol·mg−1) | t1/2 (min) | [NO]m (ppb·mg−1) | tm (min) | td (h) | |
|---|---|---|---|---|---|---|---|---|
| typed | mol%e | |||||||
| TEOS | AHAP3 | 10 | 161 ± 23 | 0.56 | 18 | 3400 | 3 | 6 |
| 25 | 142 ± 15 | 1.59 | 18 | 9500 | 3 | 8 | ||
| 35 | 128 ± 11 | 2.58 | 24 | 14500 | 5 | 10 | ||
| 45 | 136 ± 15 | 3.77 | 18 | 21700 | 8 | 11 | ||
| AEAP3 | 17 | 95 ± 9 | 0.75 | 66 | 1200 | 7 | 8 | |
| 25 | 90 ± 7 | 1.13 | 90 | 1600 | 8 | 11 | ||
| 35 | 74 ± 8 | 1.48 | 108 | 1400 | 8 | 13 | ||
| 45 | 65 ± 6 | 1.66 | 132 | 1300 | 8 | 15 | ||
| AEMP3 | 20 | 27 ± 2 | 0.57 | 119 | 500 | 12 | 12 | |
| 40 | 21 ± 2 | 0.74 | 192 | 400 | 12 | 20 | ||
| 55 | 25 ± 3 | 1.05 | 253 | 600 | 13 | 30 | ||
| DET3 | 10 | 29 ± 3 | 1.29 | 66 | 1600 | 11 | 22 | |
| 35 | 24 ± 4 | 2.99 | 72 | 6400 | 5 | 55 | ||
| 40 | 24 ± 5 | 3.55 | 66 | 7900 | 3 | 58 | ||
| AHAM3 | 35 | 46 ± 5 | 4.25 | 3 | 106000 | 2 | 6 | |
| 45 | 38 ± 4 | 5.36 | 4 | 175000 | 2 | 8 | ||
| 55 | 31 ± 2 | 6.48 | 3 | 180000 | 3 | 13 | ||
| 65 | 28 ± 4 | 7.78 | 4 | 216000 | 3 | 18 | ||
| 75 | 21 ± 2 | 11.20 | 4 | 321000 | 3 | 21 | ||
| AEAUD3 | 25 | 80 ± 10 | 0.50 | 58 | 1300 | 3 | 10 | |
| 35 | 63 ± 7 | 0.75 | 85 | 2400 | 3 | 16 | ||
| 45 | 49 ± 4 | 1.83 | 108 | 6600 | 2 | 32 | ||
| TMOS | AHAP3 | 45 | 120 ± 11 | 3.50 | 13 | 22700 | 3 | 19 |
| 55 | 103 ± 8 | 4.21 | 18 | 33600 | 5 | 23 | ||
| DET3 | 40 | 47 ± 6 | 6.25 | 95 | 12400 | 5 | 101 | |
| AHAM3 | 65 | 45 ± 4 | 9.33 | 4 | 286000 | 3 | 6 | |
| 75 | 41 ± 4 | 11.26 | 4 | 357000 | 3 | 8 | ||
| TEOS | AHAP3g | 10 | 20 ± 2 | 0.38 | 54 | 370 | 21 | 6 |
| AEAP3g | 17 | 92 ± 16 | 0.60 | 204 | 140 | 126 | 15 | |
| TMOS | AHAP3g | 77 | 65 ± 5 | 1.78 | 54 | 2800 | 7 | 11 |
Values were determined using a Sievers chemiluminescence NO analyzer (NOA280i) in deoxygenated phosphate-buffered saline (PBS; 0.01 M, pH 7.4) at 37 °C.
t[NO], total amount of NO released; t1/2, half-life of NO release; [NO]m, maximum flux of NO release; tm, time necessary to reach [NO]m; td, duration of NO release, sustained fluxes of NO ≥10 ppb·mg−1 (or ≥1.5 pmol·mg−1).
TEOS, tetraethoxysilane; TMOS, tetramethoxysilane.
AHAP3, N-(6-aminohexyl)aminopropyltrimethoxysilane; AEAP3, N-(2-aminoethyl)-3-aminopropyltrimethoxysilane; AEMP3, (aminoethylaminomethyl)-phenethyltrimethoxysilane; DET3, (3-trimethoxysilyl)diethylenetriamine; AHAM3, N-(6-aminohexyl)aminomethyltrimethoxysilane; AEAUD3, N-(2-aminoethyl)-11-aminoundecyltrimethoxysilane.
Balance TEOS or TMOS.
In diameter.
Ref. 25. N-Diazeniumdiolate-modified silica nanoparticles were prepared via a “post-formation” method.
Figure 4.

Contact mode AFM images of (A) 45 mol% N-diazeniumdiolate-modified AHAP3, (B) 35 mol% N-diazeniumdiolated AEAP3, and (C) 45 mol% N-diazeniumdiolated AHAM3 silica particles (all balance TEOS) on a mica surface. (D) Enlargement of a single particle from (C).
Solid-state 29Si NMR spectroscopy was used to (1) confirm the incorporation of N-diazeniumdiolate-modified aminoalkyl functionalities within the silica network, (2) calculate the surface coverage of such ligands, and (3) determine the degree of silica polymerization (condensation.44,45 Cross polarization and magic angle spinning (CP/MAS) NMR techniques were employed to increase the signal resolution and sensitivity of the silicon atoms in proximity to protons.45 The spectra for NO donor functionalized silica nanoparticles prepared from 35 to 75 mol% of N-diazeniumdiolated AHAM3 (balance TEOS) are shown in Figure 5, and positions and relative intensities of peaks are detailed in Table 3. The chemical environments of silicon atoms in the spectra may be characterized into two distinctive groups (i.e., Qn and Tn structures). Traditionally, the peaks at −90, −98, and −108 ppm are assignedto Q2 (geminal silanol; –O2Si(OH)2), Q3 (single silanol; –O3Si(OH)), and Q 4 (siloxane; –O4Si) structures, respectively, indicating the inorganic networks of the silica (Figure 5B).44,45 Peaks at chemical shifts of approximately −56, −64, and −73 ppm are representatives of silicon atoms connected to T1 (–OSi(OH)2R), T 2 (–O2Si(OH)R), and T3 (–O3SiR) structures, respectively (where R is an N-diazeniumdiolate-conjugated aminohexylamino-methyl group).44,45 The presence of Tn bands suggests the existence of covalent linkages between the organic group and the silica backbone (i.e., Si–C bridges).
Figure 5.

(A) Solid-state 29Si CP/MAS NMR spectra of functionalized silica nanocomposite materials with various amounts of N-diazeniumdiolate-modified AHAM3: (a) 35, (b) 55, and (c) 75 mol% (balance TEOS). (B) Schematic illustration of silicon chemical environments at the surface of N-diazeniumdiolate-conjugated AHAM3/TEOS silica nanoparticles. (C) Plot of percent surface coverage (%SC) of functional ligands vs content of AHAM3/NO adduct precursors loaded during the silica polymerization.
Table 3.
29Si chemical shifts (δSi in ppm from tetramethylsilane), relative concentrations of Tn and Qn structures, surface coverage (%SC), immobilization efficiency (Eimmo), and degree of condensation (%DC) for silica particles prepared from N-diazeniumdiolate-modified AHAM3a
| aminoalkoxysilane
|
Si structure (int%)
|
%SCc | Eimmod | %DCe | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ligand type | mol%b | T 1 (−56) | T2 (−64) | T 3 (−73) | Q 2 (−90) | Q 3 (−98) | Q 4 (−108) | |||
| AHAM3f | 35 | 6 | 17 | 10 | 12 | 35 | 20 | 41.3 | 1.18 | 75.6 |
| 55 | 11 | 30 | 15 | 7 | 27 | 10 | 62.2 | 1.13 | 72.5 | |
| 75 | 9 | 43 | 20 | 3 | 19 | 6 | 76.6 | 1.02 | 73.5 | |
Data were obtained by deconvolution of 29Si CP/MAS NMR spectra.
Balance tetraethoxysilane (TEOS).
Surface coverage was calculated with the equation of SC = (T1 + T 2 + T3)/(−T1 + T2 + T3 + Q2 + Q3).
Immobilization efficiency (Eimmo) was estimated as the ratio of %SC and mol% of N-diazeniumdiolated AHAM3 loaded during the silica formation.
The degree of condensation (%DC) was calculated from the relative intensity of Tn and Qn species.
AHAM3, N-(6-aminohexyl)aminomethyltrimethoxysilane.
As the N-diazeniumdiolate-modified AHAM3 content was increased from 35 to 75 mol%, the surface coverage of such ligands [SC = (T1 + T2 + T3)/(−T1 + T2 + T3 + Q2 + Q3)]45,46 increased from 41 to 77% (Figure 5C). The ligand immobilization efficiency (Eimmo), defined as the ratio of %SC and N-diazeniumdiolate-modified AHAM3 concentration (mol%) incorporated in the starting sol,25 was 1.02 – 1.18, indicating effective immobilization of the NO-donor functional groups. Furthermore, the stability of the silica structures was assessed by determining the degree of condensation (%DC) according to the following equation47,48
| (Eq. 1) |
where qn represents the relative intensity (I) of the Qn or Tn species, and f refers to the connectivity of the silane monomer, determined by the number of reactive alkoxy sites (i.e., f = 4 and 3 for Qn and Tn structures, respectively). The %DC of N-diazeniumdiolated AHAM3 silica particles slightly decreased from 75.6 to 72.5 as the concentration of the NO donor-modified silanes was increased from 35 to 75 mol%. However, the overall degree of condensation for these inorganic-organic hybrid silica remained similar to that of TEOS alone (%DC = 78.7), indicating that the presence of the bulky organic groups does not significantly compromise the stability of the resulting silica networks. Of note, the integration and quantitative analyses of these structures are complicated because the intensity of each peak depends on the efficiency of cross polarization and the proton relaxation time.49
As determined via nitrogen adsorption-desorption isotherms,45 the N-diazeniumdiolate-modified silica was nonporous with surface areas (SBET) of only 10 – 20 m2·g−1 (at p/p0 = 0.75 – 0.98). Such low surface areas were expected based on previous work. Shin et al.,25 Sayari and Hamoudi,34 and Stein et al.35 have each characterized organically-modified silica synthesized by the co-condensation method indicated highly dense, nonporous, and amorphous structures.
Nitric Oxide Release Characteristics
Nitric oxide storage and release properties of silica nanoparticles prepared via the pre-formation strategy were evaluated under physiological conditions (PBS; pH 7.4 at 37 °C) as a function of N-diazeniumdiolated aminoalkoxy- and tetraalkoxysilane structure and concentration. The NO release profiles of two representative silica nanoparticles (40 mol% DET3/NO, balance TMOS, and 55 mol% AHAM3/NO, balance TEOS) are shown in Figure 6. These representative particles illustrate the drastic effects of silica composition on NO release properties, including the total amount of NO (t[NO]), half-life of NO release (t1/2), maximum concentration of NO release ([NO]m), time necessary to reach [NO]m (tm), and NO release duration (td). The NO release properties for a series of N-diazeniumdiolate-modified silica nanoparticles prepared by the pre-formation approach are summarized in Table 2. Both the NO payload and release characteristics were significantly affected by the concentration and chemical identity of the N-diazeniumdiolate-modified silanes (derived from parent silanes of AHAP3, AEAP3, AEMP3, DET3, AHAM3, and AEAUD3) and backbone silanes (i.e., TEOS and TMOS) used to prepare the silica nanoparticles.
Figure 6.

Real-time NO release profiles (A) and total NO release amount (B) for 40 mol% N-diazeniumdiolated DET3 (balance TMOS, solid red curves) and 55 mol% N-diazeniumdiolated AHAM3 (balance TEOS, dashed blue curves) silica nanoparticles in deoxygenated PBS (0.01 M, pH 7.4) at 37 °C. Inset plots (A′) and (B′) represent the expansion of graphs (A) and (B) at <100 ppb·mg−1 NO release at extended periods and at <1 h, respectively.
Both t[NO] and [NO]m of the silica particles synthesized by the pre-formation strategy were markedly increased compared to the silica prepared by the post-formation method25 at identical aminoalkoxysilane concentrations (Table 2). For example, t[NO] and [NO]m for 10 mol% AHAP3 silica (balance TEOS) were increased from 0.38 to 0.56μmol·mg−1 and 370 to 3400 ppb·mg−1 for the post- and pre-formation strategies, respectively. For 17 mol% AEAP3 silica, similar trends in t[NO] and [NO]m were observed between the post- and pre-formation approaches. The elevated quantities of NO release are attributed to a more homogeneous distribution of the N-diazeniumdiolate NO donors throughout the silica particle, as depicted in Figure 1B. More importantly, the pre-formation approach enabled the synthesis of particles containing AHAP3 and AEAP3 concentrations up to 45 mol% without aggregation, resulting in concomitant increases in t[NO] and [NO]m of 3.77 and 1.66 μmol·mg−1, and 21700 and 1300 ppb·mg−1, respectively (Table 2 and Supporting Information).
In the previous post-formation synthesis of NO-releasing nanoparticles,25 the utility of TMOS as a backbone silane was evaluated as a strategy for increasing the aminoalkoxysilane concentration as the rate of TMOS hydrolysis is faster than TEOS.50,51 Indeed, the accelerated hydrolysis and condensation reactions (i.e., rapid production and consumption of silanols) via TMOS enabled the use of aminoalkoxysilane sol concentrations up to 77 and 87 mol% for AHAP3 and AEAP3, respectively.25 However, elemental analysis indicated that the levels of aminoalkoxy ligands (Camine) in the formed silica particles were not proportional to the aminoalkoxysilane concentrations (mol%) in the starting sol (Table 4). For example, while Camine of 77 mol% AHAP3/TMOS silica prepared by the post-formation route was 1.72 μmol·mg−1,25 Camine for 45 mol% AHAP3/TEOS silica synthesized by the pre-formation method was 1.91 μmol·mg−1. The less efficient immobilization of AHAP3 via the post-formation approach is attributed to the divergent hydrolytic polycondensation rates for AHAP3 and TMOS.25 Others have reported previously importance of synchronizing the hydrolysis and condensation rates of bi- and multicomponent silane systems in preparing well-ordered, porous silica materials with homogeneous distribution of active functionalities.50–53
Table 4.
Nitrogen content (%N), amine concentration (Camine), N-diazeniumdiolate concentration (Cdiaz), and amine to N-diazeniumdiolate conversion efficiency (%Econv) of various NO-releasing silica nanoparticles with different tetraalkoxy-and aminoalkoxysilane precursors synthesized via a “pre-formation” strategy
| tetraalkoxy-silanea | aminoalkoxysilane
|
%Ne | Caminef (μmol·mg−1) | Cdiazg (μmol·mg−1) | %Econv h | |
|---|---|---|---|---|---|---|
| typeb,c | mol%d | |||||
| TEOS | AHAP3 | 45 | 5.34 | 1.91 | 1.89 | 99.0 |
| AEAP3 | 45 | 3.53 | 1.26 | 0.83 | 65.9 | |
| DET3 | 10 | 2.40 | 1.14 | 0.65 | 57.0 | |
| 35 | 4.66 | 2.22 | 1.50 | 67.6 | ||
| 40 | 6.32 | 3.01 | 1.78 | 59.1 | ||
| AHAM3 | 35 | 6.91 | 2.47 | 2.13 | 86.2 | |
| 45 | 8.62 | 3.08 | 2.68 | 87.0 | ||
| 55 | 10.37 | 3.70 | 3.24 | 87.6 | ||
| 65 | 13.06 | 4.66 | 3.89 | 83.5 | ||
| 75 | 18.14 | 6.48 | 5.60 | 86.4 | ||
| AEAUD3 | 45 | 3.95 | 1.41 | 0.92 | 65.2 | |
| TMOS | AHAP3 | 45 | 5.66 | 2.02 | 1.75 | 86.6 |
| 55 | 6.79 | 2.42 | 2.11 | 87.2 | ||
| DET3 | 40 | 10.73 | 5.11 | 3.13 | 61.3 | |
| AHAM3 | 65 | 14.87 | 5.31 | 4.67 | 87.9 | |
| 75 | 18.22 | 6.51 | 5.63 | 86.5 | ||
| TMOS | AHAP3i | 77 | 4.81 | 1.72 | 0.89 | 51.7 |
| AEAP3i | 87 | 3.22 | 1.15 | 0.17 | 14.8 | |
TEOS, tetraethoxysilane; TMOS, tetramethoxysilane.
AHAP3, N-(6-aminohexyl)amino-propyltrimethoxysilane; AEAP3, N-(2-aminoethyl)-3-aminopropyltrimethoxysilane; DET3, (3-trimethoxysilyl)diethylenetriamine; AHAM3, N-(6-aminohexyl)aminomethyltrimethoxysilane; AEAUD3, N-(2-aminoethyl)-11-aminoundecyltrimethoxysilane.
Aminoalkoxysilanes employed consist of one primary and one secondary amines except DET3, which contains one primary and two secondary amines.
Balance TEOS or TMOS.
Obtained by elemental analyses within 0.3% error.
Amine concentration (Camine) is the concentration of secondary amines.
Determined by measuring the total NO concentration (t[NO]) released from the particles using a chemiluminescence nitric oxide analyzer in deoxygenated PBS (0.01 M, pH 7.4) at 37 °C.
Econv = Cdiaz/Camine.
Ref. 25. N-Diazeniumdiolate-modified silica nanoparticles were prepared via a “post-formation” method.
Silica nanoparticles prepared using N-diazeniumdiolated AHAM3 exhibited unprecedented NO storage capacity (up to t[NO] = 11.20μmol·mg−1), surpassing all macromolecular NO donors reported to date.16–25 The use of AHAM3 allowed for the synthesis of silica with N-diazeniumdiolated AHAM3 concentrations up to 75 mol% while maintaining high amine ligand immobilization efficiencies up to Camine = 6.48 μmol·mg−1 (see Tables 2 and 4). As expected, increasing the mol% of AHAM3 from 35 to 75 mol% (balance TEOS) led to a corresponding increase in the NO release characteristics (e.g., t[NO] increased from 4.25 to 11.20μmol·mg−1). The NO release from AHAM3-based silica nanoparticles was characterized by a significantly initial burst of NO ([NO]m = 106000 – 321000 ppb·mg−1) and short NO release half-life (~4 min). Such bolus release of NO indicated that AHAM3 silica represents a nanoparticle system capable of delivering the micromolar levels of NO required to serve as an antibacterial or anti-cancer agent.6–10 In contrast, N-diazeniumdiolated DET3 (TMOS) silica may prove beneficial as a vasodilatory agent where pico- to nanomolar levels of NO are required for extended periods (e.g., days). For example, t[NO], [NO]m, t1/2, and td for 40 mol% DET3 are 6.25 μmol·mg−1, 12400 ppb·mg−1, 95 min, and 101 h, respectively. Studies evaluating the effects of nanoparticles that release NO at different rates and amounts are currently underway for multiple pharmacological indicators.
The NO release characteristics of N-diazeniumdiolate-modified silica nanoparticles prepared via the pre-formation strategy are significantly expanded compared to both small molecule N-diazeniumdiolates and the silica prepared by the post-formation method. The greatest t[NO], 11.26 μmol·mg−1, was achieved with 75 mol% AHAM3 (TMOS) silica. Such NO release is a concentration roughly 6 times greater than that achieved with 77 mol% AHAP3 (TMOS) silica (t[NO] = 1.78 μmol·mg−1) prepared by the post-formation manner.25 The greatest td was 101 h for 40 mol% DET (TMOS) silica, a value significantly longer than the previous maximum (td = 15 h for 17 mol% AEAP3 (TEOS) synthesized by post-formation).
Macromolecular NO Release Kinetics
The “apparent” reaction orders (n) of the N-diazeniumdiolate-modified silica nanoparticles were determined under physiological conditions from the slope of the lnυ – lnCdiaz plots fitted with a linear regression (R2 ≥ 0.995).24 The term “apparent” refers to the reaction orders of compounds (i.e., silica) having limited solubility in PBS (heterogeneous suspensions) and therefore not exhibiting classical homogeneous kinetic behavior.24 As illustrated in Figure 7A, the “apparent” n for 35 mol% N-diazeniumdiolated AHAM3 silica nanoparticles was calculated to be 1.61, a significant deviation from 1, indicating the dissociation of N-diazeniumdiolates immobilized in the silica scaffold departs from the typical first-order kinetics. The considerable deviation from the fitting line observed at the initial dissociation events is attributed to water uptake/hydration of the particles in aqueous solution.24 The increased reaction order may be attributed to elevated local pH. As N-diazeniumdiolate dissociation proceeds, free secondary amines are generated that concomitantly increase the pH of the particles. As would be expected, the “apparent” reaction orders of the silica nanoparticles increased from 1.61 to 1.94 (Figure 7B) upon increasing the concentration of N-diazeniumdiolate-modified AHAM3 silanes from 35 to 75 mol%, respectively.
Figure 7.

(A) Dissociation kinetic plot of ln υ – lnCdiaz for 35 mol% N-diazeniumdiolated AHAM3 silica particles (balance TEOS) in deoxygenated PBS (0.01 M, pH 7.4) at 37 °C. The fitting line (dashed red) was determined from a linear regression of the experimental data (solid black). (B) Plot of dissociation reaction order (n) versus N-diazeniumdiolated AHAM3 concentration loaded during the silica polymerization.
Conclusions
The synthesis of NO-releasing silica nanoparticles represents an important step toward developing NO storage and delivery scaffolds for pharmacological applications. In contrast to the previous post-formation method25 in which the amine-functionalized silica particles were first synthesized and then reacted with NO to form N-diazeniumdiolate NO donor-modified silica, N-diazeniumdiolate-modified silane monomers were synthesized prior to particle formation. Such “pre-formation” approach enables the synthesis of nanoparticles with greater concentrations of aminoalkoxysilanes, and thus remarkably improved NO storage and release capabilities. As demonstrated herein, control over both the structure and concentration of N-diazeniumdiolate-modified aminoalkoxy- and tetraalkoxysilane precursors allows for the preparation of NO delivery silica scaffolds of variable sizes (d = 21 – 161 nm) and NO release properties (e.g., NO payloads of 0.50 – 11.26μmol·mg−1, maximum NO concentrations of 400 – 357000 ppb·mg−1, half-lives of 3 – 253 min, and NO release durations of 6 – 101 h). Silica nanoparticles prepared via the pre-formation approach exhibit both increased NO payload and NO release duration up to 6.3 and 6.7 times greater, respectively, than silica synthesized via post-formation method.25 Future studies aim to label silica nanoparticles with fluorescent dyes such as fluorescein isothiocyanate (FITC) to enable particle imaging/tracking.
Supplementary Material
Acknowledgments
The authors thank Prof. George Roberts of North Carolina State University for surface area/pore size analysis of the particles. The helpful discussion and technical assistance of Marc ter Horst of the University of North Carolina at Chapel Hill are gratefully acknowledged. This research was supported by the National Institutes of Health (NIH EB000708) and the Carolina Center of Cancer Nanotechnology Excellence.
Footnotes
Supporting Information Available: Reaction scheme of N-diazeniumdiolate formation and NO release properties of silica particles with different concentrations of AHAP3 prepared via the post- and pre-formation strategies (PDF). This material is available free of charge via the Internet http://pubs.acs.org.
References
- 1.Butler AR, Nicholson R. Life, Death and Nitric Oxide. Royal Society of Chemistry; Cambridge: 2003. [Google Scholar]
- 2.Ignarro LJ. Nitric Oxide: Biology and Pathobiology. Academic Press; San Diego: 2000. [Google Scholar]
- 3.Ignarro LJ, Bugga GM, Wood KS, Byrns RE, Chaudhuri G. Proc Natl Acad Sci USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moncada S, Higgs A. N Engl J Med. 1993;30:2002–2011. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 5.Radomski MW, Palmer RMJ, Moncada S. Br J Pharmacol. 1987;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albina JE, Reichner JS. Canc Metas Rev. 1998;17:19–53. doi: 10.1023/a:1005904704618. [DOI] [PubMed] [Google Scholar]
- 7.Nablo BJ, Chen T-Y, Schoenfisch MH. J Am Chem Soc. 2001;123:9712–9713. doi: 10.1021/ja0165077. [DOI] [PubMed] [Google Scholar]
- 8.Cobbs CS, Brenman JE, Aldape KD, Bredt DS, Israel MA. Cancer Res. 1995;55:727–730. [PubMed] [Google Scholar]
- 9.Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S. Proc Natl Acad Sci USA. 1995;92:4392–4396. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomsen LL, Miles DW, Happerfield L, Bobrow LG, Knowles RG, Moncada S. Br J Cancer. 1995;72:41–44. doi: 10.1038/bjc.1995.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135–1154. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 12.Napoli C, Ignarro LJ. Annu Rev Pharmacol Toxicol. 2003;43:97–123. doi: 10.1146/annurev.pharmtox.43.100901.140226. [DOI] [PubMed] [Google Scholar]
- 13.Keefer LK. Annu Rev Pharmacol Toxicol. 2003;43:585–607. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]
- 14.Wang PG, Cai TB, Taniguchi N. Nitric Oxide Donors: For Pharmaceutical and Biological Applications. Wiley-VCH; Weinheim, German: 2005. [Google Scholar]
- 15.Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, Janczuk AJ. Chem Rev. 2002;102:1091–1134. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 16.Hrabie JA, Saavedra JE, Roller PP, Southan GJ, Keefer LK. Bioconjugate Chem. 1999;10:838–842. doi: 10.1021/bc990035s. [DOI] [PubMed] [Google Scholar]
- 17.Jeh HS, Lu S, George SC. J Microencapsulation. 2004;21:3–13. doi: 10.1080/02652040310001619767. [DOI] [PubMed] [Google Scholar]
- 18.Pulfer SK, Ott D, Smith DJ. J Biomed Mater Res. 1997;37:182–189. doi: 10.1002/(sici)1097-4636(199711)37:2<182::aid-jbm6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 19.Parzuchowski PG, Frost MC, Meyerhoff ME. J Am Chem Soc. 2002;124:12182–12191. doi: 10.1021/ja020268l. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Z, Meyerhoff ME. Biomacromolecules. 2005;6:780–789. doi: 10.1021/bm049462l. [DOI] [PubMed] [Google Scholar]
- 21.Rothrock AR, Donkers RL, Schoenfisch MH. J Am Chem Soc. 2005;127:9362–9363. doi: 10.1021/ja052027u. [DOI] [PubMed] [Google Scholar]
- 22.Polizzi MA, Stasko NA, Schoenfisch MH. Langmuir. 2007;23:4938–4943. doi: 10.1021/la0633841. [DOI] [PubMed] [Google Scholar]
- 23.Stasko NA, Schoenfisch MH. J Am Chem Soc. 2006;128:8265–8271. doi: 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Annich GM, Miskulin J, Stankiewicz K, Osterholzer K, Merz SI, Bartlett RH, Meyerhoff ME. J Am Chem Soc. 2003;125:5015–5024. doi: 10.1021/ja0291538. [DOI] [PubMed] [Google Scholar]
- 25.Shin JH, Metzger SK, Schoenfisch MH. J Am Chem Soc. 2007;129:4612–4619. doi: 10.1021/ja0674338. [DOI] [PubMed] [Google Scholar]
- 26.Kumar CSSR, Hormes J, Leuschner C. Nanofabrication Towards Biomedical Applications: Techniques, Tools, Applications, and Impact. Wiley-VCH; Weinheim, German: 2005. [Google Scholar]
- 27.Lai C-Y, Trewyn BG, Jeftinija DM, Jeftinija K, Xu S, Jeftinija S, Lin VS-Y. J Am Chem Soc. 2003;125:4451–4459. doi: 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]
- 28.Munoz B, Ramila A, Perez-Pariente J, Diaz I, Vallet-Regi M. Chem Mater. 2003;15:500–503. [Google Scholar]
- 29.Roy I, Ohulchanskyy TY, Bharali DJ, Pudavar HE, Mistretta RA, Kaur N, Prasad PN. Proc Natl Acad Sci USA. 2005;102:279–284. doi: 10.1073/pnas.0408039101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trewyn BG, Whitman CM, Lin VS-Y. Nano Lett. 2004;4:2139–2143. [Google Scholar]
- 31.Yoshitake H. New J Chem. 2005;29:1107–1117. [Google Scholar]
- 32.Kim JS, Yoon TJ, Yu KN, Kim BG, Park SJ, Kim HW, Lee KH, Park SB, Lee JK, Cho MH. Toxicol Sci. 2006;89:338–347. doi: 10.1093/toxsci/kfj027. [DOI] [PubMed] [Google Scholar]
- 33.Kneuer C, Sameti M, Haltner EG, Schiestel T, Schirra H, Schmidt H, Lehr CM. Int J Pharm. 2000;196:257–261. doi: 10.1016/s0378-5173(99)00435-4. [DOI] [PubMed] [Google Scholar]
- 34.Sayari A, Hamoudi S. Chem Mater. 2001;13:3151–3168. [Google Scholar]
- 35.Stein A, Melde BJ, Schroden RC. Adv Mater. 2000;12:1403–1419. [Google Scholar]
- 36.McKittrick MW, Jones CW. Chem Mater. 2003;15:1132–1139. [Google Scholar]
- 37.Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 38.Davies KM, Wink DA, Saavedra JE, Keefer LK. J Am Chem Soc. 2001;123:5473–5481. doi: 10.1021/ja002899q. [DOI] [PubMed] [Google Scholar]
- 39.Konter J, Abuo-Rahma GE-DAA, El-Eman A, Lehmann J. Eur J Org Chem. 2007:616–624. [Google Scholar]
- 40.Hrabie JA, Klose JR, Wink DA, Keefer LK. J Org Chem. 1993;58:1472–1476. [Google Scholar]
- 41.Theoretically determined values obtained from ScienceFinder 2007.
- 42.Chithrani BD, Ghazani AA, Chen WCW. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 43.Schwartzberg AM, Olson TY, Talley CE, Zhang JZ. J Phys Chem B. 2006;110:19935–19944. doi: 10.1021/jp062136a. [DOI] [PubMed] [Google Scholar]
- 44.Albert K, Bayer E. J Chromatogr. 1991;544:345–370. doi: 10.1016/s0021-9673(01)95957-6. [DOI] [PubMed] [Google Scholar]
- 45.Huh S, Wiench JW, Yoo J-C, Pruski M, Lin VS-Y. Chem Mater. 2003;15:4247–4256. [Google Scholar]
- 46.Radu DR, Lai C-Y, Wiench JW, Pruski M, Lin VS-Y. J Am Chem Soc. 2004;126:1640–1641. doi: 10.1021/ja038222v. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez SA, Colon LA. Chem Mater. 1999;11:754–762. [Google Scholar]
- 48.Armelao L, Gross S, Muller K, Pace G, Tondello E, Tsetsgee O, Zattin A. Chem Mater. 2006;18:6019–6030. [Google Scholar]
- 49.Bruch MD, Fatunmbi HO. J Chromatogr A. 2003;1021:61–70. doi: 10.1016/j.chroma.2003.08.093. [DOI] [PubMed] [Google Scholar]
- 50.Lim MH, Stein A. Chem Mater. 1999;11:3285–3295. [Google Scholar]
- 51.Wight AP, Davis ME. Chem Rev. 2002;102:3589–3614. doi: 10.1021/cr010334m. [DOI] [PubMed] [Google Scholar]
- 52.Pagliaro M, Ciriminna R, Man MWC, Campestrini S. J Phys Chem B. 2006;110:1976–1988. doi: 10.1021/jp055697v. [DOI] [PubMed] [Google Scholar]
- 53.Melero JA, van Grieken R, Morales G. Chem Rev. 2006;106:3790–3812. doi: 10.1021/cr050994h. [DOI] [PubMed] [Google Scholar]
- 54.Liu J, Yang Q, Kapoor MP, Setoyama N, Inagaki S, Yang J, Zhang L. J Phys Chem B. 2005;109:12250–12256. doi: 10.1021/jp0509109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.