

Abstract
The human nuclear xenobiotic receptor PXR recognizes a range of potentially harmful drugs and endobiotic chemicals, but must complex with the nuclear receptor RXRα to control the expression of numerous drug metabolism genes. To date, the structural basis and functional consequences of this interaction have remained unclear. Here we present 2.8 Å resolution crystal structures of the heterodimeric complex formed between the ligand binding domains (LBDs) of human PXR and RXRα. These structures establish that PXR and RXRα form a heterotetramer unprecedented in the nuclear receptor family of ligand-regulated transcription factors. We further show that both PXR and RXRα bind to the transcriptional coregulator SRC-1 with higher affinity when they are part of the PXR-RXRα heterotetramer complex than they do when each LBD is examined alone. Furthermore, we purify the full-length forms of each receptor from recombinant bacterial expression systems, and characterize their interactions with a range of direct and everted repeat DNA elements. Taken together, these data advance our understanding of PXR, the master regulator of drug metabolism gene expression in humans, in its functional partnership with RXRα.
Keywords: transcription factor, nuclear receptor, DNA, x-ray crystallography, CYP3A4
INTRODUCTION
The human biotransformation of xenobiotics is conducted by a large array of enzymes classified as being part of Phase I (functionalization), Phase II (conjugation), or Phase III (excretion) drug metabolism 1; 2; 3; 4; 5. These systems are clinically significant because they impact drug efficacy and can lead to drug interactions and toxicity. The nuclear pregnane x receptor (PXR) is a member of the nuclear receptor subfamily 1, group I, member 2 sub-section of ligand-dependent transcriptional factors 6; 7; 8. It is a master regulator of the expression of genes involved in xenobiotic and endobiotic metabolism, although it was originally identified as critical in detecting endogenous pregnanes 9. Since then, it has subsequently been classified as a central xenobiotic sensor that responds to a number of pharmaceutically relevant compounds and drugs. The promiscuity of PXR’s ligand binding has proven to be unique to this nuclear receptor 6; 10, and x-ray crystallographic studies 11; 12; 13 reveal a novel insert in the ligand binding domain (LBD), as well as a highly malleable ligand binding pocket. PXR was found to interact with SRC-1 (nuclear receptor coactivator 1) 9, a member of the p160 family of coactivators that binds in a ligand-dependent fashion to various nuclear receptors utilizing a Leu-x-x-Leu-Leu repeat (where x=any amino acid) 14. Importantly, like many former orphan nuclear receptors, PXR forms a complex with the retinoid X receptor-α (RXRα) and binds to DNA response elements to control gene expression 6; 10. RXRα appears to respond to retinoid ligands including 9-cis-retinoic acid and all-trans retinoic acid 15.
A number of PXR LBD crystal structures have been resolved to date in complex with a range of distinct ligands 12; 13; 16; 17; 18; 19. Crystal structures of RXRα LBD alone, in liganded complexes, and in heterodimeric complexes with other nuclear receptor LBD’s, such as LXR, CAR, PPARγ, RAR, and others 20; 21; 22; 23; 24 have also been reported. However, only one crystal structure of a full-length, intact nuclear receptor complex (PPARγ/RXRα) has been presented, which revealed a great deal about the orientation of the DNA and DNA-binding domains (DBDs) of the heterodimeric complex relative to the LBDs 25. In contrast to most nuclear receptors, PXR was shown to form a functionally essential homodimer not seen previously for this superfamily of transcriptional factors 26. It was established in vitro and cell-based studies that the PXR homodimer interface impacted the recruitment of the steroid receptor coactivator-1 (SRC-1), but did not affect ligand binding, DNA binding or subcellular localization 27. SRC-1, also termed nuclear receptor coactivator-1 (NCOA1), enhances transcription by serving as a histone acetyltransferase and by recruiting the basal machinery 14. SRC-1 interacts with nuclear receptors via domains with multiple leucine (L) residues arranged in LxxLL motifs 28.
To date, the three-dimensional structures of PXR have included only PXR, so it remained unclear whether the presence of RXRα would impact PXR’s unique homodimer complex. Further understanding of how PXR regulates xenobiotic metabolism requires a more in-depth knowledge of the interactions made between PXR and its binding partner RXRα, and how this nuclear receptor complex interacts with various hormone response elements through their DBD. Thus, we elucidated crystal structures of the PXR/RXRα LBD heterotetramer complex in the presence and absence of PXR ligands. In addition, we conducted in vitro studies to determine SRC-1 coactivator binding affinities for the individual nuclear receptors and for the receptors in complex with one another. Finally, methods for the expression and purification of appreciable quantities of full-length PXR and RXRα are provided, along with their DNA binding characteristics.
RESULTS
Crystal Structures of the PXR/RXRα LBD Complex
Structures of the apo-PXR/RXRα and SR12813-bound PXR/RXRα LBD complexes were determined to 2.8 Å resolution (Table 1). These complexes reveal a heterotetramer in the asymmetric unit composed of two PXR and two RXRα LBD monomers (Figure 1A). The two PXR monomers form a homodimer, which has been seen in previous PXR-only LBD structures, and two RXRα monomers are bound to PXR, one per monomer. Both PXR and RXRα have been fused at their C-termini to the following region of human steroid receptor coactivator-1 (SRC-1): 678-SSHSSLTERHKILHRLLQEGSPS-700, with the LxxLL motif in bold. In PXR, the region corresponding to 681-698 of hSRC-1 is ordered, and is numbered 443-460; in RXRα, the region corresponding to 686-696 of hSRC-1 is ordered, and is numbered 476-486. The PXR residues 130-140, 178-192, 312-314 are disordered in both structures; in addition, the non-native linker-to-SRC-1 peptide residues 433-440 are also disordered. The RXRα LBD residues 246-260 are disordered in the apo-structure, but ordered in the SR12813-bound structure; similar to the PXR LBD, the non-native linker-to-SRC-1 residues 458-476 are disordered in RXRα LBD structure. The SR12813-bound LBD complex contains one conformation of the SR12813 molecule in each PXR ligand binding pocket (Figure 1B). Residues lining the binding pocket located within 4 Å of the SR12813 molecule are L206, L209, L240, M243, M246, S257, Q285, F288, W299, Y306, M323, H327, H407, R410, L411, I414, and M425. These residues have been established as common amino acids that surround bound ligands in the PXR LBD 29. Importantly, the structure confirms the PXR-PXR homodimer interface observed in PXR LBD-only structures resolved to date, formed by the β1 strands of each PXR LBD monomer (Figure 1A). Note that β1 is located on a ~50-residue insert unique to the PXRs relative to other nuclear receptors 27. Thus, these structures establish that PXR and RXRα form a dimer of PXR-RXRα heterodimers, creating a crystallographically observed heterotetramer not observed previously for members of the nuclear receptor family of transcription factors.
Table 1.
Crystallographic Statistics
| Data Collection | ||
|---|---|---|
| Apo-PXR/RXR LBD Complex | SR12813-bound PXR/RXR LBD Complex | |
| X-ray source | APS GM/CA-CAT 23-ID | |
| Space group | P212121 | P212121 |
| Unit cell: a, b, c (Å); α=β=γ=90° | 70.26, 109.6, 169.9 | 70.09, 120.3, 175.8 |
| Resolution (Å) (highest shell) | 50.0-2.77 (2.84-2.77) | 50.0-2.80 (2.85-2.80) |
| I/σ | 4.70 (1.76) | 6.68 (2.34) |
| Rsym (highest shell) | 0.132 (0.321) | 0.144 (0.440) |
| Completeness (%) | 96.1 (86.8) | 99.4 (98.7) |
| Redundancy | 3.6 (2.4) | 3.9 (2.0) |
| Refinement | ||
| Resolution (Å) (highest shell) | 50.0-2.80 (2.85-2.80) | 50.0-2.80 (2.85-2.80) |
| No. of unique reflections | 31748 | 37160 |
| Rwork | 0.250 (0.310) | 0.245 (0.315) |
| Rfree | 0.298 (0.300) | 0.298 (0.345) |
| Molecules/asymmetric unit (AU) | 2 PXR; 2 RXRα | 2 PXR; 2 RXRα |
| No. of amino acids per AU | 1044 | 1063 |
| No. of waters per AU | 175 | 160 |
| Average B-factors | 35.4 | 54.3 |
| Macrolmolecule | 35.5 | 54.3 |
| Solvent | 29.4 | 47.2 |
| R.M.S. deviations | ||
| Bond lengths (Å) | 0.004 | 0.005 |
| Bond angles (°) | 0.880 | 1.00 |
| Ramachandran (%) | ||
| Favored/Allowed | 99.3 | 99.2 |
| Outliers | 0.69 | 0.77 |
Figure 1. Structure of the PXR-RXR LBD Complex.

A. 2.8 Å resolution structure of the complex formed between the ligand binding domains (LBDs) of human PXR (cyan, green) and RXRα (magenta, yellow), with the SRC-1 peptides bound to each LBD indicated. B. In the ligand bound complex of PXR and RXRα, the cholesterol-lowering compound SR12813 is contacted by the amino acid side chains indicated.
The PXR/RXRα LBD heterodimer interface buries ~1200 Å2 total surface area, and is composed of several electrostatic and polar interactions found on the four helices that make up the binding surface (Figure 2). Eleven probable electrostatic interactions (<4 Å) were observed: Q409-E434 (PXR residue-RXRα residue), E399-R426, H359-E352, H359-K356, D363-K356, D363-R421, Q366-R421, R360-D359, E367-R421, E367-K417, and K374-E401. In addition, six potential polar contacts were observed: S402-R426 (PXR residue-RXRα residue), E399-R426, H359-E352, D363-R421, K374-E401, and E367-K417. Furthermore, hydrophobic contacts were observed involving RXRα’s L420 and PXR’s I371, and M394. Finally, a number of water-mediated (R353-R348 [PXR residue-RXRα residue], R353-S427, R360-K356, D363-E352, D363-R421, R401-R348, and R401-S427), side chain-main chain (K325-D379), and intramolecular interactions (PXR: K332, S350, E378, T398, Q406, and R413; RXRα: T351, S355, N377, K381, R393, E394, K405, and K431) were noted that strengthen the dimer interface. Compared with RXRα heterodimer complexes resolved previously, the interface formed between PXR and RXRα is largely similar. For example, a comparison of the PXR-RXRα binding interface to the CAR-RXRα interface reveals similar electrostatic and polar contacts 22; 24. Furthermore, the RXRα LBDs is located in a similar position in its complexes with the LBDs of PXR, CAR 22; 24, PPARγ 21, LXR 23 and RAR 20, but some structural differences are evident (Figure 3). Notably, translations up to 6 Å and rotations up to 29° are observed between secondary structural elements of RXRα (Figure 4).
Figure 2. PXR-RXR LBD Complex Interface.

Regions of α5, α9 and α10 of human PXR (cyan) contact regions of α7, α9 and α10 of human RXRα (magenta) using the residues listed below. The specific residue-residue interactions are detailed with connecting lines (blue= electrostatic, red= polar, purple=electrostatic and/or polar, green= hydrophobic, black= side chain-main chain), an asterisk (*= water-mediated), and italics (italics= intramolecular).
Figure 3. Comparison of RXRα Heterodimers.

The LBD of PXR was superimposed on those of CAR, PPARγ, LXR and RAR, and the positions of the RXRα LBD in these heterodimers are shown and colored as indicated.
Figure 4. RXRα Positional Shifts.

Specific structural translations (indicated in Å) or rotations (in °) for secondary structural elements of the RXRα LBD from heterodimer complexes with PXR, CAR, PPARγ, LXR and RAR when the non- RXRα LBDs are superimposed (e.g., Fig. 3). Viewed rotated roughly 30° about the vertical axis relative to Figure 3.
SR12813-bound PXR/RXRα versus apo-PXR/RXRα
An overlay of the unliganded 30 form of PXR in the PXR/RXRα complex with the SR12813-bound complex reveals that, while the all-atom root-mean-square deviation (r.m.s.d.) is 1.49 Å over 998 atoms, a number of small changes in the secondary structural elements have occurred (Figure 5A). Notable shifts in secondary structural elements of PXR include residues 145-162 (1.5 Å), 193-209 (1.5 Å), 211-230 (1.4 Å), 241-260 (1.0 Å), 270-289 (1.4 Å), and 360-375 (1.4 Å) (Figure 5B). Furthermore, shifts are noted in the RXRα LBDs, including 233-242 (1.1 Å), 264-274 (2.7 Å), 275-285 (1.5 Å), 294-318 (1.6 Å), 333-341 (4.0 Å), 343-360 (1.8 Å), 387-396 (1.3 Å), 430-441 (1.5 Å), and 450-456 (2.7 Å). The addition of a ligand to PXR facilitates the structural ordering of the entire complex, as evident by the now ordered RXRα residues 246-257. These residues form a helical motif on each end of the complex, leading to a slightly larger unit cell volume (SR12813/Apo volume ratio of 1.13; Figure 5A, Table 1). Interestingly, comparing the same residues from PXR in each complex shows that the addition of the SR12813 molecule compresses the overall structure of the ligand-bound complex relative the apo complex. For example, the length of the SR12813-PXR heterotetramer complex (vertical in Figure 5A) is ~4 Å less than the length of the corresponding apo-PXR heterotetramer (apo≈130 Å; SR12813≈126 Å). This observation supports the general belief that the binding of activating ligands serves to stabilize nuclear receptor complexes in a manner similar to the addition of coactivator fragments 11; 29. In the structures presented here, though, the mean thermal displacement parameter (B-factor) for the ligand-bound complex (54 Å2) is higher than the same value for the apo complex (35 Å2) (Table 1), while the overall Wilson B-factors on the data show the opposite trend, 37 Å2 for ligand bound and 43 Å2 for apo (data not shown). This may be a product of the 23 additional residues modeled in the SR12813-bound complex structure.
Figure 5. Comparison of the Apo and SR12813 Bound PXR-RXR Complexes.

A. Superposition of the unliganded 30 human PXR homodimer from the PXR-RXRα LBD complex (cyan) with the structure of the human PXR homodimer from the PXR-RXRα LBD complex in which PXR contains bound SR12813 (magenta) (viewed ~90 degrees rotated about the vertical relative to Figure 1A). B. Rotational and translational shifts in position between of the PXR LBDs (cyan, magenta) and the RXRα LBDs (green, yellow) in the apo and SR12813-bound PXR complexes.
Nuclear Receptor Oligomerization and Coactivator Binding
It had been previously shown that the PXR LBD contacts 15 residues of the LxxLL motif of SRC-1 11, which is approximately five additional amino acids relative to other NR LBDs crystal structures examined. The structures reported here confirm the previous data, with one turn of α-helix preceding the LxxLL helices of SRC-1 in PXR relative to RXRα (Figure 6A). In both cases, though, the LxxLL motifs of SRC-1 were fused to the C-termini of the LBDs to improve purification yield. We next purified PXR and RXRα LBDs without fused LxxLL motifs to examine the impact that nuclear receptor LBD oligomerization state plays in coactivator binding (see Experimental Procedures). Table 2 details the results of these experiments, where SRC-1 binding to each LBD is examined when the LBD is alone and when the LBD is in the PXR/RXRα heterotetramer complex. In all cases, no PXR or RXRα ligands were added to these studies. SRC-1 LxxLL peptide motif binding to the RXRα LBD alone exhibits a Kd of 1.56 μM (Table 2). In contrast, SRC-1 peptide binding to the RXRα LBD in the PXR/RXRα heterotetramer complex exhibits a Kd of 780 nM, which has a p-value of 0.005 when compared to the RXRα LBD alone (Table 2). Similarly, SRC-1 peptide binding to the PXR LBD alone exhibits a Kd of 1.18 μM (Table 2). Again, in contrast, SRC-1 peptide binding to the PXR LBD in the PXR/RXRα heterotetramer complex exhibits a Kd of 588 nM (Figure 6B), which has a p-value of 0.009 when compared to the PXR LBD alone (Table 2). Indeed, when comparing the Kd values of SRC-1 binding to the PXR LBD versus the RXRα LBD in the PXR/RXRα heterotetramer complex, they are statistically different with a p-value of 0.01 (Table 2). Thus, we conclude that the PXR LBD exhibits a higher affinity for the SRC-1 LxxLL motif employed relative to the RXRα LBD. In addition, these data indicate that both PXR and RXRα have significantly higher affinity for the coactivator SRC-1 when the LBDs are part of the PXR/RXRα heterotetramer complex, confirming the previous suggestion 26 that the higher-order PXR/RXRα complex structure likely impacts the control of gene expression by PXR via improved coactivator binding.
Figure 6. SRC-1-LBD Binding Interfaces.

A. Ordered regions of the transcriptional coactivators SRC-1 (orange, yellow) observed in contact with the LBDs of PXR (cyan) and RXRα (magenta). Note that the SRC-1 peptides were fused using a glycine-serine linker to the C-termini of each LBD, and are thus labeled with the corresponding residues within each construct (see Methods). The following region of human steroid receptor coactivator-1 (SRC-1): 678-SSHSSLTERHKILHRLLQEGSPS-700, with the LxxLL motif in bold, was linked to each receptor. In PXR, the region corresponding to 681-698 of hSRC-1 is ordered, and is numbered 443-460; in RXRα, the region corresponding to 686-696 of hSRC-1 is ordered, and is numbered 476-486 B. Binding curve of SRC-1 peptide binding (residues 676-700) to the PXR LBD, measured by fluorescence anisotropy. In this example, the LBD complex consists of the unbound and unfused version of the PXR LBD and the SRC-1-fused RXRα LBD. This creates a scenario that allows for free SRC-1 peptide to bind only the LxxLL motif in PXR.
Table 2.
Peptide Binding by Fluorescence Anisotropy
| SRC1 Peptide Binding | ||
|---|---|---|
| LBD(s) Employed | Kd (nM) | p-values |
| RXRα | 1560 nM ± 198 * | *0.005 |
| RXRα/PXR–SRC1 | 778 nM ± 88.8 *, ^ | |
| PXR | 1180 nM ± 92.2 # | ^ 0.01 |
| PXR/RXRα–SRC1 | 588 nM ± 29.7 #, ^ | # 0.009 |
Recombinant Full-Length PXR/RXRα Complex Purification
To our knowledge, the purification of intact PXR including the DNA-binding domain, linker region, and ligand-binding domain (DBD-linker-LBD, or “full-length”) in quantities sufficient for structural or biochemical analysis has not been reported. We successfully purified the complex of full-length human PXR (residues 1-434) and human RXRα (residues 1-462). Each protein was expressed with a hexa-histidine tag in bacterial cells using codon-optimized constructs; human PXR also contained a maltose-binding protein fusion to increase solubility during the initial stages of purification. Both proteins required the presence of ligand (SR12813 and 9-cis-retinoic acid), and a linker with an LxxLL motif located on the C-terminus of the LBD, which as we have shown produces a stabilized AF-2 motif (Figure 6A). In the absence of this linked coactivator motif, PXR, in particular, was prone to aggregation. The proteins were purified separately through affinity chromatography and gel filtration, and then combined to form a complex prior to the proteolytic removal of PXR’s MBD fusion protein. At that point, the PXR/RXRα complex was purified from a size-exclusion complex at an elution volume indicative of the ~210 kDa heterotetramer state. We obtained ~40 mgs of >95% pure full-length PXR/RXRα complex from 18 L of bacterial expression media for PXR, and 6 L of RXRα. Thus, we were able to create the physiologically relevant state of human PXR and RXRα useful for in vitro studies such as DNA binding and structural characterization. Unfortunately, because the linked coactivator motifs appear critical for expression of soluble PXR, the current states of these receptors are not amenable to coregulator binding studies like those conducted for the LBD complex (Table 2).
Full-Length PXR/RXRα DNA Binding
The full-length PXR/RXRα complex was employed in fluorescence-based DNA binding assays for in vitro characterization of various pieces of duplex DNA to this important nuclear receptor assembly. The duplex DNAs examined are shown in Table 3, and include two fluorescently-labeled minimum response element sequences (DR3 FAM and ER6 FAM) and several unlabeled DNA competitors, with flanking sequences derived from the promoter region of CYP3A4. Dissociation constant values (Kd) were first measured for the direct binding of the fluorescently DR3 FAM (Figure 7A) and ER6 FAM probes, and were found to be 206 ± 53 nM and 215 ± 36 nM, respectively (Table 4). These initial results indicate that the receptors purified recombinantly in bacteria are functionally competent to bind DNA, and that they are able to associate with both the direct (DR) and everted (ER) repeat oligos derived from known PXR/RXR response elements.
Figure 7. DNA Binding by PXR-RXRα.

A. Binding measured by fluorescence anisotropy of the complex of full-length PXR with full-length RXRα to a FAM-labeled 15-bp DNA duplex composed of a DR3 motif from the promoter of the human CYP3A4 gene product (see Tables 3, 4). B. Competition binding experiments using fluorescence anisotropy and monitoring the dissociation of the FAM-DR3 DNA duplex by increasing concentrations of unlabeled duplexes (see Tables 3, 4).
Table 4.
DNA Binding by Fluorescence Anisotropy
| DR3 Direct Binding | Kd (nM) | ER6 Direct Binding | Kd (nM) | |
|---|---|---|---|---|
| DR3 FAM | 206 ± 53 | ER6 FAM | 215 ± 36 | |
| DR3 Competitors | Ki (nM) | ER6 Competitors | Ki (nM) | p-value |
| DR3 +9/+9 | 86.5 ± 4.5 | ER6 +9/+9 | 94.8 ± 4.8 | 0.150 |
| DR3 +6/+9 | 94.4 ± 4.0 | ER6 +6/+9 | 98.1 ± 4.7 | 0.149 |
| DR3 +3/+9 | 87.2 ± 4.5 | ER6 +3/+9 | 103 ± 5.1 | 0.038* |
| DR3 +0/+9 | 131 ± 6.6 | ER6 +0/+9 | 113 ± 6.2 | 0.098 |
| DR3 +9/+6 | 88.3 ± 4.2 | ER6 +9/+6 | 96.6 ± 4.6 | 0.159 |
| DR3 +9/+3 | 82.8 ± 3.6 | ER6 +9/+3 | 104 ± 4.7 | 0.013* |
| DR3 +9/+0 | 95.6 ± 4.7 | ER6 +9/+0 | 98.4 ± 4.8 | 0.282 |
| DR3 +6/+6 | 98.0 ± 4.9 | ER6 +6/+6 | 120 ± 5.5 | 0.028* |
| DR3 +3/+3 | 95.6 ± 5.1 | ER6 +3/+3 | 131 ± 5.8 | 0.002* |
| DR3 −2/+9 | 144 ± 6.0 | ER6 −2/+9 | 155 ± 7.0 | 0.317 |
| DR3 +9/−2 | 102 ± 5.2 | ER6 +9/−2 | 146 ± 7.2 | 0.024* |
| DR3 −2/−2 | N.C. | ER6 −2/−2 | N.C. | - |
Statistically significant (p<0.05); N.C. – not a competitor
Next, a set of competition binding experiments using DR3 and ER6 sequences with varying lengths of flanking bases were performed, and Ki values were determined for each duplex oligonucleotide by examining their ability to compete away the corresponding labeled fluorescence probe (Table 4; Figure 7B). We found that the addition of flanking bases improves DNA binding for both the DR3 and ER6 sequences, with the strongest Ki values observed for the +9/+9 sequences, of 86.5 and 94.8 nM, respectively. A similar phenomenon has been described previously for the PPARγ/RXRα nuclear receptor complex25; 31. Removing flanking bases from either side of the response element half-sites has little effect on the DNA binding, as evidenced by competitors DR3 +9/+0 and DR3 +0/+9 (Table 4). This observation may be due to the strong binding by the receptor DBDs to the intact side of the DNA competitor, resulting in reduced anisotropy. However, the weakest DNA binding is observed when there are no flanking bases on either side (denoted by the +0); furthermore, binding is significantly hindered upon removing DNA bases from the response element sequence, and removal from both the 5′ and 3′ ends results in complete loss of binding (e.g., see competitors DR3 −2/−2 and ER6 −2/−2) (Table 4; Figure 7B). Importantly, we noted that DNA binding is strongest for the DR3 sequences relative to the ER6 sequences (Table 4). The addition of 6 to 9 DNA bases on either side of the HRE (+9/+9, +9/+6, +6/+9) yields similar binding affinities for both the DR3 and ER6 oligonucleotides, as evidenced by statistically non-significant P-values (Table 4). However, scaling back to only 3 additional bases (+9/+3, +3/+9) on either side proves to be more detrimental to ER6 binding versus DR3, as suggested by the significant difference in Ki values (Table 4). These observations suggest the extended spacer region between the half-sites negatively impacts DNA association for some oligonucleotides, such as those flanked by +3/+3 and +6/+6 duplex regions. Taken together, these data provide insights into the DNA binding characteristics of the full-length PXR/RXRα complex of human nuclear receptors.
DISCUSSION
PXR is the master regulator of the expression of drug and endobiotic metabolism genes in mammals and exhibits a distinctly promiscuous ability to bind to a wide variety of agonists. In this way, PXR is unique in function relative to other members of the nuclear receptor family of ligand-regulated transcription factors, of which it is a member. The PXR LBD also contains a ~50-residue insert unique to PXR relative to even closely related nuclear receptors, such as VDR, FXR and LXR. This insert encodes an additional helix and two β-strands with a tryptophan-zipper motif that creates a homodimer interface also unique to PXR. It was previously shown that the tryptophan and tyrosine residues that lock across this zipper are necessary for PXR LBD homodimerization and transcriptional activation, but do not impact the subcellular localization of PXR, or ligand and DNA association by the receptor 27. The structure of PXR in complex with its obligate heterodimer partner RXRα had not been resolved until this study.
The crystal structures of the PXR/RXRα LBD complex presented here establish that the PXR homodimer, formed by a ten-stranded intermolecular β-sheet formed between the PXR LBD monomers, is maintained in the context of bound RXRα (Figures 1–3). This structure also reveals unique aspects to the higher-order PXR/RXRα LBD complex, particularly the presence of a curved pocket, ~24 Å wide and ~15 Å deep, formed between the two PXR LBD monomers (Figure 8). The amino acid composition on this surface is highlighted in Figure 8A, and we note that it is located ~35 Å from the coactivator LxxLL motifs bound to the AF-2 sites of each PXR monomer. Charged residues, including D230, K210, D310, E309, K226, E300, R216, R303, E218, and D219, and polar residues, including S231, T311, S212, T296, T298, Y225, Y306, S305, Q214, N224, and S221, create this curved surface.
Figure 8. Surface of the PXR Homodimer.
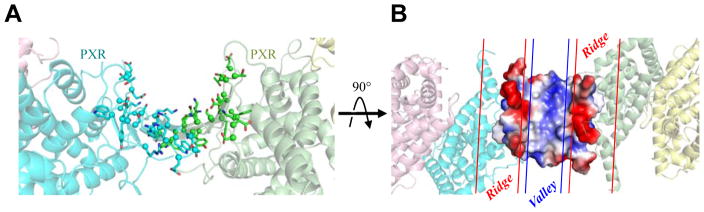
A. Ribbon representation of the homodimer of the human PXR LBDs in the PXR-RXRα heterotetramer complex reported here, with surface exposed residue shown as sticks (viewed in the same orientation as Figure 3A). B. Electrostatic surface of the homodimer interface shown in panel A, with the regions of positive (blue) and negative (red) charge indicated.
Furthermore, electrostatic considerations demonstrate that this surface exhibits two negatively charged “ridge” regions on either side of one positively charged “valley” (Figure 8B). Residues that create this electrostatic feature (namely residues 211-230 and 298-310 that comprises the PXR β-sheet dimerization interface) are highly conserved in PXRs across species (NCBI BLASTp, inputting human PXR LBD residues 130-434) 26; 32. A concave, charged and conserved face suggests this region plays important role in PXR function, possibly by forming specific macromolecular interactions. For example, this intermolecular electrostatic surface may serve as a binding site for one of the PXR DNA-binding domains (DBDs). Alternatively, or in addition, this region may act as an interface for other proteins, such as the transcriptional coactivator SRC-1. Full-length SRC-1 contains multiple LxxLL motifs that are known to bind to various nuclear receptor AF-2 regions 33; 34. As such, it is interesting to speculate that the PXR homodimer binds to multiple LxxLL motifs of a single coactivator protein, and the intervening residues between these binding motifs may be passed through the cupped-β-sheet region to stabilize a higher-order PXR/SRC-1 complex. Further work will be required to test such a hypothesis, which is expected to be unique to PXR given that it is the only member of the NR superfamily that contains these inserted structural motifs.
We also demonstrate that PXR and RXRα each associate with transcriptional coactivator LxxLL motifs with higher affinity when they are part of the PXR/RXRα complex than when each receptor is in isolation (Table 2). The increase in binding affinity for coactivators in nuclear receptor complexes versus individual receptors has previously been noted for RXR, RAR, and CAR 35; 36. The increased binding affinity (Kd) was shown to be very significant by a P-value calculation for both PXR and RXRα versus the PXR/RXRα complex. Coactivator binding affinity is increased 2-fold for both nuclear receptors upon complex formation. Although there are no significant structural changes between the structures of individual nuclear receptors versus the complexed forms, it is clear that both nuclear receptors act in a cooperative fashion to greatly improve the mechanism of coregulator recruitment and binding. Furthermore, the presence of agonist ligands for PXR (SR12813) and RXRα (9-cis-retinoic acid) in excess acts to simultaneously stabilize complex formation and to increase SRC-1 binding, which has previously been noted 35; 36. Molecular dynamics investigations of PXR both alone and with RXRα, as well as other receptors in their physiologically-relevant complexes, have indicated that the AF-2 regions of nuclear receptors move in a correlated fashion when the LBDs are complexed to their appropriate partner, either as a homodimer or heterodimer with RXRα 16. The experimental results presented here are consistent with these computational results, and further support the conclusion that protein motion is a critical factor in function.
EXPERIMENTAL PROCEDURES
Cloning and Expression of the PXR/RXRα LBD Complex
The PXR LBD (residues 130-434) was PCR amplified from the full-length, codon-optimized PXR gene (synthesized by GenScript) fused to a 5-residue glycine-serine linker (GGSGG) and the human SRC-1 coactivator fragment (residues 678-700, SSHSSLTERHKILHRLLQEGSPS). This construct was previously used in successful crystallization studies for the PXR LBD 25. The PXR LBD construct was cloned into the HIS-LIC pMCSG vector (acquired from the Sondek Lab at UNC) for protein expression. RXRα LBD (residues 227-462) was PCR amplified from the full-length, codon-optimized RXRα gene fused to the same glycine-serine linker with the SRC-1 coactivator fragment (GenScript). The coding region for the RXRα LBD was placed into pET-30b (Invitrogen) expression vector without a His-tag fused to the protein. Terrific broth media was inoculated with a saturated culture of BL21 AI cells transformed with the HIS-LIC plasmid containing the PXR LBD construct and allowed to shake at 37 °C. When the cells reach an OD600 of ~1.0 the temperature was reduced to 16 °C, at which time L-arabinose was added to each flask up to a final concentration of 0.1% and IPTG was added (final concentration of 0.2 mM) to induce protein expression. After shaking overnight, cells were collected by centrifugation at 4,500×g for 20 minutes and stored at −80 °C. The growth and expression of RXRα LBD was carried out in a similar manner as the PXR LBD described above. When the RXRα LBD growth achieved an OD600 of ~1.0, the temperature was reduced to 16 °C. A final concentration of 0.1% L-arabinose was added when temperature reached ~16 °C, and expression was induced at 16 °C with a final concentration of 0.2 mM IPTG.
LBD Complex Formation and Purification
PXR LBD and RXRα LBD proteins were co-purified to encourage complex formation. In an approximate 3:1 PXR to RXRα cell ratio, to account for the higher expression of RXRα relative to PXR, cell pellets were separately resuspended in Buffer A (50 mM HEPES pH 7.5, 30 mM imidazole, 250 mM NaCl, 5% glycerol) supplemented with a protease inhibitor tablet (Roche), DNase, and lysozyme. After sonication, the cell lysate was clarified by high-speed centrifugation at 14,500×g for 50 min. The supernatant was removed and syringe filtered through a 0.22 um filter. PXR and RXRα LBD clarified cell lysates were then combined prior to further purification. Excess amount of PXR ligand was added to the lysate to help stabilize the protein throughout the purification process. Clarified lysate was loaded onto the Äktaxpress FPLC system (Amersham Biosciences) for a gradient-elution using Buffer B (50 mM HEPES pH 7.5, 300 mM imidazole, 250 mM NaCl, 5% glycerol) on Ni His-Trap (GE Healthcare) columns. Excess RXRα LBD protein was washed away at low concentrations of Buffer B during the gradient elution as this protein was untagged. Purest fractions containing PXR-RXRα complex (determined by SDS-PAGE) were combined and concentrated as much as possible without destabilizing the protein. While concentrating the PXR/RXRα LBD complex, additional ligand was added periodically to compensate for any ligand washed away during Ni-column purification. LBD complex was dialyzed overnight into a TEV cleavage/Gel filtration buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT, and 5% glycerol) in the presence of TEV protease to cleave the His-fusion tag from the PXR LBD. Samples were concentrated and loaded on the FPLC system for size exclusion purification using a HiLoad™ 16/60 Superdex™ 200 gel filtration column. The LBD complex was eluted in 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT, and 5% glycerol. Purity of each fraction was assessed by SDS-PAGE and >95% pure fractions were combined.
LBD Complex Crystallization, Data Collection, and Structure Solution
Purified PXR/RXRα LBD complex was shipped to the Hauptman-Woodward Medical Research Institute in Buffalo, NY to screen several crystallization conditions using their automated crystallization setup 37. An initial crystal hit was identified in 20% (w/v) PEG 8000, 0.1 M magnesium chloride, and 0.1 M bis-tris propane pH 7.0 at 4 °C. Crystallization optimization was carried out in-house, by grid optimization screens around the original hit condition. Varying precipitant concentration, salt concentration, and protein to crystallant drop ratio was successful in reproducing these crystals. Additional crystallization optimization experiments were performed to improve crystal growth, as initial crystals grew as 2D plates, with only a few crystals growing successfully in the 3rd dimension. Decoupling experiments 38 as well as alternating the salt anion (MgX, where X= acetate, formate, sulfate) were employed. Optimized crystals were harvested and streaked through mother liquor supplemented with 20% (v/v) glycerol to cryo-protect the crystals for X-ray data collection.
Crystals were shipped to GM/CA-CAT (Beamline 23) at APS for X-ray data collection. Data were collected in 0.2° oscillations for each crystal. Diffraction data were obtained on crystals of apo-PXR/RXRα LBD complex and SR12813-bound PXR/RXRα LBD complex. Data collection statistics are presented in Table 1. Data from both forms were indexed in the space group P212121. Cell constants for the apo-form were a=70.26, b=109.6, c=169.9; the SR12813-bound complex cell constants were a=70.09, b=120.3, c=175.8. XDS 39 was used to process the apo-data and HKL2000 40 was utilized for data reduction of the SR12813-bound data. The SR12813-bound LBD complex was solved by molecular replacement using Phaser 41 and ensemble search methods using monomers derived from PDB ID 1NRL (PXR LBD) 11 and PDB ID 3PCU (RXRα LBD) 42. The first round of molecular replacement successfully placed two PXR monomers, which form the PXR homodimer seen the PXR LBD crystal structures 11; 12; 16; 17; 18; 27. Two consecutive rounds of molecular replacement were employed; the first located the two PXR LBD monomers forming the central PXR homodimer; the second placed an RXRα LBD monomer adjacent to each PXR LBD monomer. Utilizing the SR12813-bound LBD complex as a search model, the apo-complex was solved by molecular replacement using Phaser 41.
Structures were manually built using Coot and 2Fo-Fc and Fo-Fc electron density maps 43. The structures were refined using simulated annealing, torsion angle, and B-factor refinement using Phenix44. Furthermore, the data proved to be anisotropic and standard anisotropic refinement methods were utilized in Phenix44, which improved map quality for subsequent manual refinement. Phenix creates a modified, anisotropy-corrected, data file during each refinement run; thus, models were always refined against anisotropy-corrected data. Model quality during refinement was monitored using both the crystallographic R and cross-validating R-free statistics 45. Refinement statistics for both models are presented in Table 1.
SRC-1 Peptide Binding Assays with the LBD Complex
To compare the binding properties of the SRC-1 coactivator peptide to PXR, RXRα, and the PXR/RXRα complex, new LBD constructs were generated that allowed for the expression of wild-type LBDs without the fused coactivator peptide. PCR was used to insert a stop codon before the start of the 5-residue glycine/serine linker at the C-terminal end of both nuclear receptors. The PXR LBD is known to be unstable and express poorly in the absence of the coactivator peptide; as such, the wild-type PXR LBD was cloned into the MBP-LIC pMCSG vector (acquired from the Sondek Lab at UNC) to utilize a MBP-fusion tag to increase stability and expression. The following protein samples were purified in a similar manner as the SRC-1 fused LBDs: wild-type PXR and RXRα, and the complexes PXR-SRC-1/RXRα no SRC-1 and PXR no SRC1/RXRα-SRC-1. The combination of SRC-1 fused PXR with wild type unfused RXRα and vice versa allows for the assessment of SRC-1 binding to each of the individual nuclear receptors in a complexed state. A fluorescein labeled SRC-1 peptide (5-FAM-CPSSHSSLTERHKILHRLLQEGSPS), residues 676-700, was acquired from GenScript to utilize in fluorescence anisotropy binding experiments. SRC-1 coactivator binding reactions were composed of 50 nM FAM-SRC-1 peptide, assay buffer (25 mM HEPES pH 7.5, 100 mM NaCl, 5% glycerol, and 1 mM DTT), and various concentrations of protein. The SRC-1 peptide and protein buffer solutions were pre-diluted to yield a final assay concentration of 100 mM NaCl. Fluorescence anisotropy was measured (excitation= 485 nm, emission=520 nm) using a PHERAstar Plus microplate reader (BMG Labtech). Data acquired was analyzed using Microsoft Excel and Sigmaplot 11.0. Dissociation constants (Kd) were calculated by fitting to a nonlinear regression, one-site saturation binding model and the statistical significance of Kd values was assessed by calculating P-values in Sigmaplot 11.0.
Cloning and Expression of the Full-length PXR/RXRα Complex
Prior knowledge of PXR-LBD expression suggests that the full-length receptor would be far more difficult to express in a soluble, well-folded form and in sufficient quantities for further experimentation. The coexpression of SRC-1 peptide fragment with PXR LBD has previously been shown to facilitate PXR-LBD expression 11; 25, and so co-expression of the SRC-1 fragment was crucial for protein expression. However, instead of co-expression of the SRC-1 peptide as was previously done 11; 16; 17; 18; 27; 46, tethered versions of full-length PXR and RXR were constructed by fusing a glycine-serine linker at the end of the protein followed by the SRC-1 fragment. Literature has demonstrated that this technique is successful at producing soluble, stable protein, permanently linked to the SRC-1 coactivator 25. To increase full-length protein expression, codon optimization using the services of GenScript was utilized to construct each gene, with the addition of the tethered SRC-1 fragment.
Both genes were cloned in the MBP-LIC pMCSG vector (acquired from the Sondek Lab at UNC), as the MBP-fusion would additionally help the stability and solubility of the proteins as they are being expressed. E. coli BL21 (DE3) AI cells were transformed with either the PXR or RXRα full-length plasmid for separate protein expression. Terrific broth media was inoculated with a saturated culture of BL21 AI cells transformed with the MBP-PXR plasmid and allowed to shake at 37 °C. When the cells reach and OD600 of ~1.4–1.6 the temperature was turned down to 15 °C. At ~17 °C L-arabinose was added to each flask up to a final concentration of 0.1%. When the temperature achieved 15 °C, IPTG was added (final concentration of 1 mM) to induce protein expression. After shaking overnight, cells were collected by centrifugation at 4,500×g for 20 min and stored at −80 °C. These growths yielded on average 2–3 mg/L of full-length PXR protein. The growth and expression of RXRα was carried out in a similar manner as PXR described above. When the RXRα growth achieved an OD600 of ~1.4–1.6, the temperature was reduced to 17 °C. A final concentration of 0.1% L-arabinose was added when temperature reached ~19 °C, and expression was induced at 17 °C with a final concentration of 1 mM IPTG. With this protocol, ~10 mg/L of full-length RXRα is produced on average.
Purification and Formation of the Full-length Complex
PXR and RXRα were each purified similarly but individually prior to complex formation. Cell pellets were resuspended in Buffer A (50 mM HEPES pH 7.5, 50 mM imidazole, 500 mM NaCl, 10% glycerol) supplemented with a protease inhibitor tablet (Roche), DNase, and lysozyme. After sonication, the cell lysate was clarified by high-speed centrifugation at 14,500×g for 50 mins. The supernatant was removed and syringe filtered through a 0.22 um filter. For PXR purifications, an excess amount of ligand was added to the lysate to help stabilize the protein throughout the purification process. Clarified lysate was loaded onto the Äktaxpress FPLC system (Amersham Biosciences) for a step-purification, up to 500 mM imidazole, on Ni His-Trap (GE Healthcare) columns. Purest fractions (determined by SDS-PAGE) were combined and concentrated as much as possible without destabilizing the protein. While concentrating PXR, additional ligand was added periodically to compensate for any ligand washed away during Ni-column purification. Concentrated samples were loaded on the FPLC system for sizing purification using a HiLoad™ 16/60 Superdex™ 200 gel filtration column. Each protein was eluted into 20 mM Tris-HCl pH 7.5, 250 mM NaCl, 1 mM TCEP, and 5% glycerol. Purity of each fraction was assessed by SDS-PAGE and fractions of >95% purity were combined. After each protein was individually purified, 5 mL reactions were prepared to form the PXR/RXRα complex for a final round of gel filtration. One reaction consisted of 30 μM each of PXR and RXRα, 5-times excess of PXR ligand, 2-times excess of the RXRα ligand 9-cis-retinoic acid. TEV protease was also added to the reaction to cleave the MBP-fusion tag from each protein. The complex formation reactions were directly injected onto the S200 gel filtration column after a >8 h incubation at 4 °C to allow complete cleavage to occur. Two peaks eluted from the sizing column, the first being the PXR/RXRα complex and the second the now cleaved MBP-fusion tag. This reaction protocol for complex formation yielded the purified hetero-complex, as assessed by SDS-PAGE showing two protein bands representing full-length PXR and RXRα.
DNA Binding Studies with the Full-length Complex
The purified full-length PXR/RXRα was utilized for in vitro experiments to determine DNA binding properties of the complex. Several different types of duplex DNA containing response elements (REs) were procured to test the binding affinity for each type of RE as well as to assess the necessity of flanking bases for improved DNA binding. The sequences of each duplex DNA tested are listed in Table 3.
Table 3.
DNA Oligonucleotides Examined
| Element Type | DNA Sequence | Abbreviation |
|---|---|---|
| DR3 (CYP3A4) | 5′-FAM-TGAACTTGCTGACCC-3′ 3′-ACTTGAACGACTGGG-5′ |
DR3 FAM |
| 5′-TCAGCTGAATGAACTTGCTGACCCTCTGCTTTC-3′ 3′-AGTCGACTTACTTGAACGACTGGGAGACGAAAG-5′ |
DR3 +9/+9 | |
| 5′-GCTGAATGAACTTGCTGACCCTCTGCTTTC-3′ 3′-CGACTTACTTGAACGACTGGGAGACGAAAG-5′ |
DR3 +6/+9 | |
| 5′-GAATGAACTTGCTGACCCTCTGCTTTC-3′ 3′-CTTACTTGAACGACTGGGAGACGAAAG-5′ |
DR3 +3/+9 | |
| 5′-TGAACTTGCTGACCCTCTGCTTTC-3′ 3′-ACTTGAACGACTGGGAGACGAAAG-5′ |
DR3 +0/+9 | |
| 5′-TCAGCTGAATGAACTTGCTGACCCTCTGCT-3′ 3′-AGTCGACTTACTTGAACGACTGGGAGACGA-5′ |
DR3 +9/+6 | |
| 5′-TCAGCTGAATGAACTTGCTGACCCTCT-3′ 3′-AGTCGACTTACTTGAACGACTGGGAGA-5′ |
DR3 +9/+3 | |
| 5′-TCAGCTGAATGAACTTGCTGACCC-3′ 3′-AGTCGACTTACTTGAACGACTGGG-5′ |
DR3 +9/+0 | |
| 5′-GCTGAATGAACTTGCTGACCCTCTGCT-3′ 3′-CGACTTACTTGAACGACTGGGAGACGA-5′ |
DR3 +6/+6 | |
| 5′-GAATGAACTTGCTGACCCTCT-3′ 3′-CTTACTTGAACGACTGGGAGA-5′ |
DR3 +3/+3 | |
| 5′-AACTTGCTGACCCTCTGCTTTC-3′ 3′-TTGAACGACTGGGAGACGAAAG-5′ |
DR3 −2/+9 | |
| 5′-TCAGCTGAATGAACTTGCTGAC-3′ 3′-AGTCGACTTACTTGAACGACTG-5′ |
DR3 +9/−2 | |
| 5′-AACTTGCTGAC-3′ 3′-TTGAACGACTG-5′ |
DR3 −2/−2 | |
| ER6 (CYP3A4) | 5′-FAM-TGAACTCAAAGGAGGTCA-3′ 3′-ACTTGAGTTTCCTCCAGT-5′ |
ER6 FAM |
| 5′-CATAGAATATGAACTCAAAGGAGGTCAGTGAGTGGT-3′ 3′-GTATCTTATACTTGAGTTTCCTCCAGTCACTCACCA-5′ |
ER6 +9/+9 | |
| 5′-AGAATATGAACTCAAAGGAGGTCAGTGAGTGGT-3′ 3′-TCTTATACTTGAGTTTCCTCCAGTCACTCACCA-5′ |
ER6 +6/+9 | |
| 5′-ATATGAACTCAAAGGAGGTCAGTGAGTGGT-3′ 3′-TATACTTGAGTTTCCTCCAGTCACTCACCA-5′ |
ER6 +3/+9 | |
| 5′-TGAACTCAAAGGAGGTCAGTGAGTGGT-3′ 3′-ACTTGAGTTTCCTCCAGTCACTCACCA-5′ |
ER6 +0/+9 | |
| 5′-CATAGAATATGAACTCAAAGGAGGTCAGTGAGT-3′ 3′-GTATCTTATACTTGAGTTTCCTCCAGTCACTCA-5′ |
ER6 +9/+6 | |
| 5′-CATAGAATATGAACTCAAAGGAGGTCAGTG-3′ 3′-GTATCTTATACTTGAGTTTCCTCCAGTCAC-5′ |
ER6 +9/+3 | |
| 5′-CATAGAATATGAACTCAAAGGAGGTCA-3′ 3′-GTATCTTATACTTGAGTTTCCTCCAGT-5′ |
ER6 +9/+0 | |
| 5′-AGAATATGAACTCAAAGGAGGTCAGTGAGT-3′ 3′-TCTTATACTTGAGTTTCCTCCAGTCACTCA-5′ |
ER6 +6/+6 | |
| 5′-ATATGAACTCAAAGGAGGTCAGTG-3′ 3′-TATACTTGAGTTTCCTCCAGTCAC-5′ |
ER6 +3/+3 | |
| 5′-AACTCAAAGGAGGTCAGTGAGTGGT-3′ 3′-TTGAGTTTCCTCCAGTCACTCACCA-5′ |
ER6 −2/+9 | |
| 5′-CATAGAATATGAACTCAAAGGAGGT-3′ 3′-GTATCTTATACTTGAGTTTCCTCCA-5′ |
ER6 +9/−2 | |
| 5′-AACTCAAAGGAGGT-3′ 3′-TTGAGTTTCCTCCA-5′ |
ER6 −2/−2 |
Two DNA binding assays were utilized to measure binding of the nuclear receptor complex to REs. The first assay used fluorescein-labeled duplex DNA (Table 3) to measure the dissociation constant, Kd, for a DR3 and ER6 type DNA from the CYP3A4 promoter region. These pieces of DNA contained the minimum number of bases to create the repeat. This DNA binding assay consisted of several different concentrations of the complex, starting at a maximum of 5 μM. Twenty-five microliters of the total assay volume consisted of the nuclear receptor complex and the assay buffer, which was configured to create a final NaCl concentration of 100 mM. Another 25 μL of the DNA probe solution was added (final concentration of 50 nM), making the final reaction volume 50 μL. Fluorescence anisotropy was detected using a PHERAstar Plus microplate reader (BMG Labtech). Data acquired was analyzed using Microsoft Excel and Sigmaplot 11.0. Kd values were calculated for the DR3 FAM and ER6 FAM and subsequently used to calculate Ki values for each DNA competitor.
The second DNA binding assay is competition-based to measure the binding of several pieces of duplex DNA with different lengths of flanking bases (Table 3) in addition to the minimum RE repeats. This assay was composed of a constant protein concentration (200 nM) and a constant concentration of fluorescein-labeled duplex DNA (50 nM). A range of unlabeled DNA competitor concentrations were used in each assay, with a maximum concentration of 10 μM. Addition of each component yielded a total reaction volume of 50 μL and a final NaCl concentration of 100 mM. Data was collected and analyzed as with the first DNA binding assay described above. The inhibition constant values (Ki) were calculated using the Cheng-Prusoff equation 47. These values are presented in Table 4.
Structure of the heterotetramer formed between human PXR and RXRα
Heterotetramer unprecedented in nuclear receptor family of transcription factors
Receptors in heterotetramer exhibit enhanced association with coregulators
Purified full-length PXR and RXRα from recombinant bacterial system
DNA binding studies of full-length receptors with DR and ER response elements
Acknowledgments
Supported by NIH grant CA98468 (MRR).
Footnotes
Accession Numbers: The coordinates and structure factors for the Apo-PXR/RXRα LBD complex and SR12813-bound PXR/RXRα LBD complex structures have been submitted to the Protein Data Bank and assigned the identifier 4j5w and 4j5x, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Handschin C, Meyer UA. Induction of drug metabolism: the role of nuclear receptors. Pharmacol Rev. 2003;55:649–73. doi: 10.1124/pr.55.4.2. [DOI] [PubMed] [Google Scholar]
- 2.Miller RT, Willson TM. Regulation of xenobiotic metabolism by orphan nuclear receptors. Toxicol Pathol. 2001;29:3–5. doi: 10.1080/019262301301418801. [DOI] [PubMed] [Google Scholar]
- 3.Moore DD. Regulation of drug transport by new xenobiotic receptors. Pharmacogenomics J. 2001;1:224–5. doi: 10.1038/sj.tpj.6500058. [DOI] [PubMed] [Google Scholar]
- 4.Omiecinski CJ, Vanden Heuvel JP, Perdew GH, Peters JM. Xenobiotic metabolism, disposition, and regulation by receptors: from biochemical phenomenon to predictors of major toxicities. Toxicol Sci. 120(Suppl 1):S49–75. doi: 10.1093/toxsci/kfq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonoda J, Evans R. Biological function and mode of action of nuclear xenobiotic receptors. Pure and Applied Chemistry. 2003;75:1733–1742. [Google Scholar]
- 6.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 7.Blumberg B, Sabbagh W, Jr, Juguilon H, Bolado J, Jr, van Meter CM, Ong ES, Evans RM. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12:3195–205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A. 1998;95:12208–13. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 10.Wallace BD, Redinbo MR. Xenobiotic-sensing nuclear receptors involved in drug metabolism: a structural perspective. Drug Metab Rev. 45:79–100. doi: 10.3109/03602532.2012.740049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–28. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 12.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 A crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry. 2003;42:1430–8. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 13.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–33. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 14.Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–7. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 15.Allenby G, Bocquel MT, Saunders M, Kazmer S, Speck J, Rosenberger M, Lovey A, Kastner P, Grippo JF, Chambon P, et al. Retinoic acid receptors and retinoid X receptors: interactions with endogenous retinoic acids. Proc Natl Acad Sci U S A. 1993;90:30–4. doi: 10.1073/pnas.90.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teotico DG, Bischof JJ, Peng L, Kliewer SA, Redinbo MR. Structural basis of human pregnane X receptor activation by the hops constituent colupulone. Mol Pharmacol. 2008;74:1512–20. doi: 10.1124/mol.108.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue Y, Moore LB, Orans J, Peng L, Bencharit S, Kliewer SA, Redinbo MR. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol Endocrinol. 2007;21:1028–38. doi: 10.1210/me.2006-0323. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Redinbo MR. Activation of the human nuclear xenobiotic receptor PXR by the reverse transcriptase-targeted anti-HIV drug PNU-142721. Protein Sci. 20:1713–9. doi: 10.1002/pro.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–34. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 20.Bourguet W, Vivat V, Wurtz JM, Chambon P, Gronemeyer H, Moras D. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol Cell. 2000;5:289–98. doi: 10.1016/s1097-2765(00)80424-4. [DOI] [PubMed] [Google Scholar]
- 21.Gampe RT, Jr, Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, Kliewer SA, Willson TM, Xu HE. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cell. 2000;5:545–55. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- 22.Suino K, Peng L, Reynolds R, Li Y, Cha JY, Repa JJ, Kliewer SA, Xu HE. The nuclear xenobiotic receptor CAR: structural determinants of constitutive activation and heterodimerization. Mol Cell. 2004;16:893–905. doi: 10.1016/j.molcel.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 23.Svensson S, Ostberg T, Jacobsson M, Norstrom C, Stefansson K, Hallen D, Johansson IC, Zachrisson K, Ogg D, Jendeberg L. Crystal structure of the heterodimeric complex of LXRalpha and RXRbeta ligand-binding domains in a fully agonistic conformation. Embo J. 2003;22:4625–33. doi: 10.1093/emboj/cdg456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, Williams JD, Collins JL, Moore LB, Willson TM, Moore JT. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol Cell. 2004;16:919–28. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 25.Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-gamma-RXR-alpha nuclear receptor complex on DNA. Nature. 2008:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ray A, Siegel MD, Prefontaine KE, Ray P. Anti-inflammation: direct physical association and functional antagonism between transcription factor NF-KB and the glucocorticoid receptor. Chest. 1995;107:139S. doi: 10.1378/chest.107.3_supplement.139s. [DOI] [PubMed] [Google Scholar]
- 27.Noble SM, Carnahan VE, Moore LB, Luntz T, Wang H, Ittoop OR, Stimmel JB, Davis-Searles PR, Watkins RE, Wisely GB, LeCluyse E, Tripathy A, McDonnell DP, Redinbo MR. Human PXR forms a tryptophan zipper-mediated homodimer. Biochemistry. 2006;45:8579–89. doi: 10.1021/bi0602821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onate SA, Boonyaratanakornkit V, Spencer TE, Tsai SY, Tsai MJ, Edwards DP, O’Malley BW. The steroid receptor coactivator-1 contains multiple receptor interacting and activation domains that cooperatively enhance the activation function 1 (AF1) and AF2 domains of steroid receptors. J Biol Chem. 1998;273:12101–8. doi: 10.1074/jbc.273.20.12101. [DOI] [PubMed] [Google Scholar]
- 29.Orans J, Teotico DG, Redinbo MR. The nuclear xenobiotic receptor pregnane X receptor: recent insights and new challenges. Mol Endocrinol. 2005;19:2891–900. doi: 10.1210/me.2005-0156. [DOI] [PubMed] [Google Scholar]
- 30.Delvecchio CJ, Bilan P, Nair P, Capone JP. LXR-induced reverse cholesterol transport in human airway smooth muscle is mediated exclusively by ABCA1. Am J Physiol Lung Cell Mol Physiol. 2008;295:L949–57. doi: 10.1152/ajplung.90394.2008. [DOI] [PubMed] [Google Scholar]
- 31.Castelein H, Declercq PE, Baes M. DNA binding preferences of PPAR alpha/RXR alpha heterodimers. Biochem Biophys Res Commun. 1997;233:91–5. doi: 10.1006/bbrc.1997.6395. [DOI] [PubMed] [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 33.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–56. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gee AC, Carlson KE, Martini PG, Katzenellenbogen BS, Katzenellenbogen JA. Coactivator peptides have a differential stabilizing effect on the binding of estrogens and antiestrogens with the estrogen receptor. Mol Endocrinol. 1999;13:1912–23. doi: 10.1210/mend.13.11.0373. [DOI] [PubMed] [Google Scholar]
- 35.Pogenberg V, Guichou JF, Vivat-Hannah V, Kammerer S, Perez E, Germain P, de Lera AR, Gronemeyer H, Royer CA, Bourguet W. Characterization of the interaction between retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers and transcriptional coactivators through structural and fluorescence anisotropy studies. J Biol Chem. 2005;280:1625–33. doi: 10.1074/jbc.M409302200. [DOI] [PubMed] [Google Scholar]
- 36.Wright E, Vincent J, Fernandez EJ. Thermodynamic characterization of the interaction between CAR-RXR and SRC-1 peptide by isothermal titration calorimetry. Biochemistry. 2007;46:862–70. doi: 10.1021/bi061627i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–9. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 38.Saridakis E, Chayen NE. Improving protein crystal quality by decoupling nucleation and growth in vapor diffusion. Protein Sci. 2000;9:755–7. doi: 10.1110/ps.9.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W. Xds Acta Crystallogr D Biol Crystallogr. 66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W, Charles W, Carter . Methods in Enzymology. Vol. 276. Academic Press; 1997. [20] Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 41.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–3. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 45.Brunger AT. Free R value: cross-validation in crystallography. Methods Enzymol. 1997;277:366–96. doi: 10.1016/s0076-6879(97)77021-6. [DOI] [PubMed] [Google Scholar]
- 46.Carnahan VE, Redinbo MR. Structure and function of the human nuclear xenobiotic receptor PXR. Curr Drug Metab. 2005;6:357–67. doi: 10.2174/1389200054633844. [DOI] [PubMed] [Google Scholar]
- 47.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]