

Abstract
Patients with diabetes frequently exhibit the combined occurrence of hyperglycemia and dyslipidemia. Published data on their coexistence are often controversial. Some studies provide evidence for suboptimal lifestyle and exogenous hyperinsulinism at "mild insulin resistance" in adult diabetic patients as main pathogenic factors. In contrast, other studies confirm that visceral adiposity and insulin resistance are the basic features of dyslipidemia in type 2 diabetes (T2D). The consequence is an excess of free fatty acids, which causes hepatic gluconeogenesis to increase, metabolism in muscles to shift from glucose to lipid, beta-cell lipotoxicity, and an appearance of the classical "lipid triad", without real hypercholesterolemia. Recently, it has been proposed that cholesterol homeostasis is important for an adequate insulin secretory performance of beta-cells. The accumulation of cholesterol in beta-cells, caused by defective high-density lipoprotein (HDL) cholesterol with reduced cholesterol efflux, induces hyperglycemia, impaired insulin secretion, and beta-cell apoptosis. Data from animal models and humans, including humans with Tangier disease, who are characterized by very low HDL cholesterol levels, are frequently associated with hyperglycemia and T2D. Thus, there is a reciprocal influence of dyslipidemia on beta-cell function and inversely of beta-cell dysfunction on lipid metabolism and micro- and macrovascular complications. It remains to be clarified how these different but mutually influencing adverse effects act together to define measures for a more effective prevention and treatment of micro- and macrovascular complications in diabetes patients. While the control of circulating low-density lipoprotein (LDL) cholesterol and the level of HDL cholesterol are determinant targets for the reduction of cardiovascular risk, based on recent data, these targets should also be considered for the prevention of beta-cell dysfunction and the development of type 2 diabetes. In this review, we analyze consolidated data and recent advances on the relationship between lipid metabolism and diabetes mellitus, with particular attention to the reciprocal effects of the two features of the disease and the development of vascular complications.
Keywords: diabetes, dyslipidemia, cholesterol, lipoprotein, HDL, LDL, cardiovascular disease, atherogenic
Abbreviations: ABCA1 - ATP-binding cassette member A1; ABCG5/G8 - ATP-binding cassette transporter G5/G8; AMP - adenosine monophosphate-activated protein; Apo-AI/AII - apolipoprotein AI/AII; ATP - adenosine triphosphate; BIP - Bezafibrate Infarction Prevention study; CETP - cholesteryl ester transfer protein; CHD - coronary heart disease; CVD - cardiovascular disease; eNOS - endothelial nitric oxid synthase; FFA- free fatty acid; HbA1c - glycosylated hemoglobin A1c; HDL - high-density lipoprotein; HDL-C - high-density lipoprotein cholesterol; HL - hepatic lipase; HMG-CoA - 3-hydroxy-3-methyl-glutaryl coenzyme A; HOMA-beta - homeostasis model assessment beta; IDEAL - Incremental Decrease in End Points Through Aggressive Lipid Lowering study; IFG - impaired fasting glucose; IGT - impaired glucose tolerance; JNK - c-Jun N-terminal kinase; JUPITER - Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin trial; LDL - low-density lipoprotein; LDL-C - low-density lipoprotein cholesterol; LPL - lipoprotein lipase; LRP - LDL receptor-related protein; LXR - liver X receptor; NCP-ATP III - National Cholesterol Program-Adult Treatment Panel III; NGT - normal glucose tolerance; NO - nitric oxid; NOS - nitric oxid synthase; NPC1L1 - Niemann-Pick C1 Like-1; OGTT - oral glucose tolerance test; PROVE IT-TIMI - Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction study; PPAR-gamma - peroxisome proliferator agonist receptor gamma; sdLDL - small dense LDL; SREBP-2 - sterol regulatory element-binding protein-2; T1D - type 1 diabetes; T2D - type 2 diabetes; TC - total cholesterol; TNF-alpha - tumor necrosis factor alpha; TNT - Treating to New Targets study; Trg - triglycerides; UKPDS - United Kingdom Prospective Diabetes Study; VA-HIT - Veterans Affairs HDL Intervention Trial; VLDL - very low-density lipoproteins; WOSCOPS - West of Scotland Coronary Prevention Study
Introduction
Both type 1 and type 2 diabetes are characterized by a high risk of developing chronic complications, with micro- and macrovascular impairments in multiple organs, including retinopathy, nephropathy, neuropathy, endothelial dysfunction, and atherosclerosis. Although the pathogenic mechanisms of both forms of diabetes are basically different, the symptoms and consequences, including lack of insulin, metabolic disturbances, and impaired lipid balance, are common features. Both diseases are characterized by a highly increased mortality by cardiovascular diseases (CVD), with a direct effect of insulin resistance on lipid metabolism in type 2 diabetes (T2D). In type 1 diabetes (T1D), the mechanisms caused by insulin deficiency and dyslipidemia remain poorly understood and controversial.
Despite recent improvements in technology and advances in glycemic control, disease management, and health education, CVD is the most frequent cause of death in type 1 diabetes, with a tenfold higher CVD-related and all-cause mortality than in the general population [1]. Although chronic hyperglycemia and endothelial dysfunction are regarded as the main actors in the development of CVD, discordant results are reported concerning the association between dyslipidemia and type 1 diabetes. In a Norwegian study, total and LDL cholesterol levels were significantly higher in T1D patients than in non-T1D subjects. On the other hand, numerous studies on type 1 diabetic patients, involving juvenile and adult subjects, failed to confirm a real increase in cholesterol levels and impaired lipid metabolism.
Beta-cell dysfunction and insulin resistance contributing to dyslipidemia and vascular complications
Type 1 diabetes and dyslipidemia
Since the cholesterol turnover is the result of hepatic synthesis and intestinal absorption, specific serum markers of cholesterol absorption have been studied in T1D; among them plant sterols, cholestanol and cholesterol synthesis, i.e. cholestanol, desmosterol, and lathosterol. The results obtained in this field showed that in T1D patients the markers of cholesterol absorption were increased, while the markers of cholesterol synthesis were decreased compared with control subjects, indicating that both high cholesterol absorption and low cholesterol synthesis are present in type 1 diabetic patients [2]. This mechanism has been hypothesized to be the result of an upregulation of specific genes for the synthesis of key proteins in the process of cholesterol intestinal absorption. Among these proteins are the Niemann-Pick C1 Like-1 (NPC1L1) protein which regulates intestinal cholesterol absorption and the ATP-binding cassette transporters G5 and G8 (ABCG5 and ABCG8) which regulate cholesterol homeostasis through the excretion of enterocyte cholesterol into the intestinal lumen. In other reports, elevated total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C) have been demonstrated to be significantly higher only in females, suggesting a gender difference in lipid control.
More meaningful data are available for high-density lipoprotein cholesterol (HDL-C) levels. In fact, type 1 diabetic subjects show more large and less small HDL-C particles than non-diabetic subjects, although observed only in male patients. To confirm that there is a gender-related effect on lipid levels in T1D, it has been demonstrated that women with T1D have less large and more small dense LDL lipoproteins and reduced LDL size, and this effect on LDL size was significantly distinct in women [3]. Furthermore, in the study, a "mild insulin resistance" in T1D patients was observed, specially in adults with a prolonged history of disease. This effect is probably caused by elevated exogenous insulin supply, and this may suggest a possible role of insulin resistance in impaired lipid regulation. However, it must be considered that glucose balance also directly impacts triglyceride metabolism. In fact, the tight correlation of lipid profile with HbA1c levels is heterogeneous across the spectrum of values of glycemic control in T1D individuals, with triglycerides worsening alongside HbA1c quintiles, as observed in a large recent nationwide multicenter survey [4]. All together, these data may, in part, explain the different results reported in the literature regarding T1D and lipid control, and questioned an unified explanation for impaired lipid metabolism in these patients. Therefore, it remains unclear whether the observed lipid abnormalities in diabetic patients are caused by impaired lipid metabolism associated with T1D rather than glucose control, gender, insulin resistance, and non-regular lifestyle of these patients, or by all these factors in combination.
Type 2 diabetes and dyslipidemia
The relationship between impaired lipid metabolism and T2D is more common. T2D is associated with early and multidistrict atherosclerotic lesions. The importance of the metabolic control for the development of diabetic macroangiopathy emerged from results of the United Kingdom Prospective Diabetes Study (UKPDS); a reduction of 1% in HbA1c in patients with good metabolic control (7.0%) was associated with a significant reduction (16.0%) of cardiovascular events [5].
The close link between T2D and CVD has been confirmed by a study by Haffner et al., involving more than 1000 diabetes patients with a follow-up of 7 years. It showed that T2D patients had an increased risk of developing coronary heart disease (CHD), equal to that of a non-diabetic subjects with prior history of myocardial infarction [6]. According to international guidelines from the National Cholesterol Program-Adult Treatment Panel III (NCP-ATP III), the diabetic patient is defined as CHD risk equivalent [7]. Consequently, the need for an optimal metabolic control should be accompanied by adequate control of the lipid profile, as confirmed by lipid intervention trials with statins [8, 9]. In fact, the reduction of low density lipoprotein cholesterol (LDL-C) below 100 mg/dl in diabetic patients without other risk factors, and less than 70 mg/dl if any other CVD risk factors are present, are recommended.
Recently, it was reported about the new concept of "residual cardiovascular risk" [10]. The analysis of the Scandinavian Simvastatin Survival Study (4S trial) showed a significant reduction of CV events in patients treated with simvastatin, but there was still a 20% CVD rate in these subjects [11]. There are similar results from other important clinical studies such as the Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction study (PROVE IT-TIMI), the Incremental Decrease in End Points Through Aggressive Lipid Lowering study (IDEAL), and the Treating to New Targets study (TNT). The studies revealed that a residual CVD risk was existent even after treatment with statins, with 22.4% of patients in the PROVE IT-TIMI study, 12.0% in the IDEAL trial, and 8.7% in the TNT study [12, 13]. Despite a significant reduction of LDL-C levels through the treatment with statins, a considerable residual CVD risk remains in T2D patients. These findings suggest that other lipoprotein components, such as low HDL-C levels and hypertriglyceridemia, may be involved in atherosclerotic diseases [14]. To test this hypothesis, subjects at LDL-C target (<70 mg/dl), but with higher levels of HDL-C, were further analyzed in the TNT trial. It showed that these patients presented a lower incidence of major CHD events [15]. This showed that the link between CVD and lipoprotein levels in T2D patients is more complex than previously assumed.
Indeed, diabetic dyslipidemia seems to be very special, including increased synthesis of very low-density lipoproteins (VLDL) and a reduction of circulating HDL-C lipoproteins, in the presence of a small increase of total cholesterol [16, 17]. The elevated production of VLDL particles induces a subsequent synthesis of a particular subclass of LDL particles, the small dense LDL (sdLDL) that represent a fraction of LDL with higher atherogenic properties [18]. This lipid pattern has been called the "lipid triad", and is characterized by high circulating levels of triglyceride-rich particles, reduced synthesis of HDL lipoproteins, and enhanced production of atherogenic LDL particles [19]. The pathophysiological mechanisms underlying this multiple lipid disorder are very complex and only partially understood. A pivotal role seems to be played by excess weight, in particular visceral obesity, and the related insulin resistance [20]. Several studies have confirmed that visceral/abdominal fat depots differ from the general subcutaneous fat mass by a greater lipolytic activity and reduced response to insulin signaling (i.e. insulin resistance). The normal subcutaneous fat depots are characterized by low lipolytic activity and higher sensitivity to insulin, and tend to release free fatty acids (FFAs) into the systemic bloodstream with a "dilution effect" of these toxic molecules. In contrast, in obese patients, the abdominal adipose depots release FFAs which are exclusively released into the portal vein and then transported to the liver and pancreas, even if the proportion of the abdominal fat is small in these patients. The excessive FFAs are reassembled in the liver to form triglycerides and stored into hepatocytes, or released into the bloodstream carried by VLDL, the specific lipoproteins for endogenous triglycerides [21].
T2D patients exhibit a lack of insulin effect on hepatic lipid metabolism, which normally limits the synthesis of VLDL1, and a reduction of the endothelial lipolytic activity of lipoprotein lipase (LPL) due to insulin resistance. This scenario induces a dramatic increase of VLDL1 particles, the largest lipoproteins rich in triglycerides [22, 23]. The excess of VLDL1 in T2D patients is mainly observed in the postprandial period. This impaired metabolic pattern is maintained for many hours upon food intake, exposing the patient to a prolonged period of hypertriglyceridemia and hyperchilomicronemia [24, 25]. The consequence of this effect is an impaired exchange of cholesterol with LDL and HDL particles, with an adverse impact on the synthesis and maturation process of HDL particles [26].
Cholesteryl ester transfer protein (CETP) and hepatic lipase (HL) are pivotal enzymes involved in the metabolism of HDL particles and reverse cholesterol transport. CETP mediates the exchange of cholesterol for triglycerides between LDL and HDL and triglyceride-rich lipoprotein particles. The effect causes an increased concentration of triglycerides in HDL and LDL particles, which makes them favorable substrates for HL. This hepatic enzyme catalyses the hydrolysis of the abundant triglycerides carried by several lipoproteins, modifying the size and density of the specific carriers. The result of the synergetic activity of the two enzymes, CETP and HL, is a characteristic of the pathophysiology of insulin resistance, typical of T2D, with a lower HDL-C level, a predominance of small, dense HDL, and sdLDL particles [27-29]. These sdLDL are characterized by a reduced ratio in the concentration of cholesterol/triglycerides. They are less effective in releasing cholesterol molecules to peripheral tissues. They also have a smaller diameter than conventional LDL particles (<25.5 nm), and they are the major component of atherogenic "pattern B", associated with increased cardiovascular risk.
The more atherogenic pattern represented by the occurrence of sdLDL particles is very frequent in T2D and the metabolic syndrome, also in the presence of a good metabolic control. This is one of the central elements of diabetic dyslipidemia. In fact, despite normal or even decreased circulating LDL-C levels, small dense LDL particles prevailed in up to 50% of T2D patients in a study on LDL subfraction profiles in T2D subjects [30]. This lipoprotein subfraction has a reduced binding affinity to the LDL receptor due to a different chemical composition and electric charge. The result is a greater persistence of circulating cholesterol-rich lipoproteins which easily adhere to the endothelial surface and are more easily captured in the subendothelium space [31]. The prolonged presence of sdLDL particles in serum enables their glycation and oxidation [32]. Thus, they can be "picked up" by the scavenger receptor on the macrophage which has a much greater affinity for oxidized LDL than for non-oxidized LDL.
The modification of physiological properties of LDL to form sdLDL particles, together with the abovementioned structural impairments of HDL cholesterol, constitutes an increased CVD risk profile in T2D patients. However, several studies suggest that abnormalities in HDL cholesterol constitute a CVD risk factor epidemiologically independent of the LDL profile in T2D patients [13]. Normally, HDL particles possess several anti-atherogenic properties independent of the well-known reverse cholesterol transport from the arterial wall to the liver excretion [33]. In particular, the following properties of HDL particles have been demonstrated:
1. Anti-oxidative properties due to paraoxonase, glutathione, and selenoperoxidase enzymes
2. Anti-inflammatory properties by blockade of endothelial tumor necrosis factor alpha (TNF-alpha) production, cytokine-mediated expression adhesion molecules, and serum amyloid A protein
3. Anti-thrombotic properties by inhibition of platelet aggregation by thromboxane and platelet activating factor
4. Vasodilatory action by upregulation of endothelial NO synthase (eNOS) [34, 35].
When we study the properties and effects of HDL-C particles, we should take into account that low HDL-C levels are often accompanied by hypertriglyceridemia, especially in individuals with insulin resistance. To determine the relevance of these two lipid components, i.e. HDL-C and triglycerides, for CVD protection, different aspects need to be considered. First, in a sub-analysis of intervention trials designed to reduce triglycerides in high-risk populations (Veterans Affairs HDL Intervention Trial - VA-HIT, and Bezafibrate Infarction Prevention study - BIP), pharmacological intervention realized positive effects only in patients with visceral obesity and triglycerides/HDL ratio of >5.18 (personal data). Second, intervention trials, aimed at pharmacologically increasing HDL-C levels using CETP-inhibitors, have failed to realize cardiovascular prevention. All together these findings suggest that the vascular protective action of HDL is not only quantitative but, probably, it depends on the quality and composition of HDL particles. This was also demonstrated in a study by Khera and colleagues, where patients in secondary prevention (including 23% with T2D) had a reduced cholesterol efflux capacity independent of their HDL levels [36]. In agreement with this finding, recent data by another group showed that HDL particles obtained from T2D patients were characterized by a decreased paraxonase-1 activity, inversely correlated to HbA1c levels [37].
For the treatment of diabetic dyslipidemia, the NCP-ATP-III guidelines propose non-HDL cholesterol as another therapeutic target in individuals with insulin resistance and high triglyceride concentrations. Non-HDL cholesterol, easily calculated by total cholesterol minus HDL-C, represents the concentration of cholesterol within all lipoprotein atherogenic particles such as LDL, IDL, and VLDL. It is an important measure of these particles in the presence of high triglyceride levels. The use of non-HDL-C has been demonstrated to be a better predictor of CVD events than LDL-C alone, particularly in patients with insulin resistance and/or diabetes [38, 39]. For this reason, it is recommended that non-HDL-C should act as secondary target of therapy after targeting LDL-C levels in patients with increased Trg (>200 mg/dl), because the LDL-C level calculated by the Friedewald formula cannot be applied in the presence of hypertriglyceridemia [40].
Circulating levels of LDL-C are the result of the regulation of hepatic synthesis, intestinal absorption, and biliary excretion. Also, these cholesterol levels are influenced by its intracellular pool and the uptake of cholesterol by the activity of the specific membrane receptor for LDL lipoproteins. However, the regulation of lipid metabolism in diabetes is extremely complex, involving different carrier lipoproteins and enzymatic systems, which are only partially explored. In T2D, an increase in hepatic synthesis of cholesterol and, at the same time, a reduction of intestinal absorption have been observed [41]. Furthermore, other studies in T2D patients, involving gene expressions of specific intestinal lipids carriers (such as NPC1L1, ABCG5 and ABCG8, and the microsomal triglyceride transfer protein, which packages the chylomicron particles by assembling cholesterol, triglyceride, and apolipoprotein B48), revealed important alterations in the expression of intestinal genes that regulate cholesterol absorption and chylomicron synthesis [42]. In contrast, type 1 diabetes is associated with enhanced intestinal absorption and a reduction in hepatic synthesis of cholesterol. Thus, the regulation of lipid metabolism in diabetes is extremely complex, involving different carrier lipoproteins and enzymatic systems, which are not yet well understood. These mechanisms may affect the efficacy of lipid-lowering therapy, and the achievement and long-term maintenance of therapeutic goals. In addition, recent investigations propose another type of interaction between lipids and glucose metabolism, namely the role of lipid dysregulation as primary trigger for beta-cell impairment. These investigations are presented in the next section.
Dyslipidemia contributing to diabetes and vascular complications
Beta-cell dysfunction is one of the principal pathogenetic factors occurring in the development of T2D. A well-known mechanism associated with a significant decline in beta-cell performance is glucolipotoxicity. This term defines the chronic exposure to high concentrations of glucose and free fatty acids, typically occurring in T2D patients, may have a synergetic effect in the impairment of insulin secretion. However, new data emerged in recent years, focusing on lipid disorders as facilitating factor in the development of defective insulin secretion and altered glucose metabolism (Figure 1). This aspect of lipid metabolism and glucose intolerance has been explored by our group, showing that subjects with excess of visceral fat, associated with high cholesterol levels, presented a different response to oral glucose tolerance test (OGTT). These individuals were characterized by overweight/obesity, with a high waist circumference, and a poor glycemic profile. In addition, they had high total cholesterol levels which were associated with abnormalities in fasting glucose metabolism compared to lean or overweight subjects with normal waist circumference [43].
Figure 1. The interdependent link between dyslipidemia, beta-cell dysfunction, and type 2 diabetes.
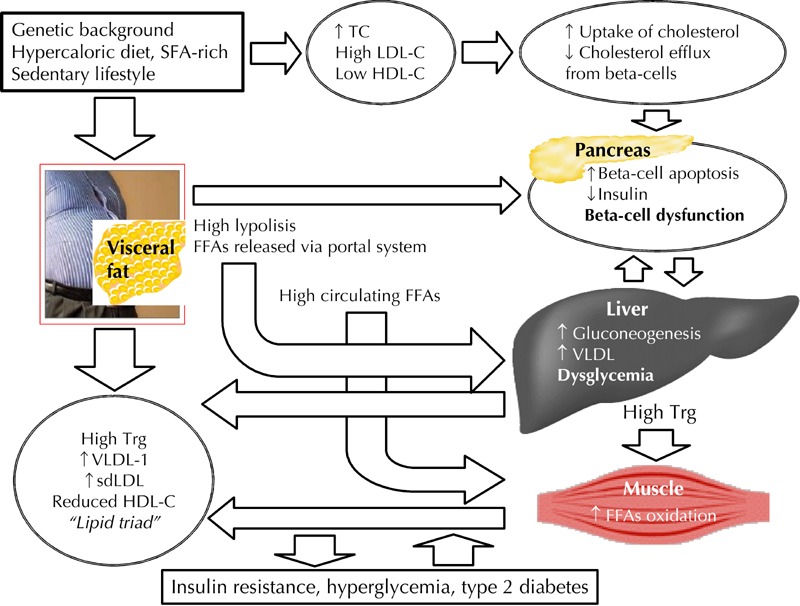
Cholesterol is a Janus-faced element in the development of dyslipidemia, diabetes, and vascular complications. Wrong lifestyle causes an accumulation of visceral fat associated with an excessive release of free fatty acids (FFAs) into the blood stream. These FFAs enter the portal vein and travel to the liver where they induce hepatic gluconeogenesis and elevated synthesis of VLDL lipoproteins. Also, they contribute to reduced glucose uptake in muscles and beta-cell lipotoxicity. These mechanisms explain the classic "lipid triad" (high VLDL1, high sdLDL, and reduced HDL particles), as observed in insulin resistance and type 2 diabetes. On the other hand, the same wrong lifestyles may impair beta-cell function through hypercholesterolemia, high LDL-C, and low HDL-C, independent of the insulin resistance status. Abbreviations: FFAs - fatty free acids, HDL-C - high-density lipoprotein cholesterol, LDL-C - low-density lipoprotein cholesterol, sdLDL - small dense LDL, SFAs - saturated fatty free acids, TC - total cholesterol, Trg – triglycerides, VLDL - very low-density lipoprotein.
Several studies addressed the role of LDL-C and low HDL-C levels as independent risk factors for beta-cell dysfunction. To this end, various researchers have clearly described specific receptors for LDL-C, namely LRPs, present in pancreatic islets, and they showed that LDL-C particles are incorporated in the metabolic pathway of beta-cells in a highly selective manner [44, 45]. However, this increased bioavailability of LDL-C in the pancreatic cell metabolism seems to have a cytotoxic effect, and may cause increased beta-cell apoptosis. This negative effect on beta-cell survival may be explained by the binding of the excessive cholesterol to the increased dimerization of the enzyme NO synthase (NOS), which downregulates the activity of glucokinase, thereby reducing the intra-cytoplasmic metabolism of glucose.
It is possible that cholesterol may also act in the exocytosis of insulin cytoplasmic granules of beta-cells. Since cholesterol is a constituent of cell membranes, it may alter the potassium channels and then impair the control of glucose-induced insulin secretion, if available in excess. Furthermore, beta-cell performance seems to be impaired in the presence of oxidized LDL particles, which is a typical feature in T2D patients, as reported above. This seems to be correlated with a decrease in insulin secretion and glucose-induced reduction of mRNA levels for preproinsulin. In this regard, observations from in vitro studies show that both treatment of cell cultures either with inhibitors of the c-Jun N-terminal kinase (JNK) pathway protein or with high concentrations of HDL particles are capable to prevent this toxic effect of oxidized LDL on insulin secretion and beta-cell survival. Several studies reported a close link between genes involved in the control of lipid metabolism and the development of impairment glucose control, thereby confirming the important impact of lipid metabolism on beta-cell activity.
The most convincing evidence is related to ATP-binding cassette member A1 (ABCA1), a particular protein involved in lipid transfer that removes excessive cholesterol from the cell to generate HDL lipoproteins. Thus, it plays a key role in the reversal cholesterol transfer pathway [46]. In this regard, the Tangier disease is an excellent model of HDL-C particle deficiency, as it is characterized by a mutation in the ABCA1 protein, which causes an impaired function, and eventually an inability of the peripheral cells to remove the excessive cholesterol. The resulting clinical picture is characterized by an increased occurrence to cardiovascular events in these patients and, surprisingly, reduced insulin secretion and hyperglycemia [47, 48]. In fact, in a Japanese study, four subjects affected by overt Tangier disease underwent OGTT and showed progressively increased plasma glucose concentrations until the diagnosis of T2D. The calculated insulinogenic index, an index of early beta-cell secretion during OGTT, was significantly lower than in controls [48]. This study suggests that the impairment of ABCA1 function may be directly involved in defective insulin secretion. It is noteworthy that these patients did not present the characteristic markers of the metabolic syndrome (including visceral obesity, hypertriglyceridemia, hypertension, and insulin resistance) nor a positive family history of diabetes.
The pathophysiology of the Tangier disease model has been confirmed in ABCA1 knock-out animals, which exhibited both an increased development of cardiovascular lesions and a progressive impairment of glucose tolerance [45]. The preserved insulin sensitivity demonstrated in these animals suggests that the impaired glucose regulation is exclusively linked to a direct toxic effect of cholesterol on the beta-cell [44]. The experimental data on ABCA1 as a modulator of insulin secretion have been confirmed by clinical observations [49]. In fact, rosiglitazone, an insulin sensitizer agonists of peroxisome proliferator agonist receptor gamma (PPAR-gamma), was demonstrated to increase the expression of ABCA1 on beta-cells [50, 51]. This indirect effect of rosiglitazone on beta-cell performance, together with the improvement of insulin resistance, was able to inhibit the development of T2D in subjects at high risk of T2D [52].
Tangier disease is not the only model for studying the association between HDL-C defects and hyperglycemia in humans. In fact, there are many data supporting the importance of ABCA1 gene mutations implicated in type 2 diabetes in several populations. In Mexican subjects for example, the polymorphous R230C variant of the ABCA1 gene was associated with hypercholesterolemia, low levels of HDL, and the metabolic syndrome. Moreover, in a Japanese population a polymorphism of intron 2 of ABCA1 was associated with 3 times increased risk of developing T2D. In the Copenhagen Heart Study, very low levels of HDL, increased cardiovascular risk, and a double incidence of T2D were observed in male carriers of the mutation K776N of ABCA1, compared to non-carriers of this polymorphism [53-55]. Further data were derived from observations in islets lacking beta-cell ABCA1. The islets showed an increased expression of another related cholesterol transporter, ABCG1, suggesting a compensatory mechanism for the lack of ABCA1 to maintain a better control of islet cholesterol concentration. Indeed, ABCG1 promotes cholesterol efflux to HDL particles and acts at the same level with ABCA1 to remove cellular cholesterol. Finally, in animal models, mice carrying a deletion of ABCG1 also presented an impaired glucose-induced insulin secretion [56]. ABCA1 and ABCG1 have complementary roles also in macrophage function. In addition to a massive lipid accumulation, loss of both ABCA1 and ABCG1 in macrophages leads to increased expression of inflammatory cytokines, with increased macrophage infiltration in pancreatic islets and enhanced susceptibility to cell apoptosis [57].
Beside Tangier disease, other genetic diseases have been proposed to disturb cholesterol metabolism. These considerations have led researchers to reexamine how cholesterol may interfere with beta-cell function in the context of such genetic diseases. One of these diseases involves dysfunction of the liver X receptor (LXR), an important activator of ABCA1. In animal models, LXR dysfunction caused an excessive accumulation of lipids in the pancreatic islets associated with impaired insulin secretion and impaired glucose tolerance. Based on this observation, other genetic studies on the modification of key proteins for cholesterol metabolism reported that patients with CHD and alterations of the liver LDL receptor-related protein 5 (LRP5) showed high levels of LDL-C, triglycerides, and fasting glucose [58]. Stearoyl coenzyme A (stearoyl-CoA) desaturase, the rate-limiting enzyme in the synthesis of monounsaturated fatty acids, has been found to be pivotal in the regulation of hepatic lipogenesis and lipid oxidation. Some mutations of this enzyme have been linked to alterations in intracellular lipid metabolism and development of obesity, metabolic syndrome, and T2D, suggesting a potential role as actor in glucose control. In support of this hypothesis, it has been demonstrated that stearoyl-CoA desaturase-deficient mice have increased energy expenditure, reduced body visceral fat masses, decreased insulin resistance, and they are not prone to high fat diet-induced obesity and liver steatosis [59].
Together with stearoyl-CoA desaturase, another important protein in hepatic lipid metabolism is the sterol regulatory element-binding protein-2 (SREBP-2), which is involved in hepatic synthesis of cholesterol. Transgenic mice, expressing high levels of SREBP-1a, showed an uncontrolled synthesis and pancreatic cholesterol accumulation, resulting in a progressive lipid infarction both in liver and beta-cells. Histological imagines of islets isolated from these transgenic animals showed insulin granules fewer in number and smaller in size, and a significant reduction of insulin granule content compared with controls [60]. Furthermore, it has been shown that the increased expression of SREBP-2 is associated with an inhibition of ABCA1 synthesis. Therefore, two potential ways of beta-cell damage and development of hyperglycemia have been proposed. Both of which can be induced by impaired cholesterol metabolism:
1. Reduction in the efflux of cholesterol from beta-cells (ABCA1 defect)
2. Enhanced synthesis and accumulation of cholesterol in beta-cells (increased SREBP-2 activity)
These considerations are derived from in vitro data and animal models only. Although it is expected that these mechanisms can occur in humans, experimental data cannot always be extrapolated to the human pathophysiology. However, since HDL-C particles represent the main acceptor of cholesterol in the reverse cholesterol transport, and cholesterol levels have a toxic effect on beta-cells, we can draw the conclusion that HDL-C lipoproteins may have an adjunctive and protective effect on beta-cells. In fact, both laboratory data and in vivo studies showed that impaired functions of HDL-C particles are associated with impaired protection from oxidative stress, a typical observation in T2D. The administration of recombinant paraoxonase-1, the principal antioxidant enzyme on the surface of HDL particles, to streptozotocin-induced diabetic mice showed a marked antioxidant and an adjunctive positive effect on insulin secretion [61].
Recently, Dullaart and colleagues explored the relationship between pancreatic beta-cell function and the functionality of HDL-C in well-controlled type 2 diabetic patients [62]. The activity of HDL-C particles, in particular the antioxidative capacity of HDL and the cellular cholesterol efflux out of cultured fibroblasts, was tested by the inhibition of LDL oxidation in vitro, while beta-cell function was defined by the homeostasis model assessment beta (HOMA-beta). The results showed that both HDL antioxidative capacity and cellular cholesterol efflux were positively correlated with HOMA-beta measures. These data demonstrated that a better functionality of HDL particles may contribute to the maintenance of a healthy beta-cell function in subjects with well-controlled T2D. In an in vivo study, intravenous recombinant HDL in T2D patients has quickly allowed to reduce blood glucose levels, with a simultaneous increase in insulin concentrations and beta-cell function and a significant improvement of insulin sensitivity [63].
Several in vitro and animal studies have shown that the effectors responsible for the actions of HDL-C on insulin secretion could be attributed to the apolipoproteins Apo-AI and Apo-AII. Also, it has been demonstrated that HDL-C particles are able to activate adenosine monophosphate-activated protein (AMP) kinase, inducing an uptake of glucose in skeletal muscles. Thus, it is conceivable that the effect of HDL-C on glucose metabolism is linked to a direct protection of beta-cells, and to an enhanced glucose uptake in peripheral muscles. To test the involvement of HDL-C in glucose metabolism, we recently examined whether insulin-resistant subjects with normal and impaired glucose tolerance at high risk of developing T2D (positive first-degree family history for T2D and/or overweight/obesity) show a positive linear correlation between indices of beta-cell function and HDL-C concentrations. We found that the insulinogenic index for early insulin secretion and the insulin secretion sensitivity for total beta-cell function were correlated to HDL-C concentrations after OGTT in patients with impaired fasting glucose (IFG) and impaired glucose tolerance (IGT), but not in subjects with normal glucose tolerance (NGT) and normal HDL-C levels [64]. This result suggests that low HDL-C levels in patients at risk of T2D may play a role in beta-cell dysfunction.
However, genetic studies, which analyzed specific mutations in the HDL-C gene, have shown equivocal results in subjects with insulin resistance, raising the question whether low levels of HDL-C particles are a consequence or a determinant of metabolic damage. In this regard, European carriers of mutations for HDL particles, who had an excessive visceral fat mass and a phenotype typical of insulin resistance, showed extremely low levels of HDL-C and increased levels of triglycerides [55]. However, in another human model of genetic defects for HDL cholesterol, low levels of these particles were observed to be associated with reduced glucose tolerance, even in subjects with normal weight and without the anthropometric characteristics of insulin resistance [65]. Also, mutations have been described in several enzymes involved in lipid control; for example polymorphisms in the CETP enzyme were associated with changes in enzyme activity, lipid concentrations, and risk of developing T2D, as observed in the Telde Study [66]. Taken together, these observations suggest that variable levels of HDL-C are associated with different phenotypes, confirming the hypothesis that low HDL-C may be a determinant and not a consequence of the state of insulin resistance.
The observation of a low incidence of T2D in patients with familial hypercholesterolemia caused concern about the importance of cholesterol in glucose metabolism. However, this apparent contradiction actually confirms the hypothesized pathogenic mechanism of cholesterol; if available in excess, it is an inducer of beta-cell dysfunction and, consequently, of impaired glucose metabolism. In fact, patients with severe hypercholesterolemia present a downregulation of LDL receptors on beta-cells, reducing the uptake of cholesterol by beta-cells, and thus preventing beta-cell apoptosis.
Some concerns appeared in the literature after publication of a meta-analysis performed on large statin trials [67]. Note that statins, the inhibitors of 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase, are the most important drugs for the control of cholesterol synthesis and reduction of total and LDL cholesterol levels. Their use has been associated with an increased incidence of T2D [68]. However, with exception of the WOSCOP study, where the use of pravastatin was associated with a reduction of approximately 30% in the incidence of new cases of T2D, therapy with high doses of statins mostly corresponded to a slight increase in T2D incidence, e.g. in the JUPITER study, with rosuvastatin, by around 0.6%.
Recently, a systematic review and meta-analysis concluded that the use of statins does not account for a significant impact on insulin sensitivity without a real "class effect" in patients without diabetes mellitus [69]. In particular, pravastatin alone seems to improve insulin sensitivity, while atorvastatin, rosuvastatin, and simvastatin rather cause an impairment in insulin resistance. The reason for this apparent discrepancy is not clear at present. It may be related to the observation that certain statins at high doses exert a negative regulation on insulin secretion in vitro, irrespective of their cholesterol-lowering effect that, in some way, counterbalance the "positive effect" consisting of cholesterol depletion in beta-cells. Another recent paper proposed an alternative explanation, focusing on the role of the number of diabetic risk factors such as hypertriglyceridemia, obesity, hypertension, and hyperglycemia present in patients with CHD as modulator of the diabetogenic effect of atorvastatin [70].
Concluding remarks
In this review, we intended to elucidate the various and interdependent mechanisms associated with impaired lipid regulation in the common forms of diabetes mellitus. In type 2 diabetes, the available data confirm the well-established pivotal association of visceral fat, insulin resistance, and FFAs with the "lipid triad", i.e. (1) increased plasma triglyceride levels, (2) decreased HDL-cholesterol concentrations, and (3) the presence of small, dense LDL particles. Whereas, the interrelation of dyslipidemia and type 1 diabetes is controversial. A series of qualitative abnormalities of lipoproteins are described in T1D patients, including increased cholesterol/triglyceride ratios within VLDL, increased triglycerides in LDL and HDL cholesterols, compositional changes in the peripheral layer of lipoproteins, glycation of apolipoproteins, increased oxidation of LDL, and increased sdLDL particles. All of these abnormalities are potentially atherogenic. In addition, HDL-C particles from T1D patients have reduced antioxidative and vasorelaxant properties, and it is assumed that they are less effective in promoting cholesterol efflux from cells.
The link between T2D and dyslipidemia may be considered a Janus-faced relation. While the impact of diabetes on abnormalities in lipid metabolism is well-established, recent data provide evidence for a reverse direction, assigning a key role for hypercholesterolemia and low HDL-C levels in the development of beta-cell dysfunction. However, more targeted studies are needed to better understand the link between dyslipidemia and diabetes. It will be important to understand how lipid disorders can be metabolically dangerous for beta-cells to define the optimal therapy.
Disclosure: The authors report no conflict of interests.
Acknowledgments
This study was supported in part by unconditioned grants from Pfizer Italy and Abiogen Pharma, Pisa, Italy.
References
- 1.Soedamah-Muthu SS, Fuller JH, Mulnier HE, Raleigh VS, Lawrenson RA, Colhoun HM. All-cause mortality rates in patients with type 1 diabetes mellitus compared with a non-diabetic population from the UK general practice research database, 1992-1999. Diabetologia. 2006;49:660–666. doi: 10.1007/s00125-005-0120-4. [DOI] [PubMed] [Google Scholar]
- 2.Miettinen TA, Gylling H, Tuominen J, Simonen P, Koivisto V. Low synthesis and high absorption of cholesterol characterize type 1 diabetes. Diabetes Care. 2004;27:53–58. doi: 10.2337/diacare.27.1.53. [DOI] [PubMed] [Google Scholar]
- 3.Maahs DM, Hokanson JE, Wang H, Kinney GL, Snell-Bergeon JK, East A, Bergman BC, Schauer IE, Rewers M, Eckel RH. Lipoprotein subfraction cholesterol distribution is proatherogenic in women with type 1 diabetes and insulin resistance. Diabetes. 2010;59:1771–1779. doi: 10.2337/db09-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giuffrida FM, Guedes AD, Rocco ER, Mory DB, Dualib P, Matos OS, Chaves-Fonseca RM, Cobas RA, Negrato CA, Gomes MB, Dib SA. Heterogeneous behavior of lipids according to HbA1c levels undermines the plausibility of metabolic syndrome in type 1 diabetes: data from a nationwide multicenter survey. Cardiovasc Diabetol. 2012;11:156. doi: 10.1186/1475-2840-11-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352(9131):837–853. [PubMed] [Google Scholar]
- 6.Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 7.National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III): final report. Circulation. 2002;106(25):3143–3421. [PubMed] [Google Scholar]
- 8.Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, Clark LT, Hunninghake DB, Pasternak RC, Smith SC Jr, Stone NJ. et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 9.Cholesterol Treatment Trialists' (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomized trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fruchart JC, Sacks F, Hermans MP, Assmann G, Brown WV, Ceska R, Chapman MJ, Dodson PM, Fioretto P, Ginsberg HN. et al. The Residual Risk Reduction Initiative: a call to action to reduce residual vascular risk in patients with dyslipidemia. Am J Cardiol. 2008;102(10 Suppl):1K–34K. doi: 10.1016/S0002-9149(08)01833-X. [DOI] [PubMed] [Google Scholar]
- 11.Pedersen TR, Kjekshus J, Berg K, Haghfelt T, Faergeman O, Faergeman G, Pyörälä K, Miettinen T, Wilhelmsen L, Olsson AG. et al. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Atheroscler Suppl. 2004;5:81–87. doi: 10.1016/j.atherosclerosissup.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 12.Olsson AG, Lindahl C, Holme I, Fayyad R, Faergeman O, Kastelein JJ, Tikkanen MJ, Larsen ML, Pedersen TR Incremental Decrease in End Points Through Aggressive Lipid Lowering Study Group. LDL cholesterol goals and cardiovascular risk during statin treatment: the IDEAL study. Eur J Cardiovasc Prev Rehabil. 2011;18:262–269. doi: 10.1177/1741826710389391. [DOI] [PubMed] [Google Scholar]
- 13.Mora S, Wenger NK, Demicco DA, Breazna A, Boekholdt SM, Arsenault BJ, Deedwania P, Kastelein JJ, Waters DD. Determinants of residual risk in secondary prevention patients treated with high- versus low-dose statin therapy: the Treating to New Targets (TNT) study. Circulation. 2012;125(16):1979–1987. doi: 10.1161/CIRCULATIONAHA.111.088591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taskinen MR. LDL-cholesterol, HDL-cholesterol or triglycerides: which is the culprit? Diabetes Res Clin Pract. 2003;61(Suppl 1):S19–S26. doi: 10.1016/s0168-8227(03)00126-8. [DOI] [PubMed] [Google Scholar]
- 15.Barter P, Gotto AM, LaRosa JC, Maroni J, Szarek M, Grundy SM, Kastelein JJ, Bittner V, Fruchart JC Treating to New Targets Investigators. HDL cholesterol, very low levels of LDL cholesterol and cardiovascular events. N Engl J Med. 2007;357:1301–1310. doi: 10.1056/NEJMoa064278. [DOI] [PubMed] [Google Scholar]
- 16.Tilly-Kiesi M, Syvänne M, Kuusi T, Lahdenperä S, Taskinen MR. Abnormalities of low density lipoproteins in normolipidemic type 2 diabetic and nondiabetic patients with coronary artery disease. J Lipid Res. 1992;33:333–342. [PubMed] [Google Scholar]
- 17.Stewart MW, Laker MF, Dyer RG, Game F, Mitcheson J, Winocour PH, Alberti KG. Lipoprotein compositional abnormalities and insulin resistance in type 2 diabetic patients with mild hyperlipidemia. Arterioscler Thromb. 1993;13:1046–1052. doi: 10.1161/01.atv.13.7.1046. [DOI] [PubMed] [Google Scholar]
- 18.Lahdenperä S, Syvänne M, Kahri J, Taskinen MR. Regulation of low-density lipoprotein particle size distribution in NIDDM and coronary disease: importance of serum triglycerides. Diabetologia. 1996;39:453–461. doi: 10.1007/BF00400677. [DOI] [PubMed] [Google Scholar]
- 19.Mooradian AD. Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5:150–159. doi: 10.1038/ncpendmet1066. [DOI] [PubMed] [Google Scholar]
- 20.Tchernof A, Despres JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93:359–404. doi: 10.1152/physrev.00033.2011. [DOI] [PubMed] [Google Scholar]
- 21.Adiels M, Taskinen MR, Boren J. Fatty liver, insulin resistance, and dyslipidemia. Curr Diabetes Rep. 2008;8:60–64. doi: 10.1007/s11892-008-0011-4. [DOI] [PubMed] [Google Scholar]
- 22.Adiels M, Olofsson SO, Taskinen MR, Boren J. Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of Very Low-Density Lipoproteins Is the Hallmark of the Dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–1236. doi: 10.1161/ATVBAHA.107.160192. [DOI] [PubMed] [Google Scholar]
- 23.Caixas A, Ordonez-Llanos J, de Leiva A, Payes A, Homs R, Perez A. Optimization of glycemic control by insulin therapy decreases the proportion of small dense LDL particles in diabetic patients. Diabetes. 1997;46:1207–1213. doi: 10.2337/diab.46.7.1207. [DOI] [PubMed] [Google Scholar]
- 24.Ginsberg HN, Illingworth R. Postprandial dyslipidemia: an atherogenic disorder common in patients with diabetes mellitus. Am J Cardiol. 2001;88(Suppl):H9–H15. doi: 10.1016/s0002-9149(01)01831-8. [DOI] [PubMed] [Google Scholar]
- 25.Panarotto D, Remillard P, Bouffard L, Maheux P. Insulin resistance affects the regulation of lipoprotein lipase in the postprandial period and in an adipose tissue-specific manner. Eur J Clin Invest. 2002;32:84–92. doi: 10.1046/j.1365-2362.2002.00945.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu M, Chung S, Shelness GS, Parks JS. Hepatic ABCA1 and VLDL triglyceride production. Biochim Biophys Acta. 2012;1821:770–777. doi: 10.1016/j.bbalip.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krauss RM. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care. 2004;27:1496–1504. doi: 10.2337/diacare.27.6.1496. [DOI] [PubMed] [Google Scholar]
- 28.Tchernof A, Lamarche B, Prud'Homme D, Nadeau A, Moorjani S, Labrie F, Lupien PJ, Despres JP. The dense LDL phenotype: association with plasma lipoprotein levels, visceral obesity and hyperinsulinemia in men. Diabetes Care. 1996;19:629–637. doi: 10.2337/diacare.19.6.629. [DOI] [PubMed] [Google Scholar]
- 29.Ambrosch A, Mühlen I, Kopf D, Augustin W, Dierkes J, König W, Luley C, Lehnert H. LDL size distribution in relation to insulin sensitivity and lipoprotein pattern in young and healthy subjects. Diabetes Care. 1998;21:2077–2084. doi: 10.2337/diacare.21.12.2077. [DOI] [PubMed] [Google Scholar]
- 30.Tan CE, Chew LS, Chio LF, Tai ES, Lim HS, Lim SC, Jayakumar L, Eng HK, Packard CJ. Cardiovascular risk factors and LDL subfraction profile in type 2 diabetes mellitus subjects with good glycaemic control. Diabetes Res Clin Pract. 2001;51:107–114. doi: 10.1016/s0168-8227(00)00211-4. [DOI] [PubMed] [Google Scholar]
- 31.Panarotto D, Remillard P, Bouffard L, Maheux P. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–754. doi: 10.1038/nature00804. [DOI] [PubMed] [Google Scholar]
- 32.Soran H, Durrington PN. Susceptibility of LDL and its subfractions to glycation. Curr Opin Lipidol. 2011;22:254–261. doi: 10.1097/MOL.0b013e328348a43f. [DOI] [PubMed] [Google Scholar]
- 33.Tan KC. Reverse cholesterol transport in type 2 diabetes mellitus. Diabetes Obes Metab. 2009;11:534–543. doi: 10.1111/j.1463-1326.2008.01012.x. [DOI] [PubMed] [Google Scholar]
- 34.deGoma EM, Leeper NJ, Heidenreich PA. Clinical significance of high-density lipoprotein cholesterol in patients with low low-density lipoprotein cholesterol. J Am Coll Cardiol. 2008;51:49–55. doi: 10.1016/j.jacc.2007.07.086. [DOI] [PubMed] [Google Scholar]
- 35.deGoma EM, Rader DJ. High-density lipoprotein particle number: a better measure to quantify high-density lipoprotein? J Am Coll Cardiol. 2012;60:517–520. doi: 10.1016/j.jacc.2012.03.058. [DOI] [PubMed] [Google Scholar]
- 36.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL. et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murakami H, Tanabe J, Tamasawa N, Matsumura K, Yamashita M, Matsuki K, Murakami H, Matsui J, Suda T. Reduction of paraoxonase-1 activity may contribute the qualitative impairment of HDL particles in patients with type 2 diabetes. Diabetes Res Clin Pract. 2013;99:30–38. doi: 10.1016/j.diabres.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 38.Cui Y, Blumenthal RS, Flaws JA, Whiteman MK, Langenberg P, Bachorik PS, Bush TL. Non-high-density lipoprotein cholesterol level as a predictor of cardiovascular disease mortality. Arch Intern Med. 2001;161:1413–1419. doi: 10.1001/archinte.161.11.1413. [DOI] [PubMed] [Google Scholar]
- 39.Lu W, Resnick HE, Jablonski KA, Jones KL, Jain AK, Howard WJ, Robbins DC, Howard BV. Non-HDL cholesterol as a predictor of cardiovascular disease in type 2 diabetes: the Strong Heart Study. Diabetes Care. 2003;26:16–23. doi: 10.2337/diacare.26.1.16. [DOI] [PubMed] [Google Scholar]
- 40.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1974;18:499–502. [PubMed] [Google Scholar]
- 41.Gylling H, Hallikainen M, Pihlajamäki J, Simonen P, Kuusisto J, Laakso M, Miettinen TA. Insulin sensitivity regulates cholesterol metabolism to a greater extent than obesity: lessons from the METSIM Study. J Lipid Res. 2010;51:2422–2427. doi: 10.1194/jlr.P006619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lally S, Tan CY, Owens D, Tomkin GH. Messenger RNA levels of genes involved in dysregulation of postprandial lipoproteins in type 2 diabetes: the role of Niemann-Pick C1-like 1, ATP-binding cassette, transporters G5 and G8, and of microsomal triglyceride transfer protein. Diabetologia. 2006;49:1008–1016. doi: 10.1007/s00125-006-0177-8. [DOI] [PubMed] [Google Scholar]
- 43.Giannini S, Bardini G, Dicembrini I, Monami M, Rotella CM, Mannucci E. Lipid levels in obese and nonobese subjects as predictors of fasting and postload glucose metabolism. J Clin Lipidol. 2012;6:132–138. doi: 10.1016/j.jacl.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Brunham LR, Kruit JK, Verchere CB, Hayden MR. Cholesterol in islet dysfunction and type 2 diabetes. Journal Clin Invest. 2008;118:403–408. doi: 10.1172/JCI33296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kruit JK, Kremer PH, Dai L, Tang R, Ruddle P, de Haan W, Brunham LR, Verchere CB, Hayden MR. Cholesterol efflux via ATP-binding cassette transporter A1 (ABCA1) and cholesterol uptake via the LDL receptor influences cholesterol-induced impairment of beta cell function in mice. Diabetologia. 2010;53:1110–1119. doi: 10.1007/s00125-010-1691-2. [DOI] [PubMed] [Google Scholar]
- 46.Oram JF, Vaughan AM. ABCA1-mediated transport of cellular cholesterol and phospholipids to HDL apolipoproteins. Curr Opin Lipidol. 2000;11(3):253–260. doi: 10.1097/00041433-200006000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Schaefer EJ, Santos RD, Asztalos BF. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol. 2010;21:289–297. doi: 10.1097/MOL.0b013e32833c1ef6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koseki M, Matsuyama A, Nakatani K, Inagaki M, Nakaoka H, Kawase R, Yuasa-Kawase M, Tsubakio-Yamamoto K, Masuda D, Sandoval JC. et al. Impaired insulin secretion in four Tangier disease patients with ABCA1 mutations. J Atheroscler Thromb. 2009;16:292–296. doi: 10.5551/jat.e599. [DOI] [PubMed] [Google Scholar]
- 49.Vergeer M, Brunham LR, Koetsveld J, Kruit JK, Verchere CB, Kastelein JJ, Hayden MR, Stroes ES. Carriers of loss-of-function mutations in ABCA1 display pancreatic beta-cell dysfunction. Diabetes Care. 2010;33:869–874. doi: 10.2337/dc09-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, Marsh BJ, Rodrigues B, Johnson JD, Parks JS, Verchere CB, Hayden MR. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med. 2007;13:340–347. doi: 10.1038/nm1546. [DOI] [PubMed] [Google Scholar]
- 51.Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, Teissier E, Minnich A, Jaye M, Duverger N. et al. PPARalpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- 52.DREAM On (Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication Ongoing Follow-up) Investigators. Gerstein HC, Mohan V, Avezum A, Bergenstal RM, Chiasson JL, Garrido M, MacKinnon I, Rao PV, Zinman B, et al. Long-term effect of rosiglitazone and/or ramipril on the incidence of diabetes. Diabetologia. 2011;54:487–495. doi: 10.1007/s00125-010-1985-4. [DOI] [PubMed] [Google Scholar]
- 53.Frikke-Schmidt R, Nordestgaard BG, Schnohr P, Steffensen R, Tybjaerg-Hansen A. Mutation in ABCA1 predicted risk of ischemic heart disease in the Copenhagen City Heart Study Population. J Am Coll Cardiol. 2005;46:1516–1520. doi: 10.1016/j.jacc.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 54.Frikke-Schmidt R, Nordestgaard BG, Stene MC, Sethi AA, Remaley AT, Schnohr P, Grande P, Tybjaerg-Hansen A. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA. 2008;299:2524–2532. doi: 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- 55.Schou J, Tybjærg-Hansen A, Moller HJ, Nordestgaard BG, Frikke-Schmidt R. ABC transporter genes and risk of type 2 diabetes: a study of 40,000 individuals from the general population. Diabetes Care. 2012;35:2600–2606. doi: 10.2337/dc12-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fryirs MA, Barter PJ, Appavoo M, Tuch BE, Tabet F, Heather AK, Rye KA. Effects of HDL on pancreatic beta-cell insulin secretion. Arterioscler Thromb Vasc Biol. 2010;30:1642–1648. doi: 10.1161/ATVBAHA.110.207373. [DOI] [PubMed] [Google Scholar]
- 57.Kruit JK, Wijesekara N, Westwell-Roper C, Vanmierlo T, de Haan W, Bhattacharjee A, Tang R, Wellington CL, LütJohann D, Johnson JD. et al. Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired beta-cell function. Diabetes. 2012;61(3):659–664. doi: 10.2337/db11-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saarinen A, Saukkonen T, Kivelä T, Lahtinen U, Laine C, Somer M, Toiviainen-Salo S, Cole WG, Lehesjoki AE, Mäkitie O. Low density lipoprotein receptor-related protein 5 (LRP5) mutations and osteoporosis, impaired glucose metabolism and hypercholesterolaemia. Clin Endocrinol. 2010;72:481–488. doi: 10.1111/j.1365-2265.2009.03680.x. [DOI] [PubMed] [Google Scholar]
- 59.Yokoyama S, Hosoi T, Ozawa K. Stearoyl-CoA Desaturase 1 (SCD1) is a key factor mediating diabetes in MyD88-deficient mice. Gene. 2012;497:340–343. doi: 10.1016/j.gene.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 60.Iwasaki Y, Iwasaki H, Yatoh S, Ishikawa M, Kato T, Matsuzaka T, Nakagawa Y, Yahagi N, Kobayashi K, Takahashi A. et al. Nuclear SREBP-1a causes loss of pancreatic beta-cells and impaired insulin secretion. Biochem Biophys Res Commun. 2009;378:545–550. doi: 10.1016/j.bbrc.2008.11.105. [DOI] [PubMed] [Google Scholar]
- 61.Koren-Gluzer M, Aviram M, Meilin E, Hayek T. The antioxidant HDL-associated paraoxonase-1 (PON1) attenuates diabetes development and stimulates beta-cell insulin release. Atherosclerosis. 2011;219:510–518. doi: 10.1016/j.atherosclerosis.2011.07.119. [DOI] [PubMed] [Google Scholar]
- 62.Dullaart RP, Annema W, de Boer JF, Tietge UJ. Pancreatic beta-cell function relates positively to HDL functionality in well-controlled type 2 diabetes mellitus. Atherosclerosis. 2012;222:567–573. doi: 10.1016/j.atherosclerosis.2012.03.037. [DOI] [PubMed] [Google Scholar]
- 63.Drew BG, Duffy SJ, Formosa MF, Natoli AK, Henstridge DC, Penfold SA, Thomas WG, Mukhamedova N, de Courten B, Forbes JM. et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation. 2009;21:2103–2111. doi: 10.1161/CIRCULATIONAHA.108.843219. [DOI] [PubMed] [Google Scholar]
- 64.Bardini G, Dicembrini I, Rotella CM, Giannini S. Correlation between HDL cholesterol levels and beta-cell function in subjects with various degree of glucose tolerance. Acta Diabetologica. 2012 doi: 10.1007/s00592-011-0339-0. In press. [DOI] [PubMed] [Google Scholar]
- 65.Soro-Paavonen A, Naukkarinen J, Lee-Rueckert M, Watanabe H, Rantala E, Soderlund S, Hiukka A, Kovanen PT, Jauhiainen M, Peltonen L, Taskinen MR. Common ABCA1 variants, HDL levels, and cellular cholesterol efflux in subjects with familial low HDL. J Lipid Res. 2007;48:1409–1416. doi: 10.1194/jlr.P600012-JLR200. [DOI] [PubMed] [Google Scholar]
- 66.Lopez-Rios L, Novoa FJ, Chirino R, Varillas F, Boronat-Cortes M, Wägner AM. Interaction between cholesteryl ester transfer protein and hepatic lipase encoding genes and the risk of type 2 diabetes: results from the Telde study. PLoS One. 2011;6:e27208. doi: 10.1371/journal.pone.0027208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rajpathak SN, Kumbhani DJ, Crandal J, Barzilai N, Alderman M, Ridker PM. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care. 2009;32:1924–1929. doi: 10.2337/dc09-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sattar N, Taskinen MR. Statins are diabetogenic - myth or reality? Atheroscler Suppl. 2012;13:1–10. doi: 10.1016/j.atherosclerosissup.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Baker WL, Talati R, White CM, Coleman CI. Differing effect of statins on insulin sensitivity in non-diabetics: a systematic review and meta-analysis. Diabetes Res Clin Pract. 2010;87:98–107. doi: 10.1016/j.diabres.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 70.Waters DD, Ho JE, Boekholdt SM, Demicco DA, Kastelein JJ, Messig M, Breazna A, Pedersen TR. Cardiovascular event reduction versus new-onset diabetes during atorvastatin therapy: effect of baseline risk factors for diabetes. Am Coll Cardiol. 2013;61:148–152. doi: 10.1016/j.jacc.2012.09.042. [DOI] [PubMed] [Google Scholar]