

Abstract
BACKGROUND: Diabetes mellitus, characterized by chronic hyperglycemia, is known to have a deleterious effect on erythrocyte structure and hemodynamic characteristics, which eventually contribute to diabetes-associated vascular complications. Protein kinase C alpha (PKCα) is a major regulator of many metabolic processes and structural changes in erythrocytes, and may play a significant role in the development of hyperglycemia-mediated cellular abnormalities. AIM: We hypothesized that acute hyperglycemic stress may affect erythrocyte structure and metabolic properties through its effect on PKCα membrane content and activity. RESULTS: Erythrocytes, from healthy individuals acutely exposed to a glucose enriched media, showed a significant decrease in the membranous fraction of PKCα and its phosphorylation (p = 0.005 and p = 0.0004, respectively). These alterations correlated with decreased affinity of PKCα to its membrane substrates (4.1R and GLUT1) and reduced RBC deformability (p = 0.017). Pre-activation of erythrocytes with PKC activator, PMA, minimized the effect of glucose on the membrane PKCα fraction and RBC deformability (p > 0.05). CONCLUSIONS: Acute glycemia-induced inhibition of PKCα membranous translocation and activation is associated with reduced erythrocyte membrane deformability.
Keywords: diabetes, protein kinase C, erythrocyte, acute, red blood cell
Abbreviations: ATP - adenosine triphosphate; BMI - body mass index; BSA - bovine serum albumin; DTT - dithiothreitol; EDTA - ethylenediaminetetraacetic acid; EGTA - ethyleneglycol-bis (β-aminoethyl ether)-N,N'-tetraacetic acid; ER - elongation ratio; FL - fluorescence; GAPDH - glyceraldehyde 3-phosphate dehydrogenase; GLUT1 - glucose transporter 1; HbA1c - glycosylated hemoglobin A1c; HDL - high-density lipoprotein; HRP - horseradish peroxidase; IB - immunoblotting; IP - immunoprecipitation; LDL - low-density lipoprotein; MCHC - mean corpuscular hemoglobin concentration; MCV - mean cellular volume; PBS - phosphate-buffered saline; PKC - protein kinase C; PMA - phorbol 12-myristate 13-acetate; PS - phosphatidylserine; PVDF - polyvinylidene difluoride; RBC - red blood cell; RDW - red cell distribution width; SDS-PAGE - sodium dodecyl sulfate polyacrylamide gel electrophoresis; SE - standard error ; TBST - Tris-buffered saline with Tween-20; Tris-HCl - Tris(hydroxymethyl)aminomethane hydrochloride
Introduction
Hyperglycemia, one of the fundamental abnormalities in type 1 and type 2 diabetes, is associated with the development of microvascular and macrovascular complications [1]. Hyperglycemia-associated vascular complications result from several pathological processes including endothelial cell dysfunction [2], abnormalities in smooth muscle properties, and changes in hemodynamic and functional features of red blood cells (RBCs, or erythrocytes) [3]. Chronic hyperglycemic stress has been proposed to lead to an increase in intra-erythrocytic levels of reactive oxygen species and carbonyl compounds [4], accompanied by an increase in non-enzymatic protein glycosylation, and a possibly associated reduction in phosphorylation or cross-linkage of membrane proteins [5-9]. Also, elevations in intracellular calcium [10], sorbitol, and diacylglycerol [11, 12] have been observed, all of which result in an imbalance of membrane lipids [13], alteration in membrane fluidity [14], and shortened RBC survival [15].
Most of these processes have been correlated with reduced membrane deformability in erythrocytes of diabetic patients [3, 8, 16]. RBC deformability is maintained by cytoskeleton proteins [17]. Members of the protein kinase C (PKC) family play a key role in the regulation of membrane stability and deformability [18-21], via activation of the membrane skeleton components [20, 22], or via phosphorylation of numerous membrane transport components [23-26], which may, in turn, influence the interactions of the cytoskeletal proteins.
Hyperglycemia and diabetes are associated with chronic alterations of PKC activity and/or level in most body tissues, a change implicated in the pathogenesis of diabetic microvascular complications, including nephropathy and neuropathy [27]. There are no reports to date documenting changes in the PKC membranous fraction or in PKC activation in RBCs from diabetic patients, in contrast to PKC changes observed in other tissues of diabetic patients. We hypothesized that PKC does play a role in human RBCs, mediating the effect of prolonged or short-term hyperglycemia to changes in RBC deformability. Therefore, in this study we examined whether RBCs from healthy individuals acutely exposed to elevated glucose levels influenced the activation and membrane content of PKC. Also, we studied simultaneous changes in cellular, metabolic and structural properties, which have been reported to be interrelated and influenced by PKC regulation.
Materials and methods
Materials
Rabbit polyclonal antibodies to cPKCα (C-20), GAPDH, and HRP-conjugated secondary antibodies were bought from Santa Cruz Biotechnology (Heidelberg, Germany). Rabbit monoclonal antibodies to T638-phosphorylated PKCα (E195), mouse monoclonal antibodies to 4.1R, and human glucose transporter 1 (SPM498) were obtained from Zotal Biologicals and Instrumentations (Tel-Aviv, Israel). All other chemicals and reagents were obtained from Sigma-Aldrich (Rehovot, Israel), unless otherwise indicated.
Cell preparation
Twenty-four healthy, non-diabetic volunteers were recruited from the local community. After obtaining informed consent, the volunteers underwent a thorough evaluation, including medical history, physical examination, and laboratory analyses (serum electrolytes, liver function tests, and a complete blood count). 50 ml of venous blood was collected in heparinized tubes. Leukocytes were removed by centrifugation at 1,000g for 5 min at 25ºC, and the erythrocytes were then washed three times with PBS buffer (137 mM NaCl, 8.9 mM Na2HPO4, 1.5 mM KH2PO4, 2.7 mM KCl, and 2 mM CaCl2 in double deionized H2O, pH 7.4). The washed cells were kept in PBS buffer at ~80% hematocrit on ice before treatment.
The erythrocytes were further exposed to physiologically high 20 mM D-glucose for 5 minutes at 37°C (denoted as 'D-glucose 20'). As a physiological control, corresponding studies with 5 mM D-glucose ('D-glucose 5') were performed. To maintain the same medium osmolarity, we supplemented the 5 mM D-glucose medium with an appropriate (15 mM) concentration of a D-glucose inactive enantiomer - L-glucose [34]. We performed corresponding studies in media in which D-glucose was completely substituted by L-glucose ('D-glucose 0') as a negative control and to mimic hypoglycemic conditions.
Erythrocyte membranes were isolated in accordance with the experimental procedure, described previously [28]. Briefly, the cells were washed twice in PBS buffer, and then incubated in 30 volumes of ice-cold hypotonic lysis medium (10 mM Tris-HCl and 2 mM EGTA, pH 7.4) for 30 min. The membranous and cytosolic fractions were separated by 27,000g centrifugation for 10 min at 4°C. Membranes were then washed, lysed, and harvested again. Finally, the isolated membranes were aliquoted and preserved at -20°C for further analysis. Protein content in the membrane fractions was determined by the commonly-used Bradford method. The cell lysate samples, or membrane fractions with equalized protein content, were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on polyacrylamide gels, and immunoblotted by Western blotting.
For immunoblotting (IB) analysis, samples were separated by SDS/PAGE using a 10% acrylamide gel, and subsequently transferred electrophoretically to PVDF membranes (Millipore, Bedford, MA, USA). Transferred membranes were incubated with 5% skimmed milk in TBST (1% 2M Tris-HCl, pH 8.0, 0.3% 5M NaCl, and 0.5% Tween 20 in double deionized H2O) for 1 hr to block non-specific binding sites. The blots were then incubated with a primary antibody (1:1,000 dilution) in TBST supplemented with 5% BSA for 1 hr at 25ºC. The PVDF membranes were then washed with TBST, and incubated for 1 hr with a secondary antibody (1:10,000 dilution). The blot was washed several times with TBST, incubated with a chemiluminescent substrate (Biological Industries, Beit Ha-Emek, Israel), and exposed to Fuji-ray film to visualize the antibody reactive bands.
Drug treatment
Activation of cellular PKCα was performed with phorbol ester [31]. Erythrocytes were incubated in PBS buffer supplemented with 25 µM phorbol 12-myristate 13-acetate (PMA) at 37°C for 10 minutes. To determine the optimal conditions for PKC activation we conducted a series of dose- and time-dependent PMA activation experiments in human erythrocytes (data not shown). PMA was prepared in accordance with the manufacturer’s protocol.
Measurement of intracellular ATP concentrations
The intracellular ATP concentration in human erythrocytes was quantified by a luciferin-luciferase assay kit (Sigma-Aldrich, Rehovot, Israel), according to the manufacturer's protocol. Briefly, following specific treatments with PMA and/or glucose, erythrocytes were centrifuged, and 50 µl of pellets (hematocrit 80%) were lysed by somatic cell ATP-releasing reagent. The ATP concentration of the aliquots was determined by luciferin/luciferase reaction using a luminometer (Berthold Technologies, Germany). A standard curve was generated from the relative light intensity peaks for serial dilutions (luminescence vs. ATP), as recommended by the manufacturer's protocol. Luminescence values of the measured samples were converted into concentrations of ATP, accordingly.
Measurement of intracellular Ca2+
To monitor the change in cytosolic calcium following exposure to physiological levels of calcium in the presence of PMA and/or glucose, erythrocytes were washed in a 10 mM D-glucose/PBS solution and loaded with Fluo-3/AM 2 µM. Fluo-3/AM was added again to achieve a final concentration of 4 µM which was maintained for additional 25 min under gentle shaking. Then, erythrocytes were incubated at 37°C for 15 min under rapid shaking. The loaded erythrocytes were washed in PBS solution, supplemented with 10 mM D glucose and 0.5% BSA, and treated according to the experimental protocol (i.e., treatments with PMA and/or glucose). Finally, Ca2+-dependent fluorescence intensity was measured in the fluorescence channel FL-1, with an excitation wavelength of 488 nm and an emission wavelength of 530 nm. Exposure time was restricted to 4 hours (to prevent loss of fluorescent dye).
Immunoprecipitation studies
Immunoprecipitation (IP) studies were performed using protein A-Sepharose beads (Santa Cruz Biotechnology, Heidelberg, Germany), and either mouse monoclonal antibody to glucose transporter 1 or mouse monoclonal antibody to 4.1R. Specifically, the antibody (0.2 mg/ml concentration) was attached to the beads by 30 minutes incubation in IP buffer (30 mM Tris-HCl, pH 7.5, 0.2 mM EDTA, 0.5 mM DTT, 0.2% Triton X-100, 150 mM NaCl) at 4ºC. Further, 1 ml cell lysate was added to 20 µl beads with the pre-attached antibodies, and the sample was incubated, with continuous shaking, for 2 hours at 4ºC. Beads were washed with the IP buffer, and the bead-attached proteins were subjected to SDS-PAGE and Western blotting.
Determination of RBC deformability
Determination of RBC deformability was performed according to a previously published protocol [33], with minimal modifications. Briefly, 50 µl RBC (1% hematocrit), under experimental conditions (i.e., exposed to PBS with D-glucose and/or L-glucose), were inserted into the flow chamber (adjusted to 200-mm gap). After 5 minutes of treatment, flow of the buffer was applied, and the deformation of adherent RBCs was monitored at shear stress (30 dyne/cm2). During the measurements, 15 to 20 randomly chosen fields (0.1 mm2 each) were collected. Image analysis of the cell shape provided the elongation ratio (ER) of individual cells, and their distribution in the RBC population (ranging from 2500 to 3500 cells in each sampling). For all cells, we estimated the major (a) and minor (b) axes, and ER was calculated by the formula ER = a/b. ER = 1 reflected round RBCs that were not deformed by the shear stress applied in this study (30 dyne/cm2). ER = 3 reflected extra-deformable erythrocytes with an elongated form.
Statistical analysis
Quantitative data were described as mean ± standard error (SE), and statistical analysis was performed using unpaired Student's t-test. Differences were considered as significant at a p-value of less than 0.05.
Results
Twenty-four healthy volunteers were recruited for the study. Their clinical and laboratory data are shown in Table 1.
Table 1. Clinical and metabolic characteristics of the study participants.
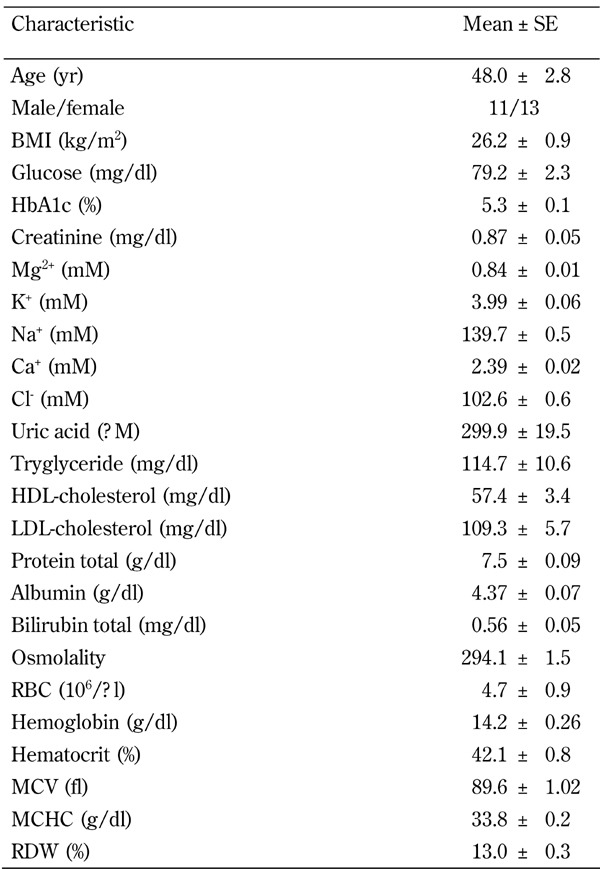
Legend: Data are mean ± SE, or percentage. Abbreviations: BMI - body mass index, HDL - high-density lipoprotein, LDL - low-density lipoprotein.
Cytosolic and membranous fractions of PKCα were measured (Figure 1). Overall, 42% of the total cellular PKCα was found in the erythrocyte's membrane. Previous studies have shown that after translocation to the membrane, PKC is activated by a sequential series of structural rearrangements and phosphorylations [29]. Therefore, it was reasonable to assume that most of the activated PKCα (66%) was found in the membranous fraction, as evidenced by a phosphorylated threonine residue at position 638 [30].
Figure 1. Total and membrane fractions of PKCα in healthy erythrocytes.
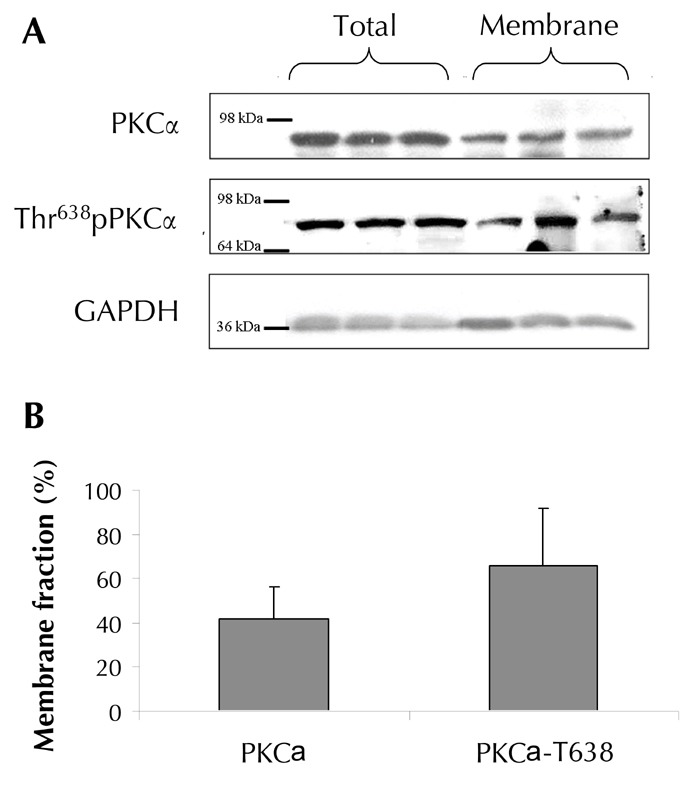
Representative Western blots (A) illustrate a comparison between the total and membranous fractions of total and activated (phosphorylated in Threonine 638) fractions of PKCα for randomly chosen three participants. The results of the Western blots were quantified by Adobe Photoshop densitometry, and presented as the percentage of the membrane fraction from the total cellular fraction of the protein kinase (B). The results represent the mean for seven individuals ± SE. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control to verify equivalent amounts of protein throughout the lanes. Molecular mass markers are indicated in kDa.
The erythrocytes were exposed to high (20 mM), physiological (5 mM), and low (0 mM) glucose concentrations, as described. The membranous fraction of total and phosphorylated PKCα was measured.
A decrease in the membranous fraction of total PKCα and Thr638-phosphorylated PKCα upon exposure to high glucose (20 mM) was observed (Figure 2). This decrease was insignificant when compared with 'D-glucose 5' (p > 0.05), but significant when compared with 'D glucose 0' (83%, p = 0.005, for total PKCα, and 65%, p = 0.0004, for phosphorylated PKCα fractions). PMA pre-exposure of RBCs, 'PMA+D-glucose 0' compared with 'D-glucose 0', showed a robust increase in the membranous fraction of the total PKC (149%, p = 0.014) and of the phosphorylated PKCα (173%, p = 0.014). This increase was observed at all glucose concentrations:
Figure 2. Changes in the membrane fraction of PKCα in erythrocytes acutely exposed to various concentrations of D-glucose.
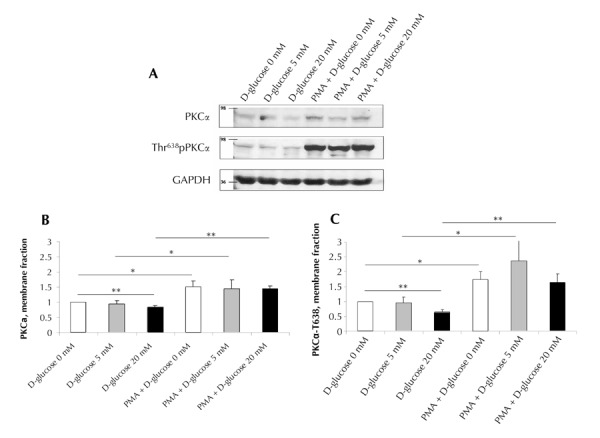
The erythrocytes were incubated in PBS supplemented with 0 mM (white bars), 5 mM (grey bars), or 20 mM D-glucose (black bars) for 5 minutes at 37ºC. When indicated, the cells were pretreated with 25 µM PMA. A representative Western blot shows the membrane fractions of PKCα and phosphorylated PKCα (PKCα-T638) (A). Molecular mass markers are indicated in kDa. Averaged results of Western blots of the membrane fractions of PKCα (B) or PKCα-T638 (C) were quantified using Adobe Photoshop, and normalized to "0 mM D-glucose"-treated cells. Data are means of 4 to 13 independent experiments ± SE. * p < 0.05; ** p < 0.01 vs. correspondent data.
1. 153% (p = 0.039), 236% (p = 0.05) 'PMA+D-glucose 5' vs. ‘D-glucose 5’
2. 173% (p = 3e-5), 208% (p = 0.0007) 'PMA+D-glucose 20' vs. 'D-glucose 20', for total and phosphorylated PKC, accordingly
Pre-exposure of erythrocytes to PMA led to a significant increase in both total and phosphorylated PKC membranous fractions, which completely prevailed over the effect of D-glucose, and caused the differences between the glucose concentrations to become insignificant (p > 0.05).
In parallel, we found an acute effect of glucose on ATP levels in erythrocytes (Figure 3). A small, but significant 11% increase in cellular ATP was observed upon 20 mM, compared with 5 mM, D-glucose exposure (p = 0.02). PMA pre-activation led to an increase in cellular ATP levels in the absence (18% 'PMA+D-glucose 0' vs. 'D-glucose 0'; p = 0.002) or in the presence of 5 mM D-glucose (18% 'PMA+D-glucose 5' vs. 'D-glucose 5'; p = 0.036). In PMA-pretreated cells, 20 mM D-glucose exposure resulted in reductions of 16% (p = 0.01) and 14% (p = 0.02) in cellular ATP levels compared with the 5 mM D-glucose or 0 mM D-glucose concentrations, respectively.
Figure 3. Changes in cellular ATP in erythrocytes acutely exposed to various concentrations of D-glucose.
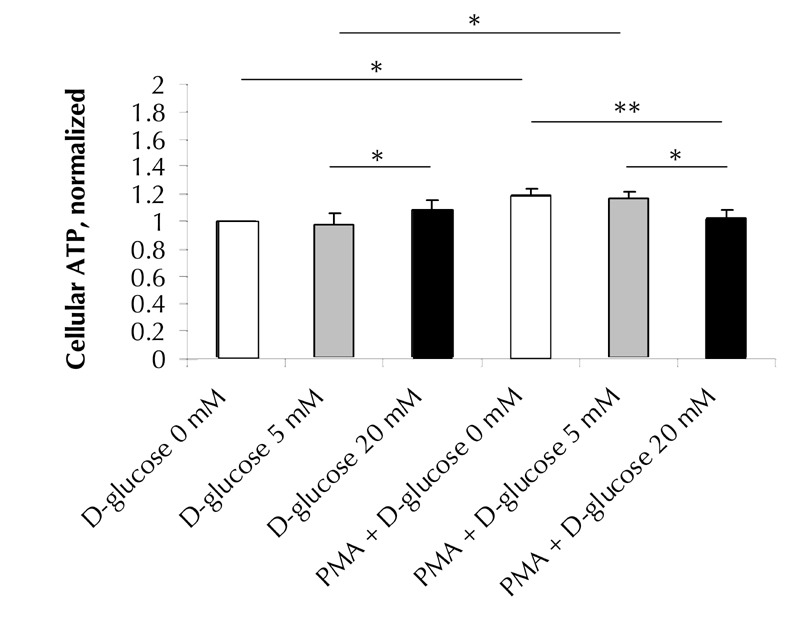
Erythrocytes, either pretreated with 25 µM PMA or untreated, were acutely incubated in PBS with 0 mM (white bars), 5 mM (grey bars), or 20 mM D-glucose (black bars). Cellular ATP concentrations were quantified by luciferin-luciferase assay, and normalized to “0 mM D-glucose”-treated cells. Data are means of 8 to 13 independent experiments ± SE. * p < 0.05; ** p < 0.01 vs. correspondent data.
Acute glucose exposure and/or PKC activation had no effect (p > 0.05) on intracellular Ca levels, mean cellular volume (MCV), or mean cellular hemoglobin concentration (a measure related to the intracellular viscosity [3, 36]), in 0, 5, or 20 mM D-glucose-exposed RBCs, with or without PMA-induced PKC activation (data not shown).
It is believed that the observed glucose-mediated reduction in the membrane fraction of PKCα may affect the interaction of kinase to its membrane substrates. We examined this hypothesis by co-immunoprecipitation of PKC with glucose transporter 1 (GLUT1), which is one of the most abundant membrane proteins [37] and the major glucose-responsive target in erythrocytes [34]. Also, we studied the affinity of PKC to 4.1R, a PKC-regulated component of the membrane skeleton [17, 20]. 20 mM glucose exposure significantly decreased the PKCα fraction bound to GLUT1 or 4.1R in comparison with the respective 'D-glucose 0' or 'D-glucose 5' studies (Figure 4). When PKCα was pre-activated with PMA, this glucose effect was mostly neutralized. To summarize these findings, the observed D-glucose-related changes in PKCα binding to its membrane substrates are mostly associated with the demonstrated reduction in the membrane fraction of PKCα.
Figure 4. Co-immunoprecipitations of PKCα with GLUT1 and 4.1R.
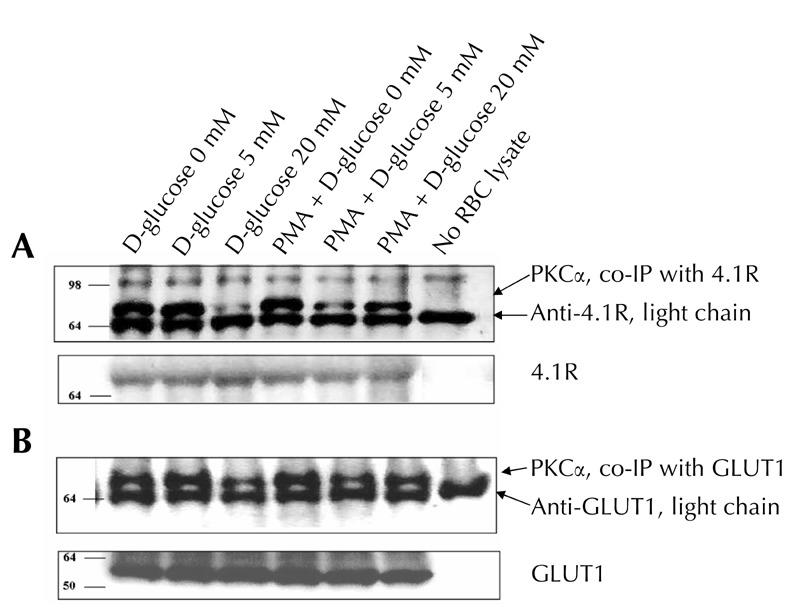
RBC lysates were exposed to protein A-Sepharose coupled to anti-GLUT1 or anti-4.1R. Immunoprecipitates were analyzed by SDS-PAGE followed by Western blot, as described under "Materials and methods". A representative result is presented in the figure. Similar results were obtained in two additional experiments. Data of 4.1R, GLUT1, and light chain of correspondent antibodies were used as controls to verify equivalent amounts of protein throughout the lanes. In the control 'no lysate' study, protein A-Sepharose coupled to an appropriate antibody was exposed to a RBC-free sample. Molecular mass markers are indicated in kDa.
It is also assumed that the glucose-mediated reduction in the membrane fraction of PKCα, and the correlative decline in PKC-4.1R link, may influence the mechanical properties of the erythrocytes. To test this hypothesis, we measured the possible effect of D-glucose on the erythrocyte deformability in the context of PKCα activation (Figure 5). Figure 5 shows the results of the median elongation ratio (ER) of the erythrocytes examined (Figure 5A), the percentages of undeformable (ER ≤ 1.1) erythrocytes (Figure 5B), and the RBC subpopulations arranged in accordance with ER ascension (0.5 ER step; Figures 5C-F), exposed to D-glucose and/or following PMA pre-activation.
Figure 5. Acute glucose effect on RBC deformability.
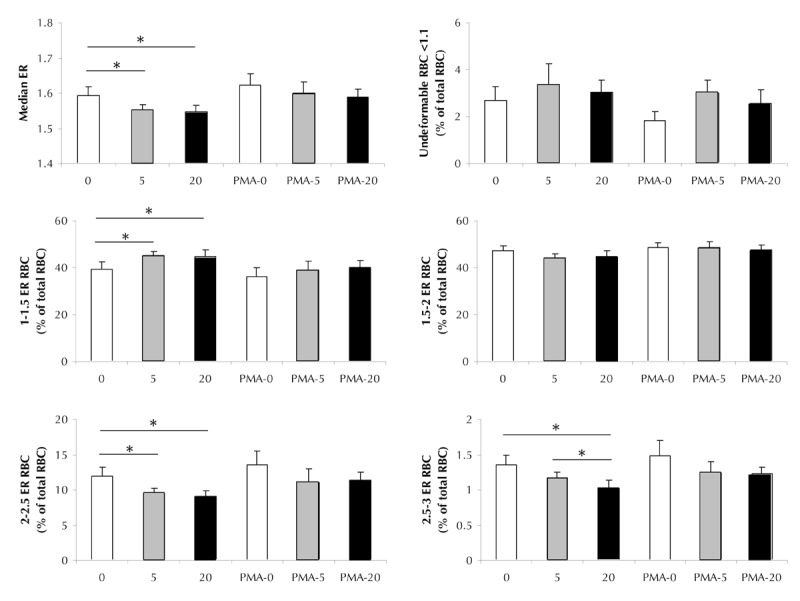
The figure shows the elongation ratio (ER) or the percentage from the total RBC population in accordance to ER of erythrocytes incubated in PBS supplemented with with 0 mM (white bars), 5 mM (grey bars), or 20 mM D-glucose (black bars) for 5 min at 37ºC. When indicated, cells were pretreated with 25 µM PMA. A. Median elongation ratio. B. Percentages of undeformable (ER ≤ 1.1) erythrocytes. C. Percentage of erythrocytes with decreased 1.1-1.5 ER deformability. D. Percentage of erythrocytes with median 1.5-2 ER deformability. E. Percentage of erythrocytes with increased 2-2.5 deformability. F. Percentage of erythrocytes with increased 2.5-3.0 deformability. Data are means of 4 to 13 independent experiments ± SE. * p < 0.05; ** p < 0.01 vs. correspondent data.
Elevation in D-glucose led to a small, but significant decrease in median RBC deformability (1.546 ± 0.02, p = 0.017, for 'D-glucose 5', and 1.552 ± 0.01, p = 0.05, for 'D-glucose 20' compared with 1.594 ± 0.02 'D-glucose 0' median ER). This is possibly a result of a pronounced elevation in the low deformability (1.1-1.5 ER) RBC portion (41.7 ± 1.2%, p = 0.05, for 'D-glucose 5', and 41.8 ± 2.3%, p = 0.024, for 'D-glucose 20' compared with 36.5 ± 2.6% 'D-glucose 0', respectively), and a drop in the relative fraction of erythrocytes with increased deformability (9.6 ± 0.6%, p = 0.04, for 'D-glucose 5', and 9.1 ± 0.7%, p = 0.006, for 'D-glucose 20' compared with 12 ± 1.2% 'D-glucose 0', respectively). These effects of glucose were minimized by pretreatment with PMA. Changes in the median ER values and RBC fractions between erythrocytes exposed to 0, 5, or 20 mM D-glucose were all insignificant (p > 0.05).
Discussion
The main finding of the study is that acute exposure of erythrocytes from healthy individuals to a glucose-enriched medium decreases the membranous distribution and activation of PKCα, PKCα's affinity to its membrane substrates, and RBC deformability. It has been reported that erythrocyte deformability is directly dependent on the phosphorylation state of its membrane skeleton proteins (such as β-spectrin, 4.1R, adducin, and dematin). Several of these proteins are phosphorylated by PKC [18-22]. Therefore, the perceived decrease in membranous PKC probably affects the biological organization of the cytoskeleton. Two independent processes contribute to the decrease in phosphorylated PKC levels:
1. A decrease in PKC translocation
2. A reduction in PKCα binding to its membrane substrates
Both processes were observed under acute hyperglycemia, and may thus be the main reasons for the observed alterations in the fraction of phosphorylated PKC and cellular deformability.
The alterations in PKC translocation can be hypothetically coupled with a cellular fluctuation of its native activators (i.e., Ca2+, diacylglycerol, or phosphatidylserine (PS) [21, 29, 38, 39]) and with possible disturbances in PKC affinity to its membrane substrates. Glucose exposure significantly reduced the affinity of PKC to GLUT1, one of the most plentiful PKC membrane substrates [24, 40], and to 4.1R, a PKC-regulated component of the membrane skeleton [17, 20]. This data allows us to assume a significant effect of acute hyperglycemia on PKC binding to the membrane skeleton proteins. However, the direct effect of glucose on protein structure and activity is not so acute. It is thus possible that the observed phenomenon is caused by glucose-mediated changes in cellular metabolic mechanisms. Therefore, the observed decrease in the membranous PKCα should have been examined in association with cellular fluctuation of native PKC activators. This hypothesis was mostly supported by a complete neutralization of the glucose effect on membranous PKC and RBC deformability in erythrocytes pre-exposed to PMA (highly efficient diacylglycerol analog). Moreover, no significant glucose-mediated changes in intracellular Ca2+ were found.
In addition, it is not expectable to find changes in intracellular diacylglycerol levels, secondary to acute changes in glucose levels [12]. Possibly, the phenomenon of a glucose-mediated decrease in membranous PKCα is linked to an ATP-dependent instability of PS in the inner part of RBC membrane bilayers [41-43]. This hypothesis implies how glucose-mediated ATP fluctuations may be involved in the correlative decrease of membranous PKC and the decrease in RBC deformability. In the near future, a major subject for research will be the study of PS redistribution in the membrane upon acute glucose exposure, and the role of PS in acute alterations of the RBC deformability.
Another key finding of this study was the fact that very short glucose exposure (5 min) may alter RBC deformability. This finding correlates with recent reports that explain how glucose exposure for hours significantly affects protein modification and inhibits RBC deformation. Riquelme et al. reported that high glucose concentrations caused significant reductions in the viscoelastic properties of the erythrocyte membrane when the erythrocytes were incubated for 2 hours [44]. Shin et al. found similar associations, with both deformability and erythrocyte aggregation decreasing in a dose- and time-dependent manner [45]. Resmi et al. demonstrated a correlation between glucose, protein oxidation, and cellular deformability with increasing incubation time (24-48 hours) [46]. Nevertheless, no correlative data of highly acute (5 min) glucose exposure and membrane deformable properties have been reported previously.
Clinically, the observed noxious effect of a minute’s glycemic surge on the RBC deformability is crucial because it may render the erythrocyte more susceptible to thrombotic phenomena. This in turn may add a possible explanation to the observed increase in morbidity and mortality associated with abnormal postprandial glucose levels [47, 48]. The proposed link between altered cellular deformability and PKC abnormalities defines PKCα as the key factor involved in these complications. Moreover, our findings may offer a possible mechanistic explanation to findings linking acute hyperglycemia in non-diabetic patients to significantly higher levels of in-hospital mortality when compared to previously diagnosed diabetic, hyperglycemic patients [49]. In spite of an absence of specific reports on diabetic RBC, we propose that most PKC-dependent processes in diabetic erythrocytes become distorted and less sensitive due to constituent PKCα dysfunction caused by the chronic hyperglycemic milieu. This could be a logical explanation for the acute glycemic surge and its more harmful effects on PKCα activation, and for the resultant rheological dysfunction in non-diabetic patients. We plan to perform studies on the deformable alterations upon acute hyperglycemia in diabetic RBC in the near future. We believe that pharmacologically targeting this process may offer a new avenue for treatment and research, aiming to protect both diabetic and non-diabetic patients from the hazardous effects of acute glycemic surges.
In this study, we limited our attention to the role of PKCα in the examined processes, the involvement of other PKC isoforms expressed in erythrocytes were not studied. In human erythrocytes, four additional PKC isoforms are expressed, PKC β, μ, ζ, and ι [20, 50]. We chose PKCα as the study target because of its sensitivity to all known native PKC activators. Moreover, PKCα is expressed in all blood cells [50-53]. Therefore, the study of PKCα in an accessible and convenient cell model such as the erythrocyte offers important information about PKC-mediated processes in other blood cells. We plan to examine the role of the other PKC isoforms in the observed cellular processes in further studies.
Disclosure: The authors report no conflict of interests.
Acknowledgments
This work was partly supported by a grant from the Joint Research Fund, established between Hebrew University Faculty of Medicine and Hadassah University Hospital, to A. Cahn and S. Yedgar. The authors thank Prof. Ehud Ziv for his very helpful discussion. We greatly appreciate Ms. Olga Foiering and Ms. Olga Fridman for technical help. Special thanks are to Ethrog Biotechnologies (Nes Ziona, Israel) for their contribution by the iBLOT instrument.
References
- 1.Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, Hadden D, Turner RC, Holman RR. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321(7258):405–412. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong WT, Wong SL, Tian XY, Huang Y. Endothelial dysfunction: the common consequence in diabetes and hypertension. J Cardiovasc Pharmacol. 2010;55:300–307. doi: 10.1097/fjc.0b013e3181d7671c. [DOI] [PubMed] [Google Scholar]
- 3.Cho YI, Mooney MP, Cho DJ. Hemorheological disorders in diabetes mellitus. J Diabetes Sci Technol. 2008;2(6):1130–1138. doi: 10.1177/193229680800200622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Constantin A, Constantinescu E, Dumitrescu M, Calin A, Popov D. Effects of ageing on carbonyl stress and antioxidant defense in RBCs of obese type 2 diabetic patients. J Cell Mol Med. 2005;9:683–691. doi: 10.1111/j.1582-4934.2005.tb00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller JA, Gravallese E, Bunn HF. Nonenzymatic glycosylation of erythrocyte membrane proteins. Relevance to diabetes. J Clin Invest. 1980;65:896–901. doi: 10.1172/JCI109743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maeda N, Kon K, Imaizumi K, Sekiya M, Shiga T. Alteration of rheological properties of human erythrocytes by crosslinking of membrane proteins. Biochim Biophys Acta. 1983;735:104–112. doi: 10.1016/0005-2736(83)90265-1. [DOI] [PubMed] [Google Scholar]
- 7.Garnier M, Attali JR, Valensi P, Delatour-Hanss E, Gaudey F, Koutsouris D. Erythrocyte deformability in diabetes and erythrocyte membrane lipid composition. Metabolism. 1990;39:794–798. doi: 10.1016/0026-0495(90)90121-r. [DOI] [PubMed] [Google Scholar]
- 8.Bryszewska M, Szosland K. Association between the glycation of erythrocyte membrane proteins and membrane fluidity. Clin Biochem. 1988;21:49–51. doi: 10.1016/s0009-9120(88)80111-5. [DOI] [PubMed] [Google Scholar]
- 9.Cohen RM, Franco RS, Khera PK, Smith EP, Lindsell CJ, Ciraolo PJ, Palascak MB, Joiner CH. Red cell life span heterogeneity in hematologically normal people is sufficient to alter HbA1c. Blood. 2008;112(10):4284–4291. doi: 10.1182/blood-2008-04-154112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujita J, Tsuda K, Takeda T, Yu L, Fujimoto S, Kajikawa M, Nishimura M, Mizuno N, Hamamoto Y, Mukai E, Adachi T, Seino Y. Nisoldipine improves the impaired erythrocyte deformability correlating with elevated intracellular free calcium-ion concentration and poor glycaemic control in NIDDM. Br J Clin Pharmacol. 1999;47(5):499–506. doi: 10.1046/j.1365-2125.1999.00934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bareford D, Jennings PE, Stone PC, Baar S, Barnett AH, Stuart J. Effects of hyperglycaemia and sorbitol accumulation on erythrocyte deformability in diabetes mellitus. J Clin Pathol. 1986;39:722–727. doi: 10.1136/jcp.39.7.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia P, Inoguchi T, Kern TS, Engerman RL, Oates PJ, King GL. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes. 1994;43:1122–1129. doi: 10.2337/diab.43.9.1122. [DOI] [PubMed] [Google Scholar]
- 13.Miller JA, Gravallese E, Bunn HF. Nonenzymatic glycosylation of erythrocyte membrane proteins. Relevance to diabetes. J Clin Invest. 1980;65:896–901. doi: 10.1172/JCI109743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Symeonidis A, Athanassiou G, Psiroyannis A, Kyriazopoulou V, Kapatais-Zoumbos K, Missirlis Y, Zoumbos N. Impairment of erythrocyte viscoelasticity is correlated with levels of glycosylated haemoglobin in diabetic patients. Clin Lab Haematol. 2001;23(2):103–109. doi: 10.1046/j.1365-2257.2001.00366.x. [DOI] [PubMed] [Google Scholar]
- 15.Umudum F, Yucel O, Sahin Y, Bakan E. Erythrocyte membrane glycation and NA(+)-K(+) levels in NIDDM. J Diabetes Complications. 2002;16:359–362. doi: 10.1016/s1056-8727(01)00223-9. [DOI] [PubMed] [Google Scholar]
- 16.Le Devehat C, Khodabandehlou T, Vimeux M. Relationship between hemorheological and microcirculatory abnormalities in diabetes mellitus. Diabetes Metab. 1994;20:401–404. [PubMed] [Google Scholar]
- 17.Baines AJ. The spectrin-ankyrin-4.1-adducin membrane skeleton: adapting eukaryotic cells to the demands of animal life. Protoplasma. 2010;244:99–131. doi: 10.1007/s00709-010-0181-1. [DOI] [PubMed] [Google Scholar]
- 18.de Oliveira S, Silva-Herdade AS, Saldanha C. Modulation of erythrocyte deformability by PKC activity. Clin Hemorheol Microcirc. 2008;39:363–373. [PubMed] [Google Scholar]
- 19.George A, Pushkaran S, Li L, An X, Zheng Y, Mohandas N, Joiner CH, Kalfa TA. Altered phosphorylation of cytoskeleton proteins in sickle red blood cells: the role of protein kinase C, Rac GTPases, and reactive oxygen species. Blood Cells Mol Dis. 2010;45(1):41–45. doi: 10.1016/j.bcmd.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manno S, Takakuwa Y, Mohandas N. Modulation of erythrocyte membrane mechanical function by protein 4. 1. phosphorylation;J Biol Chem 2005 280:7581–7587. doi: 10.1074/jbc.M410650200. [DOI] [PubMed] [Google Scholar]
- 21.Palfrey HC, Waseem A. Protein kinase C in the human erythrocyte. Translocation to the plasma membrane and phosphorylation of bands 4.1 and 4.9 and other membrane proteins. J Biol Chem. 1985;260:16021–16029. [PubMed] [Google Scholar]
- 22.Ling E, Danilov YN, Cohen CM. Modulation of red cell band 4.1 function by cAMP-dependent kinase and protein kinase C phosphorylation. J Biol Chem. 1988;263:2209–2216. [PubMed] [Google Scholar]
- 23.Ceolotto G, Sartori M, Felice M, Clari G, Bordin L, Semplicini A. Effect of protein kinase C and insulin on Na+/H+ exchange in red blood cells of essential hypertensives. J Hum Hypertens. 1999;13:321–327. doi: 10.1038/sj.jhh.1000804. [DOI] [PubMed] [Google Scholar]
- 24.Deziel MR, Lippes HA, Rampal AL, Jung CY. Phosphorylation of the human erythrocyte glucose transporter by protein kinase C: localization of the site of in vivo and in vitro phosphorylation. Int J Biochem. 1989;21:807–814. doi: 10.1016/0020-711x(89)90214-0. [DOI] [PubMed] [Google Scholar]
- 25.Enyedi A, Elwess NL, Filoteo AG, Verma AK, Paszty K, Penniston JT. Protein kinase C phosphorylates the "a" forms of plasma membrane Ca2+ pump isoforms 2 and 3 and prevents binding of calmodulin. J Biol Chem. 1997;272:27525–27528. doi: 10.1074/jbc.272.44.27525. [DOI] [PubMed] [Google Scholar]
- 26.Zancan P, Sola-Penna M. Regulation of human erythrocyte metabolism by insulin: cellular distribution of 6-phosphofructo-1-kinase and its implication for red blood cell function. Mol Genet Metab. 2005;86:401–411. doi: 10.1016/j.ymgme.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 27.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livshits L, Caduff A, Talary MS, Lutz HU, Hayashi Y, Puzenko A, Shendrik A, Feldman Y. The role of GLUT1 in the sugar-induced dielectric response of human erythrocytes. J Phys Chem B. 2009;113(7):2212–2220. doi: 10.1021/jp808721w. [DOI] [PubMed] [Google Scholar]
- 29.Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395–E402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bornancin F, Parker PJ. Phosphorylation of threonine 638 critically controls the dephosphorylation and inactivation of protein kinase Calpha. Curr Biol. 1996;6:1114–1123. doi: 10.1016/s0960-9822(02)70678-7. [DOI] [PubMed] [Google Scholar]
- 31.Goel G, Makkar HP, Francis G, Becker K. Phorbol esters: structure, biological activity, and toxicity in animals. Int J Toxicol. 2007;26:279–288. doi: 10.1080/10915810701464641. [DOI] [PubMed] [Google Scholar]
- 32.Komarova SV, Mosharov EV, Vitvitsky VM, Ataullakhanov FI. Adenine nucleotide synthesis in human erythrocytes depends on the mode of supplementation of cell suspension with adenosine. Blood Cells Mol Dis. 1999;25:170–179. doi: 10.1006/bcmd.1999.0243. [DOI] [PubMed] [Google Scholar]
- 33.Relevy H, Koshkaryev A, Manny N, Yedgar S, Barshtein G. Blood banking-induced alteration of red blood cell flow properties. Transfusion. 2008;48:136–146. doi: 10.1111/j.1537-2995.2007.01491.x. [DOI] [PubMed] [Google Scholar]
- 34.Carruthers A. Facilitated diffusion of glucose. Physiol Rev. 1990;70:1135–1176. doi: 10.1152/physrev.1990.70.4.1135. [DOI] [PubMed] [Google Scholar]
- 35.Cohen CM, Foley SF. Phorbol ester- and Ca2+-dependent phosphorylation of human red cell membrane skeletal proteins. J Biol Chem. 1986;261:7701–7709. [PubMed] [Google Scholar]
- 36.Chien S. Red cell deformability and its relevance to blood flow. Annu Rev Physiol. 1987;49:177–192. doi: 10.1146/annurev.ph.49.030187.001141. [DOI] [PubMed] [Google Scholar]
- 37.Helgerson AL, Carruthers A. Equilibrium ligand binding to the human erythrocyte sugar transporter. Evidence for two sugar-binding sites per carrier. J Biol Chem. 1987;262:5464–5475. [PubMed] [Google Scholar]
- 38.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 39.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 40.Witters LA, Vater CA, Lienhard GE. Phosphorylation of the glucose transporter in vitro and in vivo by protein kinase C. Nature. 1985;315:777–778. doi: 10.1038/315777a0. [DOI] [PubMed] [Google Scholar]
- 41.Bevers EM, Comfurius P, Dekkers DW, Harmsma M, Zwaal RF. Transmembrane phospholipid distribution in blood cells: control mechanisms and pathophysiological significance. Biol Chem. 1998;379:973–986. [PubMed] [Google Scholar]
- 42.Bevers EM, Comfurius P, Dekkers DW, Zwaal RF. Lipid translocation across the plasma membrane of mammalian cells. Biochim Biophys Acta. 1999;1439:317–330. doi: 10.1016/s1388-1981(99)00110-9. [DOI] [PubMed] [Google Scholar]
- 43.Klarl BA, Lang PA, Kempe DS, Niemoeller OM, Akel A, Sobiesiak M, Eisele K, Podolski M, Huber SM, Wieder T, Lang F. Protein kinase C mediates erythrocyte "programmed cell death" following glucose depletion. Am J Physiol Cell Physiol. 2006;290(1):C244–C253. doi: 10.1152/ajpcell.00283.2005. [DOI] [PubMed] [Google Scholar]
- 44.Riquelme B, Foresto P, D'Arrigo M, Valverde J, Rasia R. A dynamic and stationary rheological study of erythrocytes incubated in a glucose medium. J Biochem Biophys Methods. 2005;62(2):131–141. doi: 10.1016/j.jbbm.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 45.Shin S, Ku YH, Suh JS, Singh M. Rheological characteristics of erythrocytes incubated in glucose media. Clin Hemorheol Microcirc. 2008;38:153–161. [PubMed] [Google Scholar]
- 46.Resmi H, Akhunlar H, Temiz Artmann A, Guner G. In vitro effects of high glucose concentrations on membrane protein oxidation, G-actin and deformability of human erythrocytes. Cell Biochem Funct. 2005;23:163–168. doi: 10.1002/cbf.1129. [DOI] [PubMed] [Google Scholar]
- 47.DECODE Study Group, EDEG. Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care. 2003;26(3):688–696. doi: 10.2337/diacare.26.3.688. [DOI] [PubMed] [Google Scholar]
- 48.Nakagami T. Hyperglycaemia and mortality from all causes and from cardiovascular disease in five populations of Asian origin. Diabetologia. 2004;47:385–394. doi: 10.1007/s00125-004-1334-6. [DOI] [PubMed] [Google Scholar]
- 49.Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab. 2002;87:978–982. doi: 10.1210/jcem.87.3.8341. [DOI] [PubMed] [Google Scholar]
- 50.Govekar RB, Zingde SM. Protein kinase C isoforms in human erythrocytes. Ann Hematol. 2001;80:531–534. doi: 10.1007/s002770100352. [DOI] [PubMed] [Google Scholar]
- 51.Brick-Ghannam C, Ericson ML, Schelle I, Charron D. Differential regulation of mRNAs encoding protein kinase C isoenzymes in activated human B cells. Hum Immunol. 1994;41:216–224. doi: 10.1016/0198-8859(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 52.Gruber T, Hermann-Kleiter N, Pfeifhofer-Obermair C, Lutz-Nicoladoni C, Thuille N, Letschka T, Barsig J, Baudler M, Li J, Metzler B. et al. PKC theta cooperates with PKC alpha in alloimmune responses of T cells in vivo. Mol Immunol. 2009;46:2071–2079. doi: 10.1016/j.molimm.2009.02.030. [DOI] [PubMed] [Google Scholar]
- 53.Pontremoli S, Melloni E, Sparatore B, Michetti M, Salamino F, Horecker BL. Isozymes of protein kinase C in human neutrophils and their modification by two endogenous proteinases. J Biol Chem. 1990;265:706–712. [PubMed] [Google Scholar]