

Abstract
Significance: Mitochondria are the cellular energy-producing organelles and are at the crossroad of determining cell life and death. As such, the function of mitochondria has been intensely studied in metabolic disorders, including diabetes and associated maladies commonly grouped under all-inclusive pathological condition of metabolic syndrome. More recently, the altered metabolic profiles and function of mitochondria in these ailments have been correlated with their aberrant morphologies. This review describes an overview of mitochondrial fission and fusion machineries, and discusses implications of mitochondrial morphology and function in these metabolic maladies. Recent Advances: Mitochondria undergo frequent morphological changes, altering the mitochondrial network organization in response to environmental cues, termed mitochondrial dynamics. Mitochondrial fission and fusion mediate morphological plasticity of mitochondria and are controlled by membrane-remodeling mechanochemical enzymes and accessory proteins. Growing evidence suggests that mitochondrial dynamics play an important role in diabetes establishment and progression as well as associated ailments, including, but not limited to, metabolism–secretion coupling in the pancreas, nonalcoholic fatty liver disease progression, and diabetic cardiomyopathy. Critical Issues: While mitochondrial dynamics are intimately associated with mitochondrial bioenergetics, their cause-and-effect correlation remains undefined in metabolic diseases. Future Directions: The involvement of mitochondrial dynamics in metabolic diseases is in its relatively early stages. Elucidating the role of mitochondrial dynamics in pathological metabolic conditions will aid in defining the intricate form–function correlation of mitochondria in metabolic pathologies and should provide not only important clues to metabolic disease progression, but also new therapeutic targets. Antioxid. Redox Signal. 19, 415–430.
Introduction
Mitochondria are highly dynamic organelles constantly changing shape and location in the cell (6, 7). Mitochondrial shapes vary from small spherical and oval, typical textbook-depicted mitochondria, to interconnected filamentous networks. Small spherical mitochondria represent a fragmented or profission state, whereas the interconnected networks are representative of a profusion phenotype. The nature of these networks changes in response to environmental cues and varies greatly even within similar cellular lineages in the absence of stressed conditions (Fig. 1). The identification of core machinery responsible for the mechanical scission or fusion of mitochondria has allowed a rapid advance of this research field, but new mechanistic details are ever evolving through discoveries of additional players in these fundamental cellular processes. The necessity of mitochondrial dynamics is apparent by the requirement of all four mechanoenzymes in embryonic development (24, 35, 70, 138). In cell systems, their knockdown is tolerable to an extent; however, bioenergetic and cell survival deficiencies are evident. Production of adenosine triphosphate (ATP) through oxidative phosphorylation in the electron transport chain (ETC) complexes localized within the inner membrane of mitochondria is a primary ascribed function of mitochondria. Dysregulation of this highly regulated process results in bioenergetic deficiencies and/or production of damaging reactive oxygen species (ROS) contributing to organ damage. These maladies have been described in the development of metabolic pathologies.
FIG. 1.

Mitochondrial network morphology is varied between similar cell types of similar lineage and under metabolic stress. (Left panel) Primary mouse hepatocytes, under unstressed culture conditions, display a small, spherical fragmented mitochondrial morphology filling the cytoplasm, suggesting that mitochondrial fission is favored over fusion. The mitochondrial network was visualized by fluorescence microscopy after infection with adenovirus expression of mitochondria-targeted GFP (mtGFP). Image courtesy of Dr. Hakjoo Lee. (Middle panel) Mitochondria of immortalized Clone 9 hepatocytes cultured in unstressed conditions, stably expressing mtGFP, are observed as a more perinuclear interconnected network with longer tubular structures extending out toward the periphery of the cell, indicating that mitochondrial fusion is favored over fission in these cells. (Right panel) Treatment of mtGFP Clone 9 hepatic cells with 250 μM palmitate resulted in the rapid fragmentation of mitochondria. The fragmented mitochondrial morphology can be accomplished either through the increase in the frequency of fission events or through the suppression of fusion events.
The implication of mitochondrial dynamics in metabolic diseases is a relatively new concept. Given the energetic implications of these pathologies, mitochondrial involvement is obligate. The nature of changes associated with bioenergetic fluctuations and the role mitochondrial dynamic plays or responds to are an interesting new frontier in the deconvolution of the mechanisms underlying these diseases. Metabolic diseases are, at their root, a problem of excess, specifically in an insulin-resistant state, where both hyperglycemia and hyperlipidemia are present. The progression of these pathologies is dependent upon how mitochondria handle this excess. Hence, how mitochondrial dynamics govern this process is an intriguing question. We previously reviewed the implication of mitochondrial dynamics in diabetes (145), and the current review extends that discussion to commonly associated diabetic complications: nonalcoholic fatty liver disease (NAFLD) and diabetic cardiomyopathy (DCM). We will first briefly summarize mitochondrial fission and fusion machineries and further discuss recently discovered proteins implicated in these processes. We will then discuss the current understanding of mitochondrial dynamics in the context of metabolic diseases commonly associated with diabetes and insulin resistance, namely NAFLD and DCM.
The Core Machinery for Mitochondrial Fission and Fusion
Mitochondrial fission and fusion require membrane remodeling mediated by dynamin-related GTPases. In mitochondrial fission, dynamin-like/related protein 1 (DLP1/Drp1) forms oligomeric ring structures around the mitochondrial membrane, constricting and severing it in a GTPase-dependent manner (126, 148). The fused mitochondrial network observed with expression of dominant-negative (DN) DLP1 mutants or DLP1 knockdown/knockout supports its necessity in the fission process (54, 70, 81, 126). The requirement for DLP1's GTPase activity and its assembly into oligomeric complexes is supported by the DN effect of GTPase- and middle-domain mutants (22, 108, 126). Recently, cryoelectron microscopy (EM) was used to visualize the spiral ring structure of Dnm1, the yeast homolog to DLP1, and its constriction upon addition of and hydrolysis of guanosine triphosphate (GTP) (89). Structurally, DLP1 contains a middle domain, an unconserved domain, and a C-terminal coiled–coiled (CC) domain following the N-terminal GTPase domain (Fig. 2). The middle domain has been implicated in the oligomerization of DLP1 required for its membrane-constricting properties (22), and the CC domain interacts with the N-terminal domains to regulate the GTPase activity. These domains of the protein are the subject of multiple post-translational modifications, including phosphorylation (21, 31, 61, 132, 151), S-nitrosylation (25), SUMOylation (11, 51, 62, 139), and ubiquitylation (94, 144), affecting DLP1 function, interactions, stability, and subcellular localization.
FIG. 2.

Characterized domains of the core mitochondrial dynamics machinery. Dynamin-like protein 1 (DLP1), mitofusin (Mfn), and optic atrophy 1 (OPA1) are GTpases. DLP1 contains an N-terminal GTPase domain conserved with the dynamin family of proteins, a middle domain involved in higher-order self-assembly of DLP1, an unconserved domain (UC), and a coiled-coil (CC), or a GTPase effector domain (GED), functioning in protein–protein interactions. Mfn contains two transmembrane domains (TM) that cross the outer mitochondrial membrane (OMM) twice, which leaves the GTPase domain and heptad repeats 1 and 2 (HR1 and HR2) exposed to the cytosol. OPA1 contains a mitochondrial-targeting signal (MTS), which targets the protein to the inner mitochondrial membrane. The C-terminal portion of the protein containing the GTPase, CC and GED remains exposed to the intermitochondrial space. Fission protein 1 (Fis1), mitochondrial fission factor (Mff), and mitochondrial elongation factor 1 (MIEF1/MiD51) are the DLPI receptor proteins. All contain a transmembrane domain anchoring them to the OMM. No other domains of the MIEF1/MiD51 are described; however, the C-terminus was required for DLP1 interaction. Mff contains a cytosolic CC domain and two short repeats (RPT) conserved in metazoans and implicated in DLP1 interaction. Fis1 contains six α-helices in its cytosolic face with the four internal helices forming a tetratricopeptide repeat (TPR) involved in DLP1 binding. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
DLP1, unlike the other GTPases of mitochondrial dynamics, lacks a transmembrane domain and is therefore not restricted to the mitochondrial outer membrane. DLP1 is localized diffusely in the cytosol, with only a small portion localized to mitochondria (124, 126, 147), where it forms concentrated foci observed by immunofluorescence (107, 126). The punctate localization of DLP1 in foci at the mitochondrial membrane has been ascribed to the presence of a receptor in the outer mitochondrial membrane (OMM). Until recently, the role of the mitochondrial DLP1 receptor was thought to be occupied only by hFis1, a small helix-rich protein anchored in the OMM (129, 146) (Fig. 2). A recent study by Otera et al. suggested the mitochondrial fission factor (Mff) (Fig. 2) as the OMM receptor for DLP1 (103). Mff was originally identified as an OMM-bound protein with similar requirements in mitochondrial fission as hFis1 (56). Overexpression of hFis1 fragments mitochondria by increasing fission, which requires DLP1 function (129, 146). Similarly, the overexpression of Mff induced mitochondrial fragmentation in HeLa cells (103). Otera et al. indicated that the Mff-mediated mitochondrial fragmentation occurs in the absence of hFis1, suggesting that the hFis1 function is dispensable in the presence of Mff. Both hFis1 and Mff contain C-terminal transmembrane domains that allow their functional N-terminal regions exposed to the cytosol (56, 103, 129, 146). The cytosolic domain of hFis1 contains six α-helices of which the core four helices, α2–α5, form two tandem tetratricopeptide repeat (TPR) motifs that are known to participate in protein–protein interactions (40, 130). DLP1 was shown to interact transiently with the hFis1 TPR, which is regulated by the N-terminal first α-helix of hFis1 (120, 150) (Fig. 2). Mff interacts with DLP1 in a transient nature as well, requiring crosslinking to detect the interaction by immunoprecipitation (56, 103). Although this interaction is transient, coimmunoprecipitation studies in combination with hFis1 demonstrated that interaction between DLP1 and Mff was preserved in more stringent conditions than that of DLP1 with hFis1 (103). The authors suggest that the interaction requires the two conserved N-terminal repeats within the first 50 amino acid residues, as deletion of the first 50 N-terminal residues resulted in a reduction of DLP1 at the mitochondria and in mitochondrial fission (103). hFis1 has been shown to localize evenly throughout the mitochondrial surface (71, 130), and changes of hFis1 levels did not affect the DLP1 distribution/level in mitochondria (81, 130). Conversely, Mff distributed along the mitochondria in punctate clusters, colocalizing with DLP1 by immunofluorescence (103). Ribonucleic acid interference (RNAi) knockdown of Mff eliminated punctate DLP1 localization at the mitochondria, suggesting that Mff was required for DLP1 recruitment. Strengthening this argument, when Mff was mistargeted to the plasma membrane, DLP1 redistributed to the plasma membrane where they co-localized (103). These studies support a role for Mff as a receptor for DLP1 at the mitochondrial surface, but do not exclude a role for hFis1 in the mechanism of mitochondrial fission outside of targeting of DLP1 to the mitochondria. It is likely that other factors, including signal-mediated modifications in participating proteins and their relative ratios, may play a role in regulating DLP1-mediated mitochondrial fission.
Mitochondrial fusion requires sequential fusion of outer and inner membranes. Two isoforms of the dynamin-related GTPase mitofusin (Mfn) 1 and 2 exist in mammals and mediate the OMM fusion (113–115). Anchored to the OMM, their topology is unique, crossing the OMM twice, which results in a small loop within the intermembrane space leaving the GTPase domain and the two heptad-repeat (HR) domains, HR1 and HR2, in the cytosol (115). Mfn1 molecules can interact intermitochondrially to tether adjacent mitochondria together through their HR2 domains, promoting mitochondrial fusion (79). Recently, it has been shown that the HR1 domain of Mfn2 interacts with HR2 and also with the CC domain of the fission protein DLP1 (67). The interaction between HR1 and HR2 exerted an inhibitory effect on fusion, while interaction of the HR1 domain with the DLP1 CC domain appeared to promote fusion. Additionally, Mfn2 has recently also been described to serve the function of tethering mitochondria to the endoplasmic reticulum, allowing Ca2+ communication between the organelles (38). This newly ascribed function may explain the observation of enhanced levels of Mfn2 expression in skeletal and cardiac tissue (114), where Ca2+ transients are crucial to muscle motive force. In combination with Mfn1, optic atrophy 1 (OPA1) is required to complete mitochondrial fusion by mediating fusion of the inner mitochondrial membrane (IMM) (27). OPA1 is anchored in the IMM by its N-terminus with the bulk of the molecule facing the intermembrane space. This brings the loop structure of the Mfn proteins in proximity to the C-terminus of OPA1, which would allow coordinate communication in the completion of mitochondrial fusion (27, 35, 59, 99). OPA1 is extensively modified at both the transcriptional and post-translational levels, expressing eight different forms in humans through alternative splicing (39) that are further processed by multiple proteases, generating soluble forms of OPA1 (28, 44, 46, 58, 63, 69, 127). This processing gives rise to long and short forms of OPA1, both required for proper IMM fusion (127). In addition to its function in mitochondrial fusion, cleavage of OPA1 by the presenilin-associated, rhomboid-like (PARL) protease produces a soluble form of OPA1 in the intermembrane space, where it forms a heteromeric complex with the long membrane-bound form at the neck of the cristae to keep the cristae junction closed, preventing cytochrome c release (28, 55).
Mff and Mitochondrial Dynamics Protein 49/51/Mitochondrial Elongation Factor 1, New Proteins in Mitochondrial Fission/Fusion
While DLP1, hFis1, Mfn, and OPA1 are the most studied proteins for mitochondrial fission and fusion, additional proteins have been identified and implicated in mitochondrial dynamics. For the purpose of this review, proteins identified to be physically and/or functionally interactive with mitochondrial fission and fusion are summarized in a table (Table 1). The mechanisms of mitochondrial fission and fusion were initially studied in yeast, and there are still several gaps exist with mammalian systems. Yeast Ugo1 participates in mitochondrial fusion, functioning as a linker protein between Fzo1 (Mfn homolog) and Mgm1 (OPA1 homolog) (121). In mammals, however, although the required communication between Mfn and OPA1 in completing fusion of both membranes suggests its existence (27, 35, 59, 99), a mammalian Ugo1 homolog remains to be identified. The mitochondrial fission process in yeast requires an adaptor protein, Caf4 or Mdv1, that links Dnm1 to Fis1 during fission (57, 133, 155). However, homologs for these adaptor proteins have remained elusive in mammalian systems. Recent reports describing new components restricted to animal cells suggest that mitochondrial fission (and fusion as well) might have evolved differently in yeast and metazoan, which may help translating our understanding of the mitochondrial fission/fusion process from yeast to mammalian systems. As mentioned above, the recent report by Otera et al. has promoted Mff as a more-likely mammalian mitochondrial receptor for DLP1 (103). This report and that of Gandre-Babbe and van der Bliek, in their discovery of Mff (56), observed a consistent requirement for Mff in mitochondrial fission where knockdown of the protein resulted in extensive elongation of the mitochondrial network. However, the two reports contradict with each other in the ability to enhance mitochondrial fission by overexpression and to inhibit mitochondrial profission phenotypes. Gandre-Babbe and van der Bliek showed that overexpression of Mff did not induce mitochondrial fragmentation, an effect similar to DLP1 overexpression, suggesting that unlike hFis1, Mff and DLP1 are not the rate-limiting factors in mitochondrial fission. In contrast, Otera et al. indicated robust fragmentation of mitochondria upon Mff overexpression. Additionally, Otera et al. reported that Mff was required for OPA1 knockdown-induced fission, whereas Gandre-Babbe and van der Bliek observed that Mff knockdown only partially recovered from mitochondrial fragmentation induced by Mfn DN mutants (56, 103). The reports also differ in the requirement for hFis1 in the induction of apoptosis under cellular insult. While Otera et al. found it dispensable, Gandre-Babbe found both Mff and hFis1 to have similar requirements, but the stimuli used to promote cell death varied (56, 103). This raises the possibility that the recognition and induction of apoptosis may be insult specific. In support of this, Gandre-Babbe and van der Bliek reported that although both hFis1 and Mff interact, they were found in similar-sized, but separate, complexes with DLP1 (56). Although it is clear that Mff, hFis1, and DLP1 are bona fide components in mitochondrial fission, further investigation is necessary for defining their functional interactions.
Table 1.
Location and Function of Proteins Implicated in Mitochondrial Fission and Fusion
| Component | Local | Function | Altered expression | Mitochondrial phenotype |
|---|---|---|---|---|
| DLP1/Drp1 | Cyt OMM | Fission (mitochondrial membrane severing) | OX | None or elongation (67, 108, 126) or enhanced fragmentation (131) |
| KD/KO | Elongated mitochondria (70, 81) | |||
| Fis1 | OMM | Fission (possible receptor for DLP1 or other factors) | OX | Extensive fission (71, 129, 146) |
| KD | Extensive fusion (129) | |||
| Mfn1 | OMM | Outer membrane fusion (mitochondrial tethering) | OX | Clustered, fragmented mitochondria (114, 115) |
| KD/KO | Increased fission (24, 49, 79) | |||
| Mfn2 | OMM ER | Outer membrane fusion; ER–mitochondria tethering | OX | Clusters of fragmented mitochondria (68, 115) |
| KD/KO | Enhanced fragmentation (24) | |||
| OPA1 | IMM IMS | Inner membrane fusion; crista junction regulation | OX | Mitochondrial elongation (27) |
| KD/KO | Fragmentation, vacuolar cristea (99) | |||
| MTP18 | IMM | Fission regulation | OX | Fragmented mitochondria (134) |
| KD | Interconnected mitochondria (134, 136) | |||
| Endophilin B1 | Cyt | Fission (membrane curvature) | KD | OMM dissociation from IMM (75) |
| GDAP1 | OMM | Fission promotion | OX | Extensive fission (97) |
| KD/KO | Mitochondrial elongation (32) | |||
| Mff | OMM | Fission promotion; DLP1 receptor | OX | Extensive fission (103), none (56) |
| KD/KO | Mitochondrial elongation (56, 103) | |||
| MARCH-V | OMM | Ubiquitin E3 ligase modifying DLP1/Fis1/Mfn2 | KD | Fragmented mitochondria (94, 144) or elongation (76) |
| MitoPLD | OMM | Fusion promotion | KD | Fragmented mitochondria (26) |
| MIB | Cyt Mito | Negative regulator of Mfn | OX | Fragmented mitochondria (49) |
| KD | Elongated mitochondria (49) | |||
| SLP-2 | IMM | Stress-induced fusion | KD | None (135) |
| MIEF1/MiD51 | OMM | Fission decrease; DLP1 receptor | OX | Elongated mitochondria (104, 158) |
| KD | Fragmented mitochondria (158) and elongated (104) |
Cyt, cytosol; OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; ER, endoplasmic reticulum; IMS, intermembrane space; OX, overexpression; KD, knockdown; KO, knockout; DLP1, dynamin-like protein 1; Drp1, dynamin-related protein 1; Fis1, fission protein 1; OPA1, optic atrophy 1; MTP18, mitochondrial protein 18 kDa; GDAP1, ganglioside-induced differentiation-associated protein 1; Mff, mitochondrial fission factor; Mfn, mitofusin; MARCH-V, membrane-associated RING-CH-V; MitoPLD, mitochondrial phospholipase D; MIB, Mfn-binding protein; SLP-2, stomatin-like protein 2; MIEF1, mitochondrial elongation factor 1.
Additional mitochondrial membrane-bound proteins, mitochondrial dynamics protein 49 and 51 (MiD49 and MiD51)/mitochondrial elongation factor 1 (MIEF1), were independently identified by two groups (104, 158). The proteins MiD49 and 51 were discovered through a screen of uncharacterized human genes for their ability to alter mitochondrial morphology (104), whereas MIEF1 (MiD51) was identified from a screen of a green fluorescent protein (GFP)-tagged library by virtue of its mitochondrial localization (158). MiD49 and MiD51/MIEF1 share 45% homology and a highly conserved N-terminal transmembrane domain that anchors them to the OMM (104, 158). All studies carried out by Zhao et al. utilized only the protein identified as MiD51 or their nomenclature MIEF1. Zhao et al. showed that MIEF1 knockdown induced mitochondrial fragmentation (158). In contradiction to this, Palmer et al. showed that knockdown of both MiD49 and 51 by RNAi resulted in fused mitochondria (104). Conclusions drawn from these results may be difficult to interpret due to the variable nature of knockdown by RNAi. Zhao et al. knocked down MIEF1/MiD51 alone, inducing mitochondrial fragmentation, whereas Palmer et al. did both MiD49 and MiD51 simultaneously and observed elongated mitochondrial morphology. These opposite results led to differing proposed roles for the newly discovered proteins as profission by Palmer et al., and conversely profusion by Zhao et al. Despite this discrepancy, both reports demonstrated that overexpression of MiD49 or MiD51/MIEF1 resulted in a profusion phenotype with elongated mitochondrial tubules collapsing into a cluster of tubules in a time-dependent manner (104, 158). Counter intuitively, overexpression also resulted in the enhanced mitochondrial association of the fission protein DLP1, which would predict the profission phenotype. Palmer et al. suggest that this elongated mitochondrial morphology may be an artifact, arising from diffuse mislocalization of overexpressed MiD49/51 at the mitochondrial surface, which prevents the formation of fission foci. Nevertheless, MIEF1 interacts with DLP1 as judged by immunoprecipitation and yeast two-hybrid, and this interaction was independent of hFis1, Mff, and Mfn2. Recruitment of DLP1 did not require DLP1's GTPase activity, recruiting the GTPase-defective DLP1-K38A mutant to the mitochondria. However, oligomerization-defective DLP1-A395D or G363D mutants did not localize to mitochondria upon MiD49/51 MIEF1 overexpression (104, 158). The definition of the role that MiD51/MIEF1 plays in DLP1 recruitment to the mitochondrial membrane has a strong similarity to that of Mff. Although they both appear to have a DLP1 receptor quality in that respect, their functional consequence appears to be in exact opposition: Mff promoting fission and MiD51/MIEF1 promoting fusion. MIEF1 was also competent to interact with both hFis1 and DLP1, and they were found to be contained in differing complexes with MIEF1 (158). Zhao et al. speculate that the relative amounts of hFis1 and MIEF1 could regulate the fate of DLP1 in its role in fission, either profission in complex with hFis1 or profusion in complex with MIEF1. In line with this speculation, a recent study indicated functional and physical interaction between Mfn2 and DLP1, which promotes mitochondrial fusion (67). DLP1 recruitment by MiD49/50/MIEF1 would decrease the profission activity of DLP1 by inhibiting GTP binding (158), which allows DLP1 available to interact with Mfn2 for profusion effect (Fig. 3). Given contradicting mitochondrial phenotypes reported by different groups that studied the same protein, further clarification is necessary to define how mitochondrial fission and fusion are regulated through Mff and MiD49/51/MIEF1. Aforementioned, it is of note that all three of these newly described proteins are not present in yeast (56, 103, 104, 158), and may represent evolutionary changes/advances in the regulation of fission and fusion. Mammals have turned out to contain additional factors and interactions in the fission/fusion machinery. This adds to the difficulty of deciphering the obligate interactions and regulation of mitochondrial fission and fusion in mammalian cells, which likely have additional levels of complexity.
FIG. 3.

A role of DLP1 interactions controlling the balance of mitochondrial fission and fusion through alternative complex formation. The identification of multiple new receptors at the mitochondrial outer membrane has suggested that enhanced recruitment of DLP1 to the OMM may not necessarily result in enhanced fission. Localized to the OMM, both hFis1 and Mff are profission receptors for DLP1. DLP1 interaction with hFis1 in mammalian systems is associated with enhanced mitochondrial fission. Recently, a second OMM receptor Mff was reported to serve a similar role in the absence of hFis1 protein. Reports differed slightly as to the ability of their overexpression to induce mitochondrial fragmentation and in their requirement to promote apoptosis. Analysis of complexes containing DLP1 at the mitochondria determined the two profission receptors to exist in distinct complexes, suggesting that their functional promotion of fission may occur through differing pathways. MIEF1/MiD51 promotes mitochondrial fusion when overexpressed. Enhanced MIEF1/MiD51 also increases DLP1 recruitment to the OMM while still promoting fusion. DLP1 interaction with MIEF1 alters its GTPase activity, possibly defining an underlying cause. The recent report that DLP1 interaction with Mfn2 supports mitochondrial fusion could also underlie this phenotype. Taken together, recruitment of DLP1 to MIEF1/MiD51 would result in its reduced GTPase activity, still allowing its interaction with Mfn and promoting fusion. While Mff, hFis1, MIEF1/MiD51, and Mfn are bound to the OMM, DLP1 retains the ability to traffic between the cytoplasm and the OMM. DLP1 recruitment to differing specific microdomains of these proteins then represents the regulatable process, likely coordinated by the extensive post-translational modification of DLP1, in directing fission or fusion at the OMM. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Mitochondria in Metabolic Disease
Metabolic diseases are characterized by states of misregulated excess supply of energy to cells. Disruption of the body's normal energetic regulation, largely the result of insulin resistance, causes enhanced demand on mitochondria to catabolize unrestricted influx of metabolites. The crossroads of mitochondrial bioenergetics and dynamics may serve to attempt to cope with this excess, but ultimately may contribute to the disease progression, as their abnormal morphology is observed in the pathology of these diseases. While our understanding of the role mitochondrial dynamics plays in these diseases is rudimentary, we will examine corollary changes in the form and function of mitochondria in a metabolic insult. The section will highlight our current understanding of morphological changes associated with metabolic diseases, NAFLD and DCM, commonly associated with diabetes, and observations of mitochondrial dysfunction associated with them.
Mitochondria in NAFLD
NAFLD is a progressive liver disease affecting 20%–30% of the population of western industrialized countries (5, 14, 29, 87). Advancement of the disease is hypothesized to occur through a two-hit mechanism where the first hit, steatosis, the accumulation of fat in hepatocytes, enables progression to nonalcoholic steatohepatitis (NASH) via either environmental or metabolic cues, termed second hits (36). This pathological condition is commonly accompanied by metabolic disorders prevalent in insulin resistance such as obesity, hyperglycemia, and dyslipidemia. In the context of NAFLD, the associated maladies create a feed-forward detrimental cycle in which insulin resistance in combination with excess adipose tissue releases triacylglycerol (TAG) from this tissue into circulation, a process normally suppressed by insulin through inhibition of hormone-sensitive lipase. Lipids are taken up into peripheral tissues or by the liver, where the lack of insulin sensitivity is also promoting the synthesis of fatty acids, adding to the lipid overabundance, which contributes to oxidative stress for the NASH progression as a second hit.
Mitochondria have been shown to play a major role in the development of oxidative stress in NAFLD through an increased production of ROS (106, 119). Mitochondria function in the catabolic processing of carbohydrates and fatty acids, through processing glycolysis end products and β-oxidation, to produce nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) as input for the ETC. Under normal environmental conditions, this process is highly efficient or coupled with ATP production, occurring at a similar rate as O2 consumption, the final acceptor of electrons through the ETC. Hyperglycemia and hyperlipidemia, stimuli reported to contribute to enhanced mitochondrial ROS production (15, 42, 95, 98, 118, 152), create an environment of metabolic excess with enhanced ETC inputs of NADH and FADH2 at ETC complexes I and II, respectively. This could in turn cause inefficient electron transport, resulting in the production of mitochondrial ROS. In support of this, mitochondria have been observed to have a ΔΨm threshold, above which ROS are produced from the ETC (8, 78). Increases in both β-oxidation (90, 116) and ROS production from mitochondria have been reported to play a role in the development of oxidative stress conditions in NAFLD (64, 106). Enhanced cellular ROS are thought to act in close proximity to their production and oxidize proteins, lipids, and DNA, thereby altering their ascribed function or rendering them nonfunctional. ROS produced at the mitochondria would then likely modify mitochondrial proteins, such as the ETC components, which may alter their function. In support of this notion, patients who have progressed from NAFLD to NASH displayed a reduced activity at all five ETC complexes (106). This suppressed function of the ETC leads to a predictable impaired production of ATP in NASH patients (30). Although the exact mechanism by which these ROS are produced is still the subject of investigation, it is clear that their production correlates with mitochondrial dysfunction observed in NAFLD.
Another prominent question regarding the pathological progression of NAFLD is whether steatosis leads to insulin resistance or vice versa. Studies in animal models disagree with respect to the causal factor. OLETF rats have a deficiency in their saiety signaling, causing hyperphagia and obesity (92), which manifest in the pathological condition of steatosis by 20 weeks of age (109, 110). Importantly, the emergence of this pathology is associated with mitochondrial dysfunction, with hepatic triglyceride accumulation preceding hepatic insulin resistance merely through the hyperphagic phenotype (111). In this same model, a methionine–choline-feeding regimen resulted in hepatic steatosis, a condition that was aggravated by addition of high fat to the diet by promoting insulin resistance (102). The molecular mechanism by which free fatty acids (FFA), in this case palmitate, produce an insulin-resistant state, in an acute treatment, was determined to involve mitochondrial dysfunction and the production of ROS (95). Nakamura et al. demonstrated that mitochondrially derived ROS were responsible for insulin resistance through the activation of c-Jun N-terminal Kinase1/2 (JNK1/2) and subsequent suppression of insulin receptor substrate 2. The insulin-resistant phenotype was recovered by treatment with ROS-scrubbing compounds N-acetyl-l-cystiene and α-tocopherol, as well as inhibition of mitochondrial ETC complexes, or uncoupling with carbonyl cyanide m-chlorophenyl hydrazone (CCCP), indicated a causal role for ROS in activation of JNK.
Metabolic insult and mitochondrial dynamics
Morphological deformation of mitochondria has been observed in NAFLD, suggesting the involvement of mitochondrial dynamics in its pathological progression in patients (20, 116). Mitochondria were observed as round and swollen with the loss of discernable crista structure; instead, the mitochondria contained paracrystalline inclusion bodies. Sanyal et al. correlated the mitochondrial morphological defect with an environment of enhanced oxidative stress by demonstrating significant increases in nitrotyrosine in hepatocytes of patients with fatty liver disease and full-blown NASH (116). The underlying mechanism to these mitochondrial morphological changes remains to be defined, which will require the use of defined cellular systems to separate the complexity of the metabolic milieu in vivo.
Studies using in vitro cell systems have demonstrated a lipotoxic effect of high-fat incubation, particularly with the saturated fatty acid palmitate. Palmitate is the most prevalent saturated fatty acid in circulation, and its level is further elevated in patients with NASH (37). The lipotoxic effect has been observed in pancreatic β-cells (47, 93), cardiomyocytes (77, 101), and hepatic cell lines (50, 72, 84, 128, 140, 154). Molina et al., recently described a role for mitochondrial dynamics in the progression of the lipotoxic effect, or lipid-induced apoptosis, of FFA incubation in pancreatic β-cells (91). This study used mitochondrial-targeted photoactivatable GFP (mtPAGFP) and demonstrated the dynamic nature of β-cell mitochondria. The mtPAGFP allowed observation of the spread of photoactivated GFP throughout the mitochondrial network and visualized dynamic mitochondria undergoing frequent fusion and fission events. Addition of a mixture of elevated glucose and palmitate to primary islet cells resulted in significant increases in mitochondrial fragmentation within 4 h of treatment, a phenotype not observed in elevated glucose alone. The altered mitochondrial morphology was completely reversed by the RNAi suppression of hFis1, which additionally eliminated the recruitment of DLP1 to the mitochondria and further suppressed the induction of apoptosis. From a physiological aspect, treatment with elevated glucose and fatty acids inhibited the secretion of insulin, and this was not recovered by hFis1 RNAi, suggesting that a morphological alteration of mitochondria, not the apoptotic process, may perturb the insulin secretion. Previous studies overexpressing hFis1 reported mitochondrial fragmentation and promotion of apoptosis by increasing fission, a mechanism that was dependent on DLP1 function (129, 146). In contrast to this report, however, reduced levels of hFis1 did not prevent the localization of DLP1 to the mitochondrial membrane (81, 130). It cannot be ruled out that discrepancies may lie in the cell systems used, β-cells and HeLa cells. Given the recent reports of Mff (103), MiD49/50 (104), and MIEF1 (158) targeting DLP1 to the mitochondria, hFis1 may not serve as the bona fide receptor of DLP1, but rather works downstream in the fission process. This may help reconcile the observed delay in the induction of apoptosis with hFis1 and mitochondrial fission disruption under apoptotic stimuli (3, 12, 13, 54, 81), and its reported expendability in mitochondrial fission (103). Conceivably, the two receptor proteins Mff and hFis1 may function in varied capacities in the promotion of mitochondrial fission dependent upon environmental cues and normal network maintenance, but dissection of this will require further investigation. In the context of metabolic disease, enhanced levels of FFA alone are toxic to β-cells, leading to their apoptosis and subsequently reducing the available pool of insulin-secreting cells. Reduced levels of insulin in circulation could work in the feed-forward mechanism above, enhancing misregulated lipolysis and promoting lipid uptake in hepatic tissue.
In hepatic tissue or cell systems, reports are scarce regarding the mitochondrial morphology in enhanced abundance of FFA. It was shown that incubation of HepG2 cells and primary mouse hepatocytes with palmitate fragmented mitochondria (82, 154, 156). In the case of primary mouse hepatocytes, incubation with palmitate, but not unsaturated fatty acids, resulted in the loss of mitochondrial membrane potential, assessed by tetramethylrhodamine methyl ester fluorescence, and an observed release of cytochrome c into the cytoplasm (82). This was also accompanied by enhanced abundance of ROS. Human derived HepG2 cells displayed a similar fragmented profission phenotype 24 h after supplementation with palmitate (154, 156). Fragmented mitochondria corresponded to a significant decrease in the cellular ATP levels, in agreement with mouse hepatocyte studies where mitochondrial membrane potential was lost and enhanced ROS abundance was observed. Interestingly, a coordinate reduction in the abundance of Mfn2 at both transcript and protein levels was observed in the same time course (156). Simplistically, this suggests that the observed fragmented swollen mitochondria in patients with NASH could in fact be the result of decreased expression of the profusion GTPase Mfn2. In addition, the loss of the intramitochondrial structure was observed in the cristae in NASH patients (116), indicating a perturbation of the remolding of the IMM. OPA1 is bound to the IMM and appears highly concentrated at the junction of cristae (59), giving structure to invaginations of the IMM, where the ETC complexes assemble. Reduced mitochondrial membrane potential or ATP levels following palmitate treatment would promote the increased proteolytic cleavage of OPA1 (43, 46, 63). Cleavage by OMA1 of the long-form OPA1 removes the inner membrane portion of OPA1 (63) and would release crista structure. The morphological change observed in patient samples is consistent with altered OPA1 abundance and/or processing. Additionally, processing of the long forms of OPA1 is consistent with the progression of apoptosis (3, 69), a reported consequence of studies using palmitate treatment in HepG2 cells. Another consideration in the interpretation of the in vitro data is the time point in which these experiments were conducted, monitoring cells in 24 h after treatment. Insult by hyperglycemia has been shown to occur rapidly, causing mitochondrial fragmentation and increased ROS abundance within minutes of incubation with high glucose (152, 153). Furthermore, observations over a time course of 24 h demonstrated cyclic fragmentation and fusion that coordinate with ROS production (152), eventually causing cell injury in later time points (153). The reported in vivo observations show representative mitochondrial morphology of the full-blown disease (20, 116). To define whether mitochondrial morphological alterations contribute to NAFLD progression, these changes need to be examined during the progression of the pathological condition.
Previously, we defined a ROS production in metabolic excess, hyperglycemic conditions, and linked their production to a coordinate observation of mitochondrial fragmentation (152, 153). Inhibiting mitochondrial fission or increasing fusion normalized ROS levels in the high-glucose incubation (152). The ability to manipulate ROS levels through mitochondrial dynamics defined the morphological changes as upstream causal factor for the ROS production, implicating mitochondrial dynamics as a controlling factor regulating mitochondrial activity (152). While our inhibition of fission utilized the expression of DN-DLP1, another report showed that downregulation of hFis1 decreased the ROS level in acute high-glucose stimulation (137). We have observed a similar response in the suppression of palmitate-stimulated ROS production in a hepatic cell line with the overexpression of DN-DLP1 (unpublished data). The similar recovery of ROS insult in metabolically induced mitochondrial fragmentation, through excess glucose and/or palmitate, suggests a common role for mitochondrial fission in the progression in metabolic disease through oxidative stress while providing a potential therapeutic target for their treatment.
Mitochondria in DCM
Cardiac dysfunction is associated with a metabolic disease, and most commonly diabetes. Approximately 30% of diabetics develop heart disease (86). While atherosclerosis and coronary heart disease are accelerated by diabetes, DCM, characterized by decreased left ventricular function and resulting in heart failure, can arise independent of these pathologies (60, 65, 112). Dysregulation of Ca2+ handling and metabolic changes are observed early in the disease. Deficiencies in myocardial efficiency are also observed (66), where oxygen consumption does not correlate with ATP production, suggestive of an uncoupled mitochondrial phenotype. Similar to the development of NAFLD, this pathology is also correlated with accumulation of triglycerides or steatosis in the cardiac tissue (88). Given the intimate role that mitochondria play in calcium handling and cellular metabolism, their dysfunction has been a target in defining the DCM progression.
The underlying mechanism of mitochondrial dysfunction in DCM has yet to be elucidated; however, the involvement of mitochondrial ROS production has been suggested by observation of enhanced levels of ROS in cardiomyocytes of animal models of types 1 and 2 diabetes and their resulting damage in enhanced nitrosylation and lipid peroxidation of cellular components (10, 125, 142, 143). The clinical progression of diabetes and resulting pathological condition of DCM are associated with increased levels of cardiac ROS and related oxidative stress (143), reduced cardiac efficiency (66), and morphological alterations of mitochondria, appearing swollen, rounded with the loss of discernable cristae (112). More recently, patient samples from the right atrium of diabetic patients confirmed similar findings in humans (1). Anderson and colleagues determined that patient samples from diabetic subjects contained nearly twice the amount of triglyceride as nondiabetic patients, giving them a steatotic phenotype. Using permeabilized myocardial fibrils, they determined that the steatotic cardiomyocytes were deficient in respiration of fatty acid substrates independent of mitochondrial fatty acid transport (1). This suggests that the mitochondrial energetic deficiency lies within mitochondrial components and is not the result of restricted access of substrate. Respiration with glutamate as a substrate was also impaired in diabetic samples; however, the use of succinate demonstrated similar results in oxygen consumption, suggesting that complex II was functional. Importantly, although diabetic samples were deficient in respiration with their most utilized substrates, all substrates tested resulted in higher H2O2 emission, which was further amplified by the reduction in the H2O2 scavenger glutathione (1). As one would anticipate, this leads to increases in oxidative damage in enhanced protein nitrosylation and 4-hydroxynonenal adducts. In a follow-up study, the group observed an increased propensity for mPTP opening with Ca2+ stimulation and an increase in the abundance of active caspase 9 in diabetic cardiac tissue samples over that of nondiabetic patients (2). These studies were the first direct evidence that the oxidative damage in human DCM is the result of mitochondrial dysfunction, which enhanced the propensity of cardiomyoctye death and correlates the disease progression with results from animal models. While this study did not examine mitochondrial morphology in human DCM, work in animal models has observed correlations between altered mitochondrial morphology and dysfunction during DCM progression.
Various rodent models have been used to dissect the progression of DCM, including the type 1 diabetic OVE26 mice, Akita mice, and streptozotocin (STZ)-injected models, as well as the type 2 diabetic Lepob and Leprdb models. In all models tested, except Akita mice where oxidative stress was not observed, cardiac and mitochondrial functions were decreased (9, 10, 16, 18, 19, 41, 53, 73, 80, 96, 122, 123), and these decreases were associated with increases in oxidative stress (10, 125, 142, 143). The OVE26 mouse model develops a type 1 diabetic phenotype through the overexpression of calmodulin, specifically in pancreatic β-cells, leading to damage and extremely low levels of insulin secretion (48). Transmission EM of cardiac tissue of OVE26 mice demonstrated an extreme mitochondrial morphology disruption accompanied by disruption of the myofibril structure (83, 122, 143). Interfibrillar mitochondria (IFM), normally aligned between myofibers, appeared fragmented and disordered. At a higher magnification, cardiac mitochondria were less electron dense with disordered cristae (143). Changes in mitochondrial and myofibril structure were reflected in defects in contractile function. Western blotting of cardiac extracts established an environment of increased oxidative stress with enhanced levels of malondialdehyde-modified protein, and isolated cardiomyocytes produced enhanced levels of ROS observed as increased dichlorofluorescein fluorescence. The overexpression of a catalase transgene in these mice recovered the mitochondrial and myofibril structural defects in addition to the contractile defects (143), suggesting that the production of ROS is integral in the myocardial defects of this type 1 diabetic model. In a follow-up study, the Epstein Lab further implicated the role of mitochondrial ROS production in cardiac morphological and physiological defects by demonstrating that the mitochondrial-specific ROS-scrubbing enzyme manganese superoxide dismutase (MnSOD) recovered the OVE26 cardiac maladies (122). Akita mice, which become diabetic by β-cell death in response to misfolded proinsulin (149), display a similar cardiac mitochondrial defect by EM (19). Despite their abnormal cardiac pathology, physiologically, they demonstrate very subtle contractile defects and retain mitochondrial coupling while lacking enhanced production of ROS and the resulting oxidative stress (18). Subsequent studies utilizing succinate and glutamate as substrates determined that Akita mice do have deficiencies in respiration in cardiac tissue, which results in suppressed ATP production (19). This suggests that ROS production may not be required for the development of DCM; however, it may exacerbate the physiological impairment upon metabolic insults, as type 2 models of diabetes demonstrate more robust impairment of contractile function and energetic deficiencies (16, 85). Despite the absence of ROS, there is a consistent abnormal mitochondrial morphology in cardiac tissue from these type 1 diabetic genetic mouse models, which correlates with suppressed energy production.
The type 2 diabetic mouse models Lepob and Leprdb result from a hyperphagic phenotype promoted by either deletion of functional leptin (157) or the knockout of the long-form leptin receptor (23), respectively. Studies by Dong et al. monitored the mitochondrial morphology present in the Lepob cardiac tissue and observed a swollen phenotype with disordered cristae, often with vacuoles within the IMM (41). Additionally, mitochondria were reported to be shortened, suggestive of fragmentation, and again coupled with increases in ROS, assessed by dihydroethidium. Treatment of cardiomyocytes with leptin suppressed the observed increases in ROS, which is interesting in that leptin sensitizes cells to insulin treatment, and defective insulin signaling is proposed to facilitate DCM progression. It would be interesting to see whether the mitochondrial morphology correlates with cardiac recovery during leptin treatment. In these models, oxygen consumption is increased while cardiac efficiency is decreased. The enzymes associated with fatty acid oxidation are increased in these animal models (10, 16, 85). Recently, using proteomic approaches, the Hollander lab defined increases in the subsarcolemmal mitochondrial (SSM) population and decrease of IFM in Leprdb mice (33). These alterations appear sufficient to enhance the oxygen consumption through fatty acid oxidation and were mimicked by cardiac-specific overexpression of peroxisome proliferator-activated receptor α (PPARα) (52). However, the enhanced oxygen consumption did not result in increased production of ATP, defining their decreased cardiac efficiency and subsequent deficiencies in cardiac function. A proposed mechanism for this is through the overexpression of uncoupling proteins (UCPs) that relieve the buildup of the proton gradient (17). Results from Boudina et al. support this theory, demonstrating mitochondrial uncoupling in the Leprdb heart occurs through increased UCP activity (10). Importantly, ROS may play a crucial role in activating these proteins, and precedence for this is established in ROS activation of UCP3 in skeletal muscle (45). If in fact, ROS enhancement is dependent on morphological change of mitochondria, as we have previously observed (152), then this places mitochondrial dynamics upstream of the observed uncoupling-mediated energy depletion and cardiac dysfunction in DCM.
Aside from genetic models, chemical manipulation to ablate pancreatic β-cells has been used to induce a type 1 diabetic phenotype through STZ (34, 73, 96, 141). The Hollander lab examined DCM in this model 5 weeks after the development of elevated blood glucose levels (34). As anticipated, contractile function was impaired in the hearts of diabetic mice. In this study, the investigators made a distinction between subpopulations of mitochondria in cardiac tissue, separating the IFM from SSM for assessment of mitochondrial function. Using a unique light scatter approach to determine both the size and internal complexity of isolated IFM and SSM, Dabkowski and colleagues determined that IFM recovered from diabetic cardiac tissue were smaller in size and retained less internal complexity than wild-type controls (34). This was not observed in the SSM samples. The reduced size of IFM could be explained by an enhanced fission, whereas the loss of internal complexity is consistent with the findings from Akita and OVE26 genetic models (19, 143). Respiration analyses of ETC components of the augmented IFM revealed deficiencies in respiration from complexes I, II, and III. Inefficient respiration from IFM fits with the loss of contractile force, as IFM juxtaposed to the sacromere supply ATP for motive force. Coordinates with these changes in diabetic cardiac IFM were enhanced ROS production and oxidative stress in the form of lipid modification (34). We have observed a similar fragmented phenotype of cardiac IFM with STZ-induced diabetes (Fig. 4), which correlated with enhanced ROS abundance (unpublished data). In the follow-up study by Williamson et al., the same IFM were more susceptible to apoptotic cell death, with increased caspase activation, decreased mitochondrial membrane potential, and increased susceptibility to mPTP opening with enhanced proapoptotic protein expression (141). The smaller or fragmented characteristic of the IFM purports mitochondrial morphology as a pathological marker of DCM, as these mitochondria are more susceptible to mitochondrial apoptosis and subsequent tissue damage. Mitophagy, the autophagy of damaged mitochondria, has been reported to follow mitochondrial fission, selectively removing the depolarized daughter mitochondria (137). The accumulation of fragmented depolarized IFM in these studies could represent an improper control of mitophagy, possibly contributing to DCM. Alternatively, it may represent an inhibition of mitochondrial fusion, which would not allow the proper mixing of mitochondrial components. As discussed above in the context of NAFLD, while enhanced abundance of FFA can induce mitochondrial fission supporting the fragmented phenotype, whether this is the result of increased fission or decreased fusion will require further investigation (154, 156). It will be informative to examine changes in the abundance of fission and fusion factors in DCM, as studied in the in vitro palmitate treatment of HepG2 cells (156).
FIG. 4.
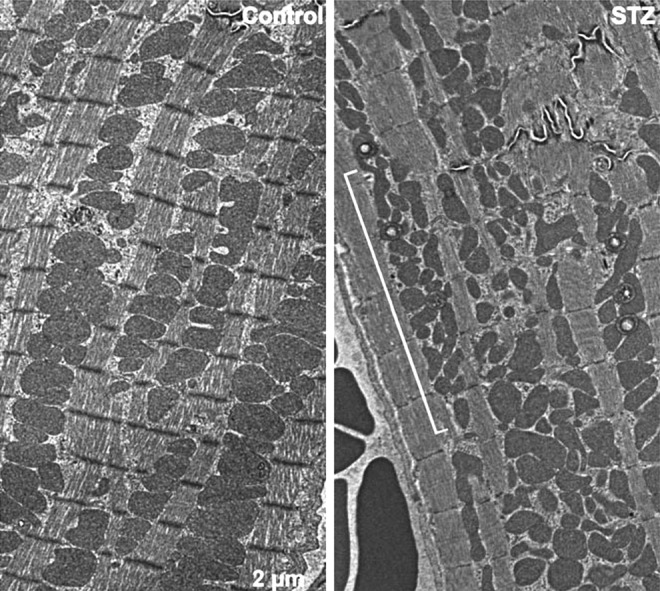
Mitochondrial morphology in left ventricular samples from a type I diabetic mouse model is altered. Control heart shows normal tightly packed interfibrillar mitochondria (IFM) with normal uniform globular appearance. Most mitochondria are observed to span one sacromere (left panel). Following 8 weeks of hyperglycemia, induced by streptozotocin (STZ) β-cell ablation, IFM are observed less uniform and more fragmented, highlighted by the bracketed region. The mitochondrial morphology is consistent with an increase in mitochondrial fission (right panel).
The suggestion of dynamic mitochondria seems to contradict a long-standing paradigm in the mitochondrial field that cardiac mitochondria were relatively static as far as morphological changes. In vivo observations support a presence of mitochondrial morphological change, but the question remains as to whether these are merely a consequence or causal to the disease progression. In addition to DCM, dysregulation of mitochondrial morphology has also been observed in dilated cardiomyopathy (117) and myocardial hibernation (74). Recently, a mutagenic screen described the first instance of a mitochondrial-remodeling protein defect, mutation of C452F in DLP1, resulting in cardiomyopathy (4). Interestingly, ischemia reperfusion injury (IRI) in cardiac tissue is also associated with mitochondrial fragmentation and can be circumvented by inhibiting mitochondrial fission with a small molecule, mdivi-1 (100). The study by Ong et al. implicates mitochondrial dynamics as a required process for cardiomyocyte damage in IRI and that may follow a scheme similar to the progression of DCM. The mechanism through which mitochondrial dynamics are altered in IRI remains unclear; however, mitochondrial fragmentation occurred upstream to apoptotic cell death placing misregulated mitochondrial dynamics as a possible causal factor in the injury. The overexpression of either the profusion GTPase Mfn2 or fission inhibitory DN-DLP1 delayed the progression of the apoptotic death in their HL-1 cell system (100). Treatment in vivo with a mitochondrial fission inhibitor, mdivi-1, was demonstrated to serve a protective role in IRI injury resulting in a smaller infarct size after the insult. Increased survival correlated with an observed increased abundance of elongated IFM, suggesting that elongated mitochondria are protective of IRI insult. In contrast to this, Papanicolaou et al. recently reported that cardiac-specific knockdown of the profusion GTPase Mfn2 conferred protective effects on mitochondria in adult cardiomyocytes under apoptosis-inducing conditions, delaying mitochondrial permeability transition (105). Within the same study, however, the authors report that the suppression of Mfn2 in neonatal cardiomyocytes has opposing effects. Despite discrepancies in the functional outcome, mitochondrial dynamics and the proteins associated with these changes play roles in cardiac insult. The paradigm of immobile, restricted mitochondria in cardiac tissue has been replaced by the implication of mitochondrial dynamics in both the progression of cardiac disease, and its modulation as possible therapeutics to combat these diseases.
Perspectives
A wealth of evidence is present for a mitochondrial role in the progression of metabolic disease, but defining the nature of this role in human physiology has been difficult. Recent evidence has confirmed that the observed oxidative damage and mitochondrial dysfunction in animal models are also present in diabetic patients (1, 2). A prevailing commonality to metabolic disease pathologies is the suppression of ATP levels, which is the manifestation of mitochondrial bioenergetic deficiencies. Given the long-standing ascribed function of mitochondria as metabolic regulators of the cell, it is not surprising that they play an intricate role in metabolic diseases associated with hyperglycemia and hyperlipidemia.
Mitochondrial fission itself resulting from a hyperglycemic insult correlated with enhanced ROS production, but is ameliorated by suppression of fission. A similar mechanism could govern the dual insult of hyperglycemic and hyperlipidemic conditions in insulin-resistance-driven metabolic diseases (Fig. 5). Changes in the abundance of Mfn2 and OPA1 have been described to alter bioenergetic properties of mitochondria, with increased Mfn2 levels in increased energy demand and suppressed OPA1 levels associated with decreased respiration. While mitochondrial dynamics are intimately linked to mitochondrial energetics, their role causative or resultant remains an avenue of investigation. In either situation, changes in mitochondrial morphology implicate proteins involved in their remolding in the etiology of metabolic disease. Their association with Bcl-2 proteins and the role of mitochondrial dynamics in apoptotic cell death also connects these proteins to the decision of cell life or death. Cyclically, this offers support for their role in maintaining or responding to energetic change. Defining the role of mitochondrial dynamics in pathological metabolic conditions will require more detailed assessments in the progression of the disease and correlations with mitochondrial bioenergetic and morphological properties. Mechanistically, defining the mitochondrial decision of fission or fusion from the perspective of functional interactions between implicated machineries will aid in defining the temporal execution in metabolic pathologies, such as NAFLD and DCM. While investigation of these disease states has been ongoing for decades, the involvement of mitochondrial dynamics is in its relatively juvenile stages and should yield important clues to their pathological progression and define new therapeutic targets.
FIG. 5.

A role of mitochondrial dynamics in the progression of metabolic disease. Nonalcoholic fatty liver disease (NAFLD) and diabetic cardiomyopathy both are associated with steatosis in their respective afflicted tissue type. This was hypothesized to be the first hit in NAFLD and is also associated with insulin resistance. Hyperglycemia and hyperlipidemia induce mitochondrial fragmentation, a process correlated with the enhanced production of mitochondrial reactive oxygen species (ROS). ROS modify cellular macromolecules, including proteins, lipids, and DNA, with functional consequences. Modification of electron transport chain (ETC) components in particular may exacerbate ROS production and bioenergetic deficiencies. These are reflected in the observed reduction of ΔΨm and lowered adenosine triphosphate (ATP) levels despite an excess of ETC input. Suppression of these properties is also associated with processing of OPA1 with implications in cell viability and mitochondrial morphology. In this way, mitochondrial dynamics appear implicated in the characterized progression of metabolic disease associated with steatosis and may be integral to the hypothesized second hit in these conditions. To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars
Abbreviations Used
- ATP
adenosine triphosphate
- CC
coiled-coil
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- DCM
diabetic cardiomyopathy
- DLP1
dynamin-like protein 1
- DN
dominant-negative
- Drp1
dynamin-related protein 1
- EM
electron microscopy
- ETC
electron transport chain
- FADH2
flavin adenine dinucleotide
- FFA
free fatty acid
- Fis1
fission protein 1
- GDAP1
ganglioside-induced differentiation-associated protein 1
- GFP
green fluorescent protein
- GTP
guanosine triphosphate
- HR
heptad repeat
- IFM
interfibrillar mitochondria
- IMM
inner mitochondrial membrane
- IRI
ischemia reperfusion injury
- JNK1/2
c-Jun N-terminal Kinase1/2
- MARCH-V
membrane-associated RING-CH-V
- Mff
mitochondrial fission factor
- Mfn
mitofusin
- MIB
Mfn-binding protein
- MiD49/51
mitochondrial dynamics protein 49/51
- MIEF1
mitochondrial elongation factor 1
- MitoPLD
mitochondrial phospholipase D
- MnSOD
manganese superoxide dismutase
- MTP18
mitochondrial protein 18 kDa
- NADH
nicotinamide adenine dinucleotide
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OMM
outer mitochondrial membrane
- OPA1
optic atrophy 1
- PARL
presenilin-associated rhomboid-like
- PPARα
peroxisome proliferator-activated receptor α
- RNAi
ribonucleic acid interference
- ROS
reactive oxygen species
- SLP-2
stomatin-like protein 2
- SSM
subsarcolemmal mitochondria
- STZ
streptozotocin
- TAG
triacylglycerol
- TPR
tetratricopeptide repeat
- UCP
uncoupling protein
Acknowledgments
This work is supported by National Institutes of Health grants DK 078618 and DK 061991 to Y.Y. and the American Heart Association Postdoctoral Fellowship 12POST9430003 to C.A.G.
References
- 1.Anderson EJ. Kypson AP. Rodriguez E. Anderson CA. Lehr EJ. Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–1898. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson EJ. Rodriguez E. Anderson CA. Thayne K. Chitwood WR. Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am J Physiol Heart Circ Physiol. 2011;300:H118–H124. doi: 10.1152/ajpheart.00932.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnoult D. Rismanchi N. Grodet A. Roberts RG. Seeburg DP. Estaquier J. Sheng M. Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 4.Ashrafian H. Docherty L. Leo V. Towlson C. Neilan M. Steeples V. Lygate CA. Hough T. Townsend S. Williams D, et al. A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000. doi: 10.1371/journal.pgen.1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellentani S. Saccoccio G. Masutti F. Croce LS. Brandi G. Sasso F. Cristanini G. Tiribelli C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med. 2000;132:112–117. doi: 10.7326/0003-4819-132-2-200001180-00004. [DOI] [PubMed] [Google Scholar]
- 6.Bereiter-Hahn J. Behavior of mitochondria in the living cell. Int Rev Cytol. 1990;122:1–63. doi: 10.1016/s0074-7696(08)61205-x. [DOI] [PubMed] [Google Scholar]
- 7.Bereiter-Hahn J. Voth M. Dynamics of mitochondria in living cells: shape changes, dislocation, fusion, and fission of mitochondria. Microsc Res Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- 8.Bodrova ME. Dedukhova VI. Mokhova EN. Skulachev VP. Membrane potential generation coupled to oxidation of external NADH in liver mitochondria. FEBS Lett. 1998;435:269–274. doi: 10.1016/s0014-5793(98)01072-2. [DOI] [PubMed] [Google Scholar]
- 9.Boudina S. Sena S. O'Neill BT. Tathireddy P. Young ME. Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 10.Boudina S. Sena S. Theobald H. Sheng X. Wright JJ. Hu XX. Aziz S. Johnson JI. Bugger H. Zaha VG, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 11.Braschi E. Zunino R. McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brooks C. Wei Q. Cho SG. Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks C. Wei Q. Feng L. Dong G. Tao Y. Mei L. Xie ZJ. Dong Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Browning JD. Szczepaniak LS. Dobbins R. Nuremberg P. Horton JD. Cohen JC. Grundy SM. Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 15.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 16.Buchanan J. Mazumder PK. Hu P. Chakrabarti G. Roberts MW. Yun UJ. Cooksey RC. Litwin SE. Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 17.Bugger H. Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res. 2010;88:229–240. doi: 10.1093/cvr/cvq239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bugger H. Boudina S. Hu XX. Tuinei J. Zaha VG. Theobald HA. Yun UJ. McQueen AP. Wayment B. Litwin SE, et al. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes. 2008;57:2924–2932. doi: 10.2337/db08-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bugger H. Chen D. Riehle C. Soto J. Theobald HA. Hu XX. Ganesan B. Weimer BC. Abel ED. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes. 2009;58:1986–1997. doi: 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caldwell SH. Swerdlow RH. Khan EM. Iezzoni JC. Hespenheide EE. Parks JK. Parker WD., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31:430–434. doi: 10.1016/s0168-8278(99)80033-6. [DOI] [PubMed] [Google Scholar]
- 21.Chang CR. Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 22.Chang CR. Manlandro CM. Arnoult D. Stadler J. Posey AE. Hill RB. Blackstone C. A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J Biol Chem. 2010;285:32494–32503. doi: 10.1074/jbc.M110.142430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H. Charlat O. Tartaglia LA. Woolf EA. Weng X. Ellis SJ. Lakey ND. Culpepper J. Moore KJ. Breitbart RE, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 24.Chen H. Detmer SA. Ewald AJ. Griffin EE. Fraser SE. Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho DH. Nakamura T. Fang J. Cieplak P. Godzik A. Gu Z. Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi SY. Huang P. Jenkins GM. Chan DC. Schiller J. Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 27.Cipolat S. Martins de Brito O. Dal Zilio B. Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cipolat S. Rudka T. Hartmann D. Costa V. Serneels L. Craessaerts K. Metzger K. Frezza C. Annaert W. D'Adamio L, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 29.Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40(Suppl 1):S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 30.Cortez-Pinto H. Chatham J. Chacko VP. Arnold C. Rashid A. Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659–1664. doi: 10.1001/jama.282.17.1659. [DOI] [PubMed] [Google Scholar]
- 31.Cribbs JT. Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuesta A. Pedrola L. Sevilla T. Garcia-Planells J. Chumillas MJ. Mayordomo F. LeGuern E. Marin I. Vilchez JJ. Palau F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet. 2002;30:22–25. doi: 10.1038/ng798. [DOI] [PubMed] [Google Scholar]
- 33.Dabkowski ER. Baseler WA. Williamson CL. Powell M. Razunguzwa TT. Frisbee JC. Hollander JM. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol. 2010;299:H529–H540. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dabkowski ER. Williamson CL. Bukowski VC. Chapman RS. Leonard SS. Peer CJ. Callery PS. Hollander JM. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am J Physiol Heart Circ Physiol. 2009;296:H359–H369. doi: 10.1152/ajpheart.00467.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davies VJ. Hollins AJ. Piechota MJ. Yip W. Davies JR. White KE. Nicols PP. Boulton ME. Votruba M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. 2007;16:1307–1318. doi: 10.1093/hmg/ddm079. [DOI] [PubMed] [Google Scholar]
- 36.Day CP. James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 37.de Almeida IT. Cortez-Pinto H. Fidalgo G. Rodrigues D. Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr. 2002;21:219–223. doi: 10.1054/clnu.2001.0529. [DOI] [PubMed] [Google Scholar]
- 38.de Brito OM. Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 39.Delettre C. Lenaers G. Griffoin JM. Gigarel N. Lorenzo C. Belenguer P. Pelloquin L. Grosgeorge J. Turc-Carel C. Perret E, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 40.Dohm JA. Lee SJ. Hardwick JM. Hill RB. Gittis AG. Cytosolic domain of the human mitochondrial fission protein Fis1 adopts a TPR fold. Proteins. 2004;54:153–156. doi: 10.1002/prot.10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong F. Zhang X. Yang X. Esberg LB. Yang H. Zhang Z. Culver B. Ren J. Impaired cardiac contractile function in ventricular myocytes from leptin-deficient ob/ob obese mice. J Endocrinol. 2006;188:25–36. doi: 10.1677/joe.1.06241. [DOI] [PubMed] [Google Scholar]
- 42.Du XL. Edelstein D. Rossetti L. Fantus IG. Goldberg H. Ziyadeh F. Wu J. Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duvezin-Caubet S. Jagasia R. Wagener J. Hofmann S. Trifunovic A. Hansson A. Chomyn A. Bauer MF. Attardi G. Larsson NG, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- 44.Duvezin-Caubet S. Koppen M. Wagener J. Zick M. Israel L. Bernacchia A. Jagasia R. Rugarli EI. Imhof A. Neupert W, et al. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol Biol Cell. 2007;18:3582–3590. doi: 10.1091/mbc.E07-02-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Echtay KS. Roussel D. St-Pierre J. Jekabsons MB. Cadenas S. Stuart JA. Harper JA. Roebuck SJ. Morrison A. Pickering S, et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 46.Ehses S. Raschke I. Mancuso G. Bernacchia A. Geimer S. Tondera D. Martinou JC. Westermann B. Rugarli EI. Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eitel K. Staiger H. Brendel MD. Brandhorst D. Bretzel RG. Haring HU. Kellerer M. Different role of saturated and unsaturated fatty acids in beta-cell apoptosis. Biochem Biophys Res Commun. 2002;299:853–856. doi: 10.1016/s0006-291x(02)02752-3. [DOI] [PubMed] [Google Scholar]
- 48.Epstein PN. Overbeek PA. Means AR. Calmodulin-induced early-onset diabetes in transgenic mice. Cell. 1989;58:1067–1073. doi: 10.1016/0092-8674(89)90505-9. [DOI] [PubMed] [Google Scholar]
- 49.Eura Y. Ishihara N. Oka T. Mihara K. Identification of a novel protein that regulates mitochondrial fusion by modulating mitofusin (Mfn) protein function. J Cell Sci. 2006;119:4913–4925. doi: 10.1242/jcs.03253. [DOI] [PubMed] [Google Scholar]
- 50.Feldstein AE. Werneburg NW. Canbay A. Guicciardi ME. Bronk SF. Rydzewski R. Burgart LJ. Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 51.Figueroa-Romero C. Iniguez-Lluhi JA. Stadler J. Chang CR. Arnoult D. Keller PJ. Hong Y. Blackstone C. Feldman EL. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 2009;23:3917–3927. doi: 10.1096/fj.09-136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Finck BN. Lehman JJ. Leone TC. Welch MJ. Bennett MJ. Kovacs A. Han X. Gross RW. Kozak R. Lopaschuk GD, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flarsheim CE. Grupp IL. Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Physiol. 1996;271:H192–H202. doi: 10.1152/ajpheart.1996.271.1.H192. [DOI] [PubMed] [Google Scholar]
- 54.Frank S. Gaume B. Bergmann-Leitner ES. Leitner WW. Robert EG. Catez F. Smith CL. Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 55.Frezza C. Cipolat S. Martins de Brito O. Micaroni M. Beznoussenko GV. Rudka T. Bartoli D. Polishuck RS. Danial NN. De Strooper B, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 56.Gandre-Babbe S. van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Griffin EE. Graumann J. Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. 2005;170:237–248. doi: 10.1083/jcb.200503148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griparic L. Kanazawa T. van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Griparic L. van der Wel NN. Orozco IJ. Peters PJ. van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. 2004;279:18792–18798. doi: 10.1074/jbc.M400920200. [DOI] [PubMed] [Google Scholar]
- 60.Hamby RI. Zoneraich S. Sherman L. Diabetic cardiomyopathy. JAMA. 1974;229:1749–1754. [PubMed] [Google Scholar]
- 61.Han XJ. Lu YF. Li SA. Kaitsuka T. Sato Y. Tomizawa K. Nairn AC. Takei K. Matsui H. Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harder Z. Zunino R. McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 63.Head B. Griparic L. Amiri M. Gandre-Babbe S. van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hensley K. Kotake Y. Sang H. Pye QN. Wallis GL. Kolker LM. Tabatabaie T. Stewart CA. Konishi Y. Nakae D, et al. Dietary choline restriction causes complex I dysfunction and increased H(2)O(2) generation in liver mitochondria. Carcinogenesis. 2000;21:983–989. doi: 10.1093/carcin/21.5.983. [DOI] [PubMed] [Google Scholar]
- 65.Ho KK. Pinsky JL. Kannel WB. Levy D. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol. 1993;22:6A–13A. doi: 10.1016/0735-1097(93)90455-a. [DOI] [PubMed] [Google Scholar]
- 66.How OJ. Aasum E. Severson DL. Chan WY. Essop MF. Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes. 2006;55:466–473. doi: 10.2337/diabetes.55.02.06.db05-1164. [DOI] [PubMed] [Google Scholar]
- 67.Huang P. Galloway CA. Yoon Y. Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS One. 2011;6:e20655. doi: 10.1371/journal.pone.0020655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang P. Yu T. Yoon Y. Mitochondrial clustering induced by overexpression of the mitochondrial fusion protein Mfn2 causes mitochondrial dysfunction and cell death. Eur J Cell Biol. 2007;86:289–302. doi: 10.1016/j.ejcb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 69.Ishihara N. Fujita Y. Oka T. Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ishihara N. Nomura M. Jofuku A. Kato H. Suzuki SO. Masuda K. Otera H. Nakanishi Y. Nonaka I. Goto Y, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 71.James DI. Parone PA. Mattenberger Y. Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 72.Ji J. Zhang L. Wang P. Mu YM. Zhu XY. Wu YY. Yu H. Zhang B. Chen SM. Sun XZ. Saturated free fatty acid, palmitic acid, induces apoptosis in fetal hepatocytes in culture. Exp Toxicol Pathol. 2005;56:369–376. doi: 10.1016/j.etp.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 73.Kajstura J. Fiordaliso F. Andreoli AM. Li B. Chimenti S. Medow MS. Limana F. Nadal-Ginard B. Leri A. Anversa P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50:1414–1424. doi: 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- 74.Kalra DK. Zoghbi WA. Myocardial hibernation in coronary artery disease. Curr Atheroscler Rep. 2002;4:149–155. doi: 10.1007/s11883-002-0039-x. [DOI] [PubMed] [Google Scholar]
- 75.Karbowski M. Jeong SY. Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–1039. doi: 10.1083/jcb.200407046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karbowski M. Neutzner A. Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kong JY. Rabkin SW. Palmitate-induced apoptosis in cardiomyocytes is mediated through alterations in mitochondria: prevention by cyclosporin A. Biochim Biophys Acta. 2000;1485:45–55. doi: 10.1016/s1388-1981(00)00028-7. [DOI] [PubMed] [Google Scholar]
- 78.Korshunov SS. Skulachev VP. Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 79.Koshiba T. Detmer SA. Kaiser JT. Chen H. McCaffery JM. Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 80.Lashin OM. Szweda PA. Szweda LI. Romani AM. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic Biol Med. 2006;40:886–896. doi: 10.1016/j.freeradbiomed.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 81.Lee YJ. Jeong SY. Karbowski M. Smith CL. Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Z. Berk M. McIntyre TM. Gores GJ. Feldstein AE. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology. 2008;47:1495–1503. doi: 10.1002/hep.22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liang Q. Carlson EC. Donthi RV. Kralik PM. Shen X. Epstein PN. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes. 2002;51:174–181. doi: 10.2337/diabetes.51.1.174. [DOI] [PubMed] [Google Scholar]
- 84.Malhi H. Bronk SF. Werneburg NW. Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 85.Mazumder PK. O'Neill BT. Roberts MW. Buchanan J. Yun UJ. Cooksey RC. Boudina S. Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 86.McCarron P. Davey Smith G. Commentary: incubation of coronary heart disease—recent developments. Int J Epidemiol. 2005;34:248–250. doi: 10.1093/ije/dyi057. [DOI] [PubMed] [Google Scholar]
- 87.McCullough AJ. The epidemiology and risk factors of NASH. In: Farrell GC, editor; Hall PM, editor; George J, editor; McCullough AJ, editor. Fatty Liver Disease: NASH and Related Disorders. Malden, MA: Blackwell Publishing; 2005. pp. 23–37. [Google Scholar]
- 88.McGavock JM. Lingvay I. Zib I. Tillery T. Salas N. Unger R. Levine BD. Raskin P. Victor RG. Szczepaniak LS. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 89.Mears JA. Lackner LL. Fang S. Ingerman E. Nunnari J. Hinshaw JE. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol. 2011;18:20–26. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miele L. Grieco A. Armuzzi A. Candelli M. Forgione A. Gasbarrini A. Gasbarrini G. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am J Gastroenterol. 2003;98:2335–2336. doi: 10.1111/j.1572-0241.2003.07725.x. [DOI] [PubMed] [Google Scholar]
- 91.Molina AJ. Wikstrom JD. Stiles L. Las G. Mohamed H. Elorza A. Walzer G. Twig G. Katz S. Corkey BE, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–2315. doi: 10.2337/db07-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moran TH. Bi S. Hyperphagia and obesity of OLETF rats lacking CCK1 receptors: developmental aspects. Dev Psychobiol. 2006;48:360–367. doi: 10.1002/dev.20149. [DOI] [PubMed] [Google Scholar]
- 93.Morgan NG. Dhayal S. Diakogiannaki E. Welters HJ. The cytoprotective actions of long-chain mono-unsaturated fatty acids in pancreatic beta-cells. Biochem Soc Trans. 2008;36:905–908. doi: 10.1042/BST0360905. [DOI] [PubMed] [Google Scholar]
- 94.Nakamura N. Kimura Y. Tokuda M. Honda S. Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–1022. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nakamura S. Takamura T. Matsuzawa-Nagata N. Takayama H. Misu H. Noda H. Nabemoto S. Kurita S. Ota T. Ando H, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284:14809–14818. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nielsen LB. Bartels ED. Bollano E. Overexpression of apolipoprotein B in the heart impedes cardiac triglyceride accumulation and development of cardiac dysfunction in diabetic mice. J Biol Chem. 2002;277:27014–27020. doi: 10.1074/jbc.M203458200. [DOI] [PubMed] [Google Scholar]
- 97.Niemann A. Ruegg M. La Padula V. Schenone A. Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol. 2005;170:1067–1078. doi: 10.1083/jcb.200507087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nishikawa T. Edelstein D. Du XL. Yamagishi S. Matsumura T. Kaneda Y. Yorek MA. Beebe D. Oates PJ. Hammes HP, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 99.Olichon A. Baricault L. Gas N. Guillou E. Valette A. Belenguer P. Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 100.Ong SB. Subrayan S. Lim SY. Yellon DM. Davidson SM. Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 101.Ostrander DB. Sparagna GC. Amoscato AA. McMillin JB. Dowhan W. Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J Biol Chem. 2001;276:38061–38067. doi: 10.1074/jbc.M107067200. [DOI] [PubMed] [Google Scholar]
- 102.Ota T. Takamura T. Kurita S. Matsuzawa N. Kita Y. Uno M. Akahori H. Misu H. Sakurai M. Zen Y, et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology. 2007;132:282–293. doi: 10.1053/j.gastro.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 103.Otera H. Wang C. Cleland MM. Setoguchi K. Yokota S. Youle RJ. Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Palmer CS. Osellame LD. Laine D. Koutsopoulos OS. Frazier AE. Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Papanicolaou KN. Khairallah RJ. Ngoh GA. Chikando A. Luptak I. O'Shea KM. Riley DD. Lugus JJ. Colucci WS. Lederer WJ, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Perez-Carreras M. Del Hoyo P. Martin MA. Rubio JC. Martin A. Castellano G. Colina F. Arenas J. Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 107.Pitts KR. McNiven MA. Yoon Y. Mitochondria-specific function of the dynamin family protein DLP1 is mediated by its C-terminal domains. J Biol Chem. 2004;279:50286–50294. doi: 10.1074/jbc.M405531200. [DOI] [PubMed] [Google Scholar]
- 108.Pitts KR. Yoon Y. Krueger EW. McNiven MA. The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol Biol Cell. 1999;10:4403–4417. doi: 10.1091/mbc.10.12.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rector RS. Thyfault JP. Laye MJ. Morris RT. Borengasser SJ. Uptergrove GM. Chakravarthy MV. Booth FW. Ibdah JA. Cessation of daily exercise dramatically alters precursors of hepatic steatosis in Otsuka Long-Evans Tokushima Fatty (OLETF) rats. J Physiol. 2008;586:4241–4249. doi: 10.1113/jphysiol.2008.156745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rector RS. Thyfault JP. Morris RT. Laye MJ. Borengasser SJ. Booth FW. Ibdah JA. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol. 2008;294:G619–G626. doi: 10.1152/ajpgi.00428.2007. [DOI] [PubMed] [Google Scholar]
- 111.Rector RS. Thyfault JP. Uptergrove GM. Morris EM. Naples SP. Borengasser SJ. Mikus CR. Laye MJ. Laughlin MH. Booth FW, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52:727–736. doi: 10.1016/j.jhep.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Regan TJ. Lyons MM. Ahmed SS. Levinson GE. Oldewurtel HA. Ahmad MR. Haider B. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest. 1977;60:884–899. doi: 10.1172/JCI108843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rojo M. Legros F. Chateau D. Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115:1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 114.Santel A. Frank S. Gaume B. Herrler M. Youle RJ. Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci. 2003;116:2763–2774. doi: 10.1242/jcs.00479. [DOI] [PubMed] [Google Scholar]
- 115.Santel A. Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- 116.Sanyal AJ. Campbell-Sargent C. Mirshahi F. Rizzo WB. Contos MJ. Sterling RK. Luketic VA. Shiffman ML. Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 117.Schaper J. Froede R. Hein S. Buck A. Hashizume H. Speiser B. Friedl A. Bleese N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83:504–514. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- 118.Schonfeld P. Wojtczak L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med. 2008;45:231–241. doi: 10.1016/j.freeradbiomed.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 119.Seki S. Kitada T. Yamada T. Sakaguchi H. Nakatani K. Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol. 2002;37:56–62. doi: 10.1016/s0168-8278(02)00073-9. [DOI] [PubMed] [Google Scholar]
- 120.Serasinghe MN. Yoon Y. The mitochondrial outer membrane protein hFis1 regulates mitochondrial morphology and fission through self-interaction. Exp Cell Res. 2008;314:3494–3507. doi: 10.1016/j.yexcr.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sesaki H. Jensen RE. Ugo1p links the Fzo1p and Mgm1p GTPases for mitochondrial fusion. J Biol Chem. 2004;279:28298–28303. doi: 10.1074/jbc.M401363200. [DOI] [PubMed] [Google Scholar]
- 122.Shen X. Zheng S. Metreveli NS. Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- 123.Shen X. Zheng S. Thongboonkerd V. Xu M. Pierce WM., Jr. Klein JB. Epstein PN. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab. 2004;287:E896–E905. doi: 10.1152/ajpendo.00047.2004. [DOI] [PubMed] [Google Scholar]
- 124.Shin HW. Shinotsuka C. Torii S. Murakami K. Nakayama K. Identification and subcellular localization of a novel mammalian dynamin-related protein homologous to yeast Vps1p and Dnm1p. J Biochem. 1997;122:525–530. doi: 10.1093/oxfordjournals.jbchem.a021784. [DOI] [PubMed] [Google Scholar]
- 125.Singh VP. Le B. Khode R. Baker KM. Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57:3297–3306. doi: 10.2337/db08-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Smirnova E. Griparic L. Shurland DL. van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Song Z. Chen H. Fiket M. Alexander C. Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Srivastava S. Chan C. Hydrogen peroxide and hydroxyl radicals mediate palmitate-induced cytotoxicity to hepatoma cells: relation to mitochondrial permeability transition. Free Radic Res. 2007;41:38–49. doi: 10.1080/10715760600943900. [DOI] [PubMed] [Google Scholar]
- 129.Stojanovski D. Koutsopoulos OS. Okamoto K. Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. 2004;117:1201–1210. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- 130.Suzuki M. Jeong SY. Karbowski M. Youle RJ. Tjandra N. The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J Mol Biol. 2003;334:445–458. doi: 10.1016/j.jmb.2003.09.064. [DOI] [PubMed] [Google Scholar]
- 131.Szabadkai G. Simoni AM. Chami M. Wieckowski MR. Youle RJ. Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 132.Taguchi N. Ishihara N. Jofuku A. Oka T. Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 133.Tieu Q. Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. 2000;151:353–366. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tondera D. Czauderna F. Paulick K. Schwarzer R. Kaufmann J. Santel A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. 2005;118:3049–3059. doi: 10.1242/jcs.02415. [DOI] [PubMed] [Google Scholar]
- 135.Tondera D. Grandemange S. Jourdain A. Karbowski M. Mattenberger Y. Herzig S. Da Cruz S. Clerc P. Raschke I. Merkwirth C, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tondera D. Santel A. Schwarzer R. Dames S. Giese K. Klippel A. Kaufmann J. Knockdown of MTP18, a novel phosphatidylinositol 3-kinase-dependent protein, affects mitochondrial morphology and induces apoptosis. J Biol Chem. 2004;279:31544–31555. doi: 10.1074/jbc.M404704200. [DOI] [PubMed] [Google Scholar]
- 137.Twig G. Elorza A. Molina AJ. Mohamed H. Wikstrom JD. Walzer G. Stiles L. Haigh SE. Katz S. Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wakabayashi J. Zhang Z. Wakabayashi N. Tamura Y. Fukaya M. Kensler TW. Iijima M. Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wasiak S. Zunino R. McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wei Y. Wang D. Topczewski F. Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 141.Williamson CL. Dabkowski ER. Baseler WA. Croston TL. Alway SE. Hollander JM. Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. Am J Physiol Heart Circ Physiol. 2010;298:H633–H642. doi: 10.1152/ajpheart.00668.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wold LE. Ren J. Streptozotocin directly impairs cardiac contractile function in isolated ventricular myocytes via a p38 map kinase-dependent oxidative stress mechanism. Biochem Biophys Res Commun. 2004;318:1066–1071. doi: 10.1016/j.bbrc.2004.04.138. [DOI] [PubMed] [Google Scholar]
- 143.Ye G. Metreveli NS. Donthi RV. Xia S. Xu M. Carlson EC. Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes. 2004;53:1336–1343. doi: 10.2337/diabetes.53.5.1336. [DOI] [PubMed] [Google Scholar]
- 144.Yonashiro R. Ishido S. Kyo S. Fukuda T. Goto E. Matsuki Y. Ohmura-Hoshino M. Sada K. Hotta H. Yamamura H, et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006;25:3618–3626. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yoon Y. Galloway CA. Jhun BS. Yu T. Mitochondrial dynamics in diabetes. Antioxid Redox Signal. 2011;14:439–457. doi: 10.1089/ars.2010.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yoon Y. Krueger EW. Oswald BJ. McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yoon Y. Pitts KR. Dahan S. McNiven MA. A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J Cell Biol. 1998;140:779–793. doi: 10.1083/jcb.140.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yoon Y. Pitts KR. McNiven MA. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell. 2001;12:2894–2905. doi: 10.1091/mbc.12.9.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yoshioka M. Kayo T. Ikeda T. Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- 150.Yu T. Fox RJ. Burwell LS. Yoon Y. Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J Cell Sci. 2005;118:4141–4151. doi: 10.1242/jcs.02537. [DOI] [PubMed] [Google Scholar]
- 151.Yu T. Jhun BS. Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal. 2011;14:425–437. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yu T. Robotham JL. Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yu T. Sheu SS. Robotham JL. Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79:341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang L. Seitz LC. Abramczyk AM. Chan C. Synergistic effect of cAMP and palmitate in promoting altered mitochondrial function and cell death in HepG2 cells. Exp Cell Res. 2010;316:716–727. doi: 10.1016/j.yexcr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhang Y. Chan DC. Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proc Natl Acad Sci U S A. 2007;104:18526–18530. doi: 10.1073/pnas.0706441104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang Y. Jiang L. Hu W. Zheng Q. Xiang W. Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by omega-3 fatty acid-induced up-regulation of mitofusin 2. Metabolism. 2011;60:767–775. doi: 10.1016/j.metabol.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 157.Zhang Y. Proenca R. Maffei M. Barone M. Leopold L. Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 158.Zhao J. Liu T. Jin S. Wang X. Qu M. Uhlen P. Tomilin N. Shupliakov O. Lendahl U. Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]